Rho GTPases as Key Molecular Players within Intestinal Mucosa and GI Diseases

Abstract
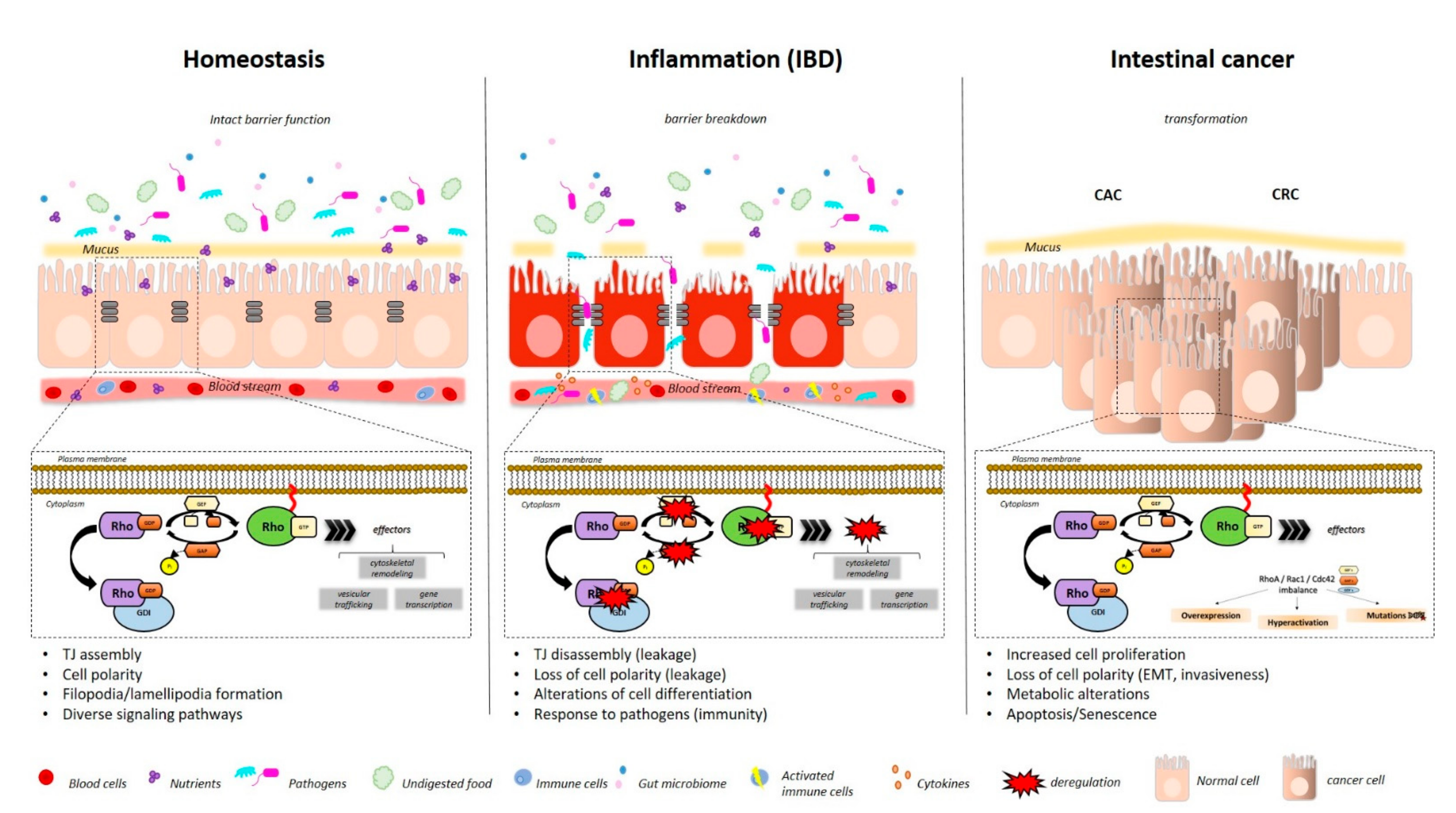
:1. Gut
1.1. Cellular Players within the Intestinal Mucosa
1.2. IBD
1.3. CRC
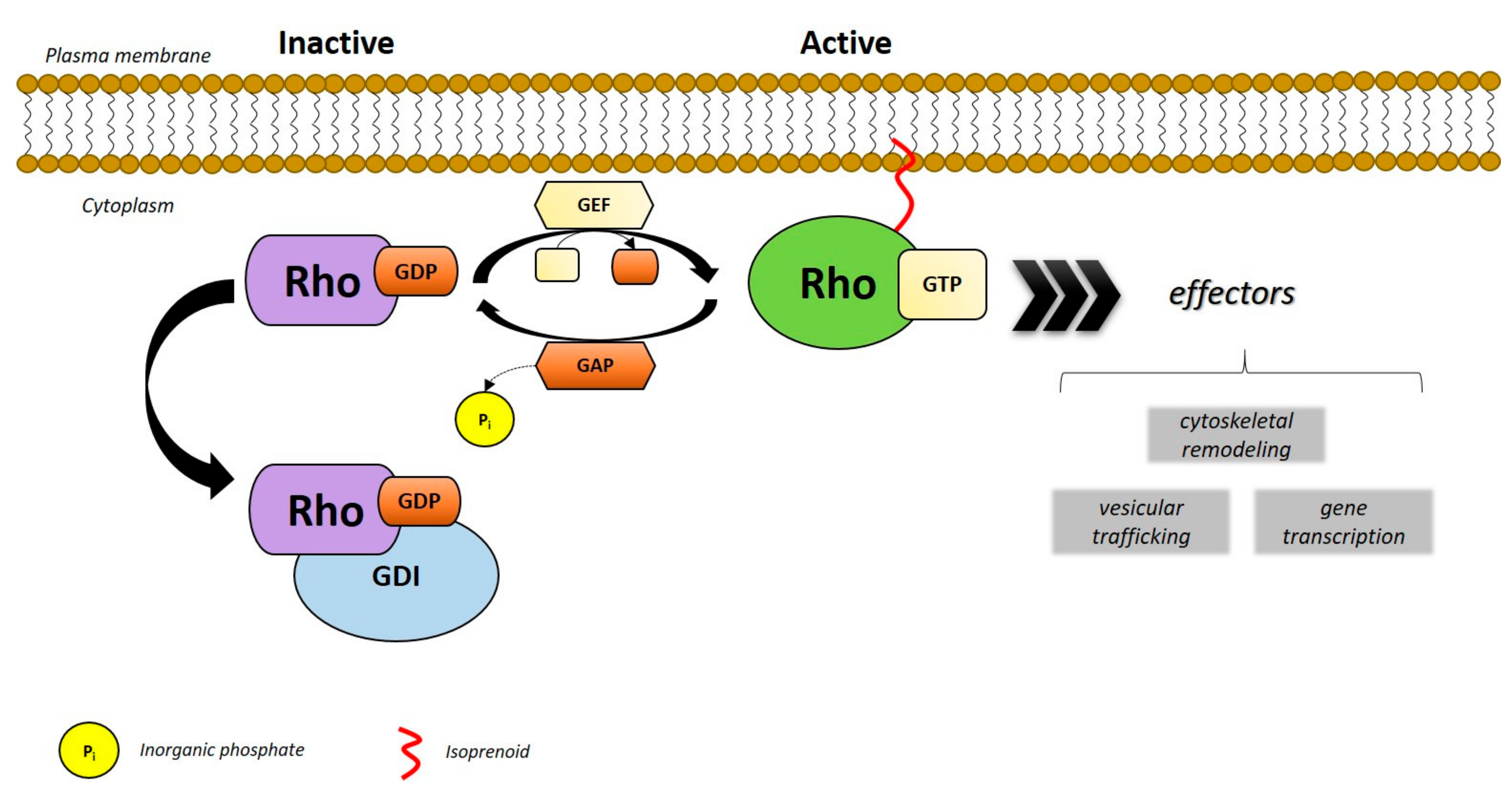
2. Rho GTPases
3. RHOA (Ras Homology Family Member A)
3.1. RHOA Function and Intestinal Inflammation
3.2. RHOA Function and Cancer
3.3. Targeting RHOA in the GI Tract
4. RAC1 (Ras-Related C3 Botulinum Toxin Substrate 1)
4.1. RAC1 and Intestinal Inflammation
4.2. RAC1 and Cancer
4.3. Targeting RAC1 in the GI Tract
5. CDC42 (Cell Division Control Protein 42)
5.1. CDC42 and Intestinal Inflammation
5.2. CDC42 and Cancer
5.3. Targeting CDC42 in the GI Tract
6. Other Rho GTPases
6.1. Inflammation
6.2. Cancer
7. Conclusive Remarks
Funding
Conflicts of Interest
References
- Weaver, L.T.; Austin, S.; Cole, T.J. Small Intestinal Length: A Factor Essential for Gut Adaptation. Gut 1991, 32, 1321–1323. [Google Scholar] [CrossRef]
- Phillips, S.F. Functions of the Large Bowel: An Overview. Scand. J. Gastroenterol. Suppl. 1984, 93, 1–12. [Google Scholar]
- Leppkes, M.; Siegmund, B.; Becker, C. Editorial: Immune-Epithelial Crosstalk in Inflammatory Bowel Diseases and Mucosal Wound Healing. Front. Immunol. 2018, 9, 1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newberry, R.D.; Gustafsson, J.K. Anatomy and Function of the Gut Immune System. In Encyclopedia of Immunobiology; Michael, J., Ratcliffe, H., Eds.; Academic Press: Oxford, UK, 2016; pp. 427–433. [Google Scholar]
- Hansson, G.C.; Johansson, M.E.V. The Inner of the Two Muc2 Mucin-Dependent Mucus Layers in Colon Is Devoid of Bacteria. Gut Microbes 2010, 1, 51–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokiyoshi, A.; Satchell, D.P.; Wilson, C.L.; Parks, W.C.; Selsted, M.E.; Ouellette, A.J. Secretion of Microbicidal A-Defensins by Intestinal Paneth Cells in Response to Bacteria. Nat. Immunol. 2000, 1, 113–118. [Google Scholar]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, N.; Clevers, H. Paneth Cells Constitute the Niche for Lgr5 Stem Cells in Intestinal Crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternini, C.; Anselmi, L.; Rozengurt, E. Enteroendocrine Cells: A Site of ‘Taste’ in Gastrointestinal Chemosensing. Curr. Opin. Endocrinol. Diabetes Obes. 2008, 15, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft Cells, Taste-Chemosensory Cells, Orchestrate Parasite Type 2 Immunity in the Gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V. Chapter 3—Epithelial Cell Contributions to Intestinal Immunity. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 129–172. [Google Scholar]
- Maloy, K.J.; Powrie, F. Intestinal Homeostasis and Its Breakdown in Inflammatory Bowel Disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Winther, K.V.; Jess, T.; Langholz, E.; Munkholm, P.; Binder, V. Survival and Cause-Specific Mortality in Ulcerative Colitis: Follow-up of a Population-Based Cohort in Copenhagen County. Gastroenterology 2003, 125, 1576–1582. [Google Scholar] [CrossRef]
- Neurath, M.F. Current and Emerging Therapeutic Targets for Ibd. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, S.; Nijhuis, A.; Kumagai, T.; Lindsay, J.; Silver, A. Defects in the Adherens Junction Complex (E-Cadherin/Β-Catenin) in Inflammatory Bowel Disease. Cell Tissue Res. 2014, 360, 749–760. [Google Scholar] [CrossRef] [PubMed]
- López-Posadas, R.; Stürzl, M.; Atreya, I.; Neurath, M.F.; Britzen-Laurent, N. Interplay of Gtpases and Cytoskeleton in Cellular Barrier Defects During Gut Inflammation. Front. Immunol. 2017, 8, 1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, S.; Nusrat, A. Dynamic Regulation of Epithelial Cell Fate and Barrier Function by Intercellular Junctions. Ann. N.Y. Acad. Sci. 2009, 1165, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Saoudi, A.; Kassem, S.; Dejean, A.S.; Gaud, G. Rho-Gtpases as Key Regulators of T Lymphocyte Biology. Small GTPases 2014, 5, e983862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; Born, M.V.D.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. Marincola. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.; Schmid, B.; Wirtz, S.; Thoma, O.-M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. Stat3 Activation through Il-6/Il-11 in Cancer-Associated Fibroblasts Promotes Colorectal Tumour Development and Correlates with Poor Prognosis. Gut 2019, 69, 1269–1282. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Janssen, K.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. Apc and Oncogenic Kras Are Synergistic in Enhancing Wnt Signaling in Intestinal Tumor Formation and Progression. Gastroenterology 2006, 131, 1096–1109. [Google Scholar] [CrossRef]
- Arends, J.W. Molecular Interactions in the Vogelstein Model of Colorectal Carcinoma. J. Pathol. 2000, 190, 412–416. [Google Scholar] [CrossRef]
- Pai, P.; Rachagani, S.; Dhawan, P.; Batra, S.K. Mucins and Wnt/Beta-Catenin Signaling in Gastrointestinal Cancers: An Unholy Nexus. Carcinogenesis 2016, 7, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodde, R.; Smits, R.; Clevers, H. Apc, Signal Transduction and Genetic Instability in Colorectal Cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- van der Kraak, L.; Gros, P.; Beauchemin, N. Colitis-Associated Colon Cancer: Is It in Your Genes? World J. Gastroenterol. 2015, 21, 11688–11699. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Rutter, M.D. Colorectal Cancer in Inflammatory Bowel Disease: What Is the Real Magnitude of the Risk? World J. Gastroenterol. 2012, 18, 3839–3848. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.; Martin-Zanca, D.; Reddy, E.P.; Pierotti, A.M.; Della-Porta, G.; Barbacid, M. Malignant Activation of a K-Ras Oncogene in Lung Carcinoma but Not in Normal Tissue of the Same Patient. Science 1984, 223, 661–664. [Google Scholar] [CrossRef]
- Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and Predictive Roles of Kras Mutation in Colorectal Cancer. Int. J. Mol. Sci. 2012, 13, 12153–12168. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.; Bos, J.L. Genetic Alterations During Colorectal-Tumor Development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, P.; Macaya, I.; Bazzocco, S.; Mazzolini, R.; Andretta, E.; Dopeso, H.; Mateo-Lozano, S.; Bilic, J.; Carton-Garcia, F.; Nieto, R.; et al. Rhoa Inactivation Enhances Wnt Signalling and Promotes Colorectal Cancer. Nat. Commun. 2014, 5, 5458. [Google Scholar] [CrossRef] [Green Version]
- Erik, S.; Marshall, C.J. Rho–Gtpases and Cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar]
- Fritz, G.; Just, I.; Kaina, B. Rho Gtpases Are over-expressed in Human Tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Vega, F.M.; Ridley, A.M. Rho Gtpases in Cancer Cell Biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho Family of Ras-Like Gtpases in Eukaryotes. Mol. Biol. Evol. 2006, 24, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Nobes, C.D.; Lauritzen, I.; Mattei, M.-G.; Paris, S.; Hall, A.; Chardin, P. A New Member of the Rho Family, Rnd1, Promotes Disassembly of Actin Filament Structures and Loss of Cell Adhesion. J. Cell Biol. 1998, 141, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Foster, R.; Hu, K.Q.; Lu, Y.; Nolan, K.M.; Thissen, J.; Settleman, J. Identification of a Novel Human Rho Protein with Unusual Properties: Gtpase Deficiency and in Vivo Farnesylation. Mol. Cell Biol. 1996, 16, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Madaule, P.; Axel, R. A novel ras-related gene family. Cell 1985, 41, 31–40. [Google Scholar] [CrossRef]
- Cannizzaro, L.A.; Madaule, P.; Hecht, F.; Axel, R.; Croce, C.M.; Huebner, K. Chromosome Localization of Human Arh Genes, a Ras-Related Gene Family. Genomics 1990, 6, 197–203. [Google Scholar] [CrossRef]
- Murphy, C.; Saffrich, R.; Grummt, M.; Gournier, H.; Rybin, V.; Rubino, M.; Auvinen, P.; Lütcke, A.; Parton, R.G.; Zerial, M. Endosome Dynamics Regulated by a Rho Protein. Nat. Cell Biol. 1996, 384, 427–432. [Google Scholar] [CrossRef]
- Ellis, S.; Mellor, H. The Novel Rho-Family Gtpase Rif Regulates Coordinated Actin-Based Membrane Rearrangements. Curr. Biol. 2000, 10, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Didsbury, J.; Weber, R.F.; Bokoch, G.M.; Evans, T.; Snyderman, R. Rac, a Novel Ras-Related Family of Proteins That Are Botulinum Toxin Substrates. J. Biol. Chem. 1989, 264, 16378–16382. [Google Scholar]
- Haataja, L.; Groffen, J.; Heisterkamp, N. Characterization of Rac3, a Novel Member of the Rho Family. J. Biol. Chem. 1997, 272, 20384–20388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, S.; Jeanteur, P.; Fort, P. Growth-Regulated Expression of Rhog, a New Member of the Ras Homolog Gene Family. Mol. Cell. Biol. 1992, 12, 3138–3148. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P.G.; Snyderman, R.; Evans, T. Characterization of G25k, a Gtp-Binding Protein Containing a Novel Putative Nucleotide Binding Domain. Biochem. Biophys. Res. Commun. 1989, 160, 25–32. [Google Scholar] [CrossRef]
- Vignal, E.; de Toledo, M.; Comunale, F.; Ladopoulou, A.; Gauthier-Rouviere, C.; Blangy, A.; Fort, P. Characterization of Tcl, a New Gtpase of the Rho Family Related to Tc10 Andccdc42. J. Biol. Chem. 2000, 275, 36457–36464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neudauer, C.L.; Joberty, G.; Tatsis, N.; Macara, I.G. Distinct Cellular Effects and Interactions of the Rho-Family Gtpase Tc10. Curr. Biol. 1998, 8, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Pennica, D.; Xu, L.; Kalejta, R.F.; Levine, A.J. Wrch-1, a Novel Member of the Rho Gene Family That Is Regulated by Wnt-1. Genes Dev. 2001, 15, 1796–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronheim, A.; Broder, Y.C.; Cohen, A.; Fritsch, A.; Belisle, B.; Abo, A. Chp, a Homologue of the Gtpase Cdc42hs, Activates the Jnk Pathway and Is Implicated in Reorganizing the Actin Cytoskeleton. Curr. Biol. 1998, 8, 1125–1128. [Google Scholar] [CrossRef] [Green Version]
- Dallery, E.; Galiegue-Zouitina, S.; Collyn-d’Hooghe, M.; Quief, S.; Denis, C.; Hildebrand, M.P.; Lantoine, D.; Deweindt, C.; Tilly, H.; Bastard, C. Ttf, a Gene Encoding a Novel Small G Protein, Fuses to the Lymphoma-Associated Laz3 Gene by T(3;4) Chromosomal Translocation. Oncogene 1995, 10, 2171–2178. [Google Scholar]
- Rivero, F.; Dislich, H.; Glockner, G.; Noegel, A.A. The Dictyostelium Discoideum Family of Rho-Related Proteins. Nucleic Acids Res. 2001, 29, 1068–1079. [Google Scholar] [CrossRef]
- Fort, P.; Blangy, A. The Evolutionary Landscape of Dbl-Like Rhogef Families: Adapting Eukaryotic Cells to Environmental Signals. Genome Biol. Evol. 2017, 9, 1471–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, E.; Jaiswal, M.; Derewenda, U.; Reis, K.; Nouri, K.; Koessmeier, K.T.; Aspenstrom, P.; Somlyo, A.V.; Dvorsky, R.; Ahmadian, M.R. Deciphering the Molecular and Functional Basis of Rhogap Family Proteins: A Systematic Approach toward Selective Inactivation of Rho Family Proteins. J. Biol. Chem. 2016, 291, 20353–20371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovas, A.; Couchman, J.R. Couchman. Rhogdi: Multiple Functions in the Regulation of Rho Family Gtpase Activities. Biochem. J. 2005, 390, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aspenstrom, P.; Ruusala, A.; Pacholsky, D. Taking Rho Gtpases to the Next Level: The Cellular Functions of Atypical Rho Gtpases. Exp. Cell Res. 2007, 313, 3673–3679. [Google Scholar] [CrossRef] [PubMed]
- Aspenstrom, P. Fast-Cycling Rho Gtpases. Small GTPases 2020, 11, 248–255. [Google Scholar] [CrossRef]
- Berthold, J.; Schenkova, K.; Ramos, S.; Miura, Y.; Furukawa, M.; Aspenstrom, P.; Rivero, F. Characterization of Rhobtb-Dependent Cul3 Ubiquitin Ligase Complexes—Evidence for an Autoregulatory Mechanism. Exp. Cell Res. 2008, 314, 3453–3465. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.F. Rho Gtpases, Their Post-Translational Modifications, Disease-Associated Mutations and Pharmacological Inhibitors. Small GTPases 2018, 9, 203–215. [Google Scholar] [CrossRef]
- Manolaridis, I.; Kulkarni, K.; Dodd, R.B.; Ogasawara, S.; Zhang, Z.; Bineva, G.; Reilly, N.O.; Hanrahan, S.J.; Thompson, A.J.; Cronin, N.; et al. Mechanism of Farnesylated Caax Protein Processing by the Intramembrane Protease Rce1. Nature 2013, 504, 301–305. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Casey, P.J. Post-Prenylation-Processing Enzymes as New Targets in Oncogenesis. Nat. Rev. Cancer 2005, 5, 405–412. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho Gtpases and Their Regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Zverina, E.A.; Lamphear, C.L.; Wright, E.N.; Fierke, C.A. Recent Advances in Protein Prenyltransferases: Substrate Identification, Regulation, and Disease Interventions. Curr. Opin. Chem. Biol. 2012, 16, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Garcia-Mata, R.; Burridge, K. Rho Protein Crosstalk: Another Social Network? Trends Cell Biol. 2011, 21, 718–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, Y.; Hartwig, J.H.; Stossel, T.P. Filgap, a Rho- and Rock-Regulated Gap for Rac Binds Filamin a to Control Actin Remodelling. Nat. Cell Biol. 2006, 8, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-Kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Rock and Dia Have Opposing Effects on Adherens Junctions Downstream of Rho. Nat. Cell Biol. 2002, 4, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.; Settleman, J. The Prk2 Kinase Is a Potential Effector Target of Both Rho and Rac Gtpases and Regulates Actin Cytoskeletal Organization. Mol. Cell. Biol. 1997, 17, 2247–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thumkeo, D.; Watanabe, S.; Narumiya, S. Physiological Roles of Rho and Rho Effectors in Mammals. Eur. J. Cell Biol. 2013, 92, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. Rhoa as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, N.; Meir, M.; Spindler, V.; Germer, C.-T.; Waschke, J. Differential Role of Rho Gtpases in Intestinal Epithelial Barrier Regulation in Vitro. J. Cell. Physiol. 2011, 226, 1196–1203. [Google Scholar] [CrossRef]
- Segain, J.-P.; De La Blétière, D.R.; Sauzeau, V.; Bourreille, A.; Hilaret, G.; Cario-Toumaniantz, C.; Pacaud, P.; Galmiche, J.-P.; Loirand, G. Rho Kinase Blockade Prevents Inflammation Via Nuclear FactorκB Inhibition: Evidence in Crohn’S Disease and Experimental Colitis. Gastroenterology 2003, 124, 1180–1187. [Google Scholar] [CrossRef]
- Hopkins, A.M.; Walsh, S.V.; Verkade, P.; Boquet, P.; Nusrat, A. Constitutive Activation of Rho Proteins by Cnf-1 Influences Tight Junction Structure and Epithelial Barrier Function. J. Cell Sci. 2003, 116 (Pt 4), 725–742. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Nie, M.; Matter, K.; Balda, M.S. Rho Signaling and Tight Junction Functions. Physiology 2010, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Slattum, G.; McGee, K.M.; Rosenblatt, J. P115 Rhogef and Microtubules Decide the Direction Apoptotic Cells Extrude from an Epithelium. J. Cell Biol. 2009, 186, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhoffer, G.T.; Loftus, P.D.; Yoshigi, M.; Otsuna, H.; Chien, C.-B.; Morcos, P.A.; Rosenblatt, J. Crowding Induces Live Cell Extrusion to Maintain Homeostatic Cell Numbers in Epithelia. Nature 2012, 484, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Grothaus, J.S.; Ares, G.; Yuan, C.; Wood, D.R.; Hunter, C.J. Rho Kinase Inhibition Maintains Intestinal and Vascular Barrier Function by Upregulation of Occludin in Experimental Necrotizing Enterocolitis. Am. J. Physiol. Gastrointest Liver Physiol. 2018, 315, G514–G528. [Google Scholar] [CrossRef] [PubMed]
- López-Posadas, R.; Becker, C.; Günther, C.; Tenzer, S.; Amann, K.; Billmeier, U.; Atreya, R.; Fiorino, G.; Vetrano, S.; Danese, S.; et al. Rho-a Prenylation and Signaling Link Epithelial Homeostasis to Intestinal Inflammation. J. Clin. Investig. 2016, 126, 611–626. [Google Scholar] [CrossRef] [Green Version]
- Shkoda, A.; Werner, T.; Daniel, H.; Gunckel, M.; Rogler, G.; Haller, D. Differential Protein Expression Profile in the Intestinal Epithelium from Patients with Inflammatory Bowel Disease. J. Proteome Res. 2007, 6, 1114–1125. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, H.; Kim, K.; Lee, H.; Lee, S.; Lee, D. Arhgap17, a Rhogtpase Activating Protein, Regulates Mucosal and Epithelial Barrier Function in the Mouse Colon. Sci. Rep. 2016, 6, 26923. [Google Scholar] [CrossRef] [Green Version]
- Vielkind, S.; Gallagher-Gambarelli, M.; Gomez, M.; Hinton, H.J.; Cantrell, D.A. Integrin Regulation by Rhoa in Thymocytes. J. Immunol. 2005, 175, 350–357. [Google Scholar] [CrossRef]
- Zhang, S.; Konstantinidis, D.G.; Yang, J.Q.; Mizukawa, B.; Kalim, K.; Lang, R.A.; Kalfa, T.A.; Zheng, Y.; Guo, F. Gene Targeting Rhoa Reveals Its Essential Role in Coordinating Mitochondrial Function and Thymocyte Development. J. Immunol. 2014, 193, 5973–5982. [Google Scholar] [CrossRef] [Green Version]
- Manresa-Arraut, A.; Johansen, F.F.; Brakebusch, C.; Issazadeh-Navikas, S.; Hasseldam, H. Rhoa Drives T-Cell Activation and Encephalitogenic Potential in an Animal Model of Multiple Sclerosis. Front. Immunol. 2018, 9, 1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Posadas, R.; Fastancz, P.; Martinez-Sanchez, L.D.C.; Panteleev-Ivlev, J.; Thonn, V.; Kisseleva, T.; Becker, L.S.; Schulz-Kuhnt, A.; Zundler, S.; Wirtz, S.; et al. Inhibiting Pggt1b Disrupts Function of Rhoa, Resulting in T-Cell Expression of Integrin Alpha4beta7 and Development of Colitis in Mice. Gastroenterology 2019, 157, 1293–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megrelis, L.; el Ghoul, E.; Moalli, F.; Versapuech, M.; Cassim, S.; Ruef, N.; Stein, J.V.; Mangeney, M.; Delon, J. Fam65b Phosphorylation Relieves Tonic Rhoa Inhibition During T Cell Migration. Front. Immunol. 2018, 9, 2001. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Park, S.; Kim, H.; Kim, C.-J.; Ahn, T.S.; Bae, S.B.; Kim, H.J.; Kim, T.H.; Im, J.; Lee, M.S.; et al. Rhoa Is Associated with Invasion and Poor Prognosis in Colorectal Cancer. Int. J. Oncol. 2015, 48, 714–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango, D.; Laiho, P.; Kokko, A.; Alhopuro, P.; Sammalkorpi, H.; Salovaara, R.; Nicorici, D.; Hautaniemi, S.; Alazzouzi, H.; Mecklin, J.P.; et al. Gene-Expression Profiling Predicts Recurrence in Dukes’ C Colorectal Cancer. Gastroenterology 2005, 129, 874–884. [Google Scholar] [CrossRef] [Green Version]
- Ruihua, H.; Mengyi, Z.; Chong, Z.; Meng, Q.; Xin, M.; Qiulin, T.; Feng, B.; Liu, M. Rhoa Regulates Resistance to Irinotecan by Regulating Membrane Transporter and Apoptosis Signaling in Colorectal Cancer. Oncotarget 2016, 7, 87136–87146. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhong, W.W.; Srivastava, N.; Slavin, A.; Yang, J.; Hoey, T.; An, S. G Protein-Coupled Lysophosphatidic Acid Receptors Stimulate Proliferation of Colon Cancer Cells through the {Beta}-Catenin Pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 6027–6032. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhao, G.; Liu, X.; Sui, A.; Yang, K.; Yao, R.; Wang, Z.; Shi, Q. Silencing of Rhoa and Rhoc Expression by Rna Interference Suppresses Human Colorectal Carcinoma Growth in vivo. J. Exp. Clin. Cancer Res. 2010, 29, 123. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, Z.; Sampson, L.; Zhou, X.; Nalapareddy, K.; Feng, Y.; Akunuru, S.; Melendez, J.; Davis, A.K.; Bi, F.; et al. Rhoa Gtpase Controls Yap-Mediated Ereg Signaling in Small Intestinal Stem Cell Maintenance. Stem Cell Rep. 2017, 9, 1961–1975. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.G.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.E.; et al. A Recurrent Inactivating Mutation in Rhoa Gtpase in Angioimmunoblastic T Cell Lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic Rhoa Mutation in Angioimmunoblastic T Cell Lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Brown, L.; Stevenson, K.; de Souza, T.; Aster, J.C.; Louissaint, A., Jr.; Weinstock, D.M. Rhoa G17v Is Sufficient to Induce Autoimmunity and Promotes T-Cell Lymphomagenesis in Mice. Blood 2018, 132, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shou, Z.; Fan, H.; Xu, M.; Chen, Q.; Tang, Q.; Liu, X.; Wu, H.; Zhang, M.; Yu, T.; et al. Protective Effects of Oxymatrine against Dss-Induced Acute Intestinal Inflammation in Mice Via Blocking the Rhoa/Rock Signaling Pathway. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.M.; Tian, S.Y.; Wang, D.; Cui, F.; Zhang, X.J.; Zhang, Y. Elevated Expression of the Leptin Receptor Obr May Contribute to Inflammation in Patients with Ulcerative Colitis. Mol. Med. Rep. 2019, 20, 4706–4712. [Google Scholar]
- Fang, K.; Law, I.K.M.; Padua, D.; Sideri, A.; Huang, V.; Kevil, C.G.; Iliopoulos, D.; Pothoulakis, C. Microrna-31-3p Is Involved in Substance P (Sp)-Associated Inflammation in Human Colonic Epithelial Cells and Experimental Colitis. Am. J. Pathol. 2018, 188, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Chen, Z.H.; Zheng, Z.; Liu, Y.; Zhao, Q.; Liu, Y.; Qiu, H.; Long, Q.; Chen, M.; Li, L.; et al. Investigation of the Role and Mechanism of Arhgap5-Mediated Colorectal Cancer Metastasis. Theranostics 2020, 10, 5998–6010. [Google Scholar] [CrossRef]
- Liu, L.; Xie, D.; Xie, H.; Huang, W.; Zhang, J.; Jin, W.; Jiang, W.; Xie, D. Arhgap10 Inhibits the Proliferation and Metastasis of Crc Cells Via Blocking the Activity of Rhoa/Akt Signaling Pathway. OncoTargets Ther. 2019, 12, 11507–11516. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Cui, R.; Huang, Y.; Luo, Y.; Qin, S.; Zhong, M. Increased Proton-Sensing Receptor Gpr4 Signalling Promotes Colorectal Cancer Progression by Activating the Hippo Pathway. EBioMedicine 2019, 48, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Yang, T.; Tang, D.; Zhou, F.; Qian, Y.; Zou, X. Increased Expression of Gef-H1 Promotes Colon Cancer Progression by Rhoa Signaling. Pathol. Res. Pract. 2019, 215, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, D.; Wang, X.; Sun, S.; Zhang, Y.; Wang, S.; Miao, R.; Xu, X.; Qu, X. Cxcl12/Cxcr4 Promotes Inflammation-Driven Colorectal Cancer Progression through Activation of Rhoa Signaling by Sponging Mir-133a-3p. J. Exp. Clin. Cancer Res. 2019, 38, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wu, Y.; Yang, M.; Wang, Z.; Zhang, H.; Jiang, X.; Chen, M.; Jin, T.; Wang, T. Irx5 Promotes Colorectal Cancer Metastasis by Negatively Regulating the Core Components of the Rhoa Pathway. Mol. Carcinog. 2019, 58, 2065–2076. [Google Scholar] [CrossRef]
- Murali, A.; Rajalingam, K. Small Rho Gtpases in the Control of Cell Shape and Mobility. Cell Mol. Life Sci. 2014, 71, 1703–1721. [Google Scholar] [CrossRef] [PubMed]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R.; et al. Cd28-Dependent Rac1 Activation Is the Molecular Target of Azathioprine in Primary Human Cd4+ T Lymphocytes. J. Clin. Investig. 2003, 111, 1133–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Xiong, J.; Wang, J.; Wen, J.; Zhi, F. Inhibition of Rac Family Protein Impairs Colitis and Colitis-Associated Cancer in Mice. Am. J. Cancer Res. 2018, 8, 70–80. [Google Scholar]
- Xiu, M.-X.; Liu, Y.-M.; Chen, G.-Y.; Hu, C.; Kuang, B. Identifying Hub Genes, Key Pathways and Immune Cell Infiltration Characteristics in Pediatric and Adult Ulcerative Colitis by Integrated Bioinformatic Analysis. Dig. Dis. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fattouh, R.; Guo, C.-H.; Lam, G.Y.; Gareau, M.G.; Ngan, B.-Y.; Glogauer, M.; Muise, A.M.; Brumell, J.H. Rac2-Deficiency Leads to Exacerbated and Protracted Colitis in Response to Citrobacter Rodentium Infection. PLoS ONE 2013, 8, e61629. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.N.; Liu, S.V.; Zou, T.; Liu, L.; Xiao, L.; Zhang, X.; Bellavance, E.; Yuan, J.X.; Wang, J.Y. Rac1 Promotes Intestinal Epithelial Restitution by Increasing Ca2+ Influx through Interaction with Phospholipase C-(Gamma)1 after Wounding. Am. J. Physiol. Cell Physiol. 2008, 295, C1499–C1509. [Google Scholar] [CrossRef] [Green Version]
- Myant, K.B.; Scopelliti, A.; Haque, S.; Vidal, M.; Sansom, O.J.; Cordero, J.B. Rac1 Drives Intestinal Stem Cell Proliferation and Regeneration. Cell Cycle 2013, 12, 2973–2977. [Google Scholar] [CrossRef] [Green Version]
- Stappenbeck, T.S.; Gordon, J.I. Rac1 Mutations Produce Aberrant Epithelial Differentiation in the Developing and Adult Mouse Small Intestine. Development 2000, 127, 2629–2642. [Google Scholar] [PubMed]
- Sumigray, K.D.; Terwilliger, M.; Lechler, T. Morphogenesis and Compartmentalization of the Intestinal Crypt. Dev. Cell 2018, 45, 183–197.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muise, A.M.; Walters, T.; Xu, W.; Shen-Tu, G.; Guo, C.H.; Fattouh, R.; Lam, G.Y.; Wolters, V.M.; Bennitz, J.; van Limbergen, J.; et al. Single Nucleotide Polymorphisms That Increase Expression of the Guanosine Triphosphatase Rac1 Are Associated with Ulcerative Colitis. Gastroenterology 2011, 141, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muise, A.M.; Xu, W.; Guo, C.H.; Walters, T.D.; Wolters, V.M.; Fattouh, R.; Lam, G.Y.; Hu, P.; Murchie, R.; Sherlock, M.; et al. Nadph Oxidase Complex and Ibd Candidate Gene Studies: Identification of a Rare Variant in Ncf2 That Results in Reduced Binding to Rac2. Gut 2012, 61, 1028–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seinen, M.L.; Amerongen, G.P.V.; de Boer, N.K.; van Bodegraven, A.A. Rac Attack: Modulation of the Small Gtpase Rac in Inflammatory Bowel Disease and Thiopurine Therapy. Mol. Diagn Ther. 2016, 20, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Koifman, E.; Karban, A.; Mazor, Y.; Chermesh, I.; Waterman, M.; Almog, R.; Ben-Horin, S.; Eliakim, R.; Krivoy, N.; Efrati, E.; et al. Thiopurine Effectiveness in Patients with Crohn’s Disease: A Study of Genetic and Clinical Predictive Factors. Inflamm. Bowel Dis. 2013, 19, 1639–1644. [Google Scholar] [CrossRef]
- Seinen, M.L.; Amerongen, G.P.V.; de Boer, N.K.; Mulder, C.J.; van Bezu, J.; van Bodegraven, A.A. Rac1 as a Potential Pharmacodynamic Biomarker for Thiopurine Therapy in Inflammatory Bowel Disease. Ther. Drug Monit. 2016, 38, 621–627. [Google Scholar] [CrossRef]
- De, P.; Peng, Q.; Traktuev, D.O.; Li, W.; Yoder, M.C.; March, K.L.; Durden, D.L. Expression of Rac2 in Endothelial Cells Is Required for the Postnatal Neovascular Response. Exp. Cell Res. 2009, 315, 248–263. [Google Scholar] [CrossRef] [Green Version]
- Pradip, D.; Peng, X.; Durden, D.L. Rac2 Specificity in Macrophage Integrin Signaling: Potential Role for Syk Kinase. J. Biol. Chem. 2003, 278, 41661–41669. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, M.J.; Ooi, S.K.; Reynolds, L.F.; Smith, S.H.; Ruf, S.; Mathiot, A.; Vanes, L.; Williams, D.A.; Cancro, M.P.; Tybulewicz, V.L. Critical Roles for Rac1 and Rac2 Gtpases in B Cell Development and Signaling. Science 2003, 302, 459–462. [Google Scholar] [CrossRef]
- Li, B.; Yu, H.; Zheng, W.; Voll, R.; Na, S.; Roberts, A.W.; Williams, D.A.; Davis, R.J.; Ghosh, S.; Flavell, R.A. Role of the Guanosine Triphosphatase Rac2 in T Helper 1 Cell Differentiation. Science 2000, 288, 2219–2222. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Leitenberg, D.; Li, B.; Flavell, R.A. Deficiency of Small Gtpase Rac2 Affects T Cell Activation. J. Exp. Med. 2001, 194, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croker, B.A.; Handman, E.; Hayball, J.D.; Baldwin, T.M.; Voigt, V.; Cluse, L.A.; Yang, F.C.; Williams, A.D.; Roberts, A.W. Rac2-Deficient Mice Display Perturbed T-Cell Distribution and Chemotaxis, but Only Minor Abnormalities in T(H)1 Responses. Immunol. Cell Biol. 2002, 80, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Dumont, C.; Corsoni-Tadrzak, A.; Ruf, S.; de Boer, J.; Williams, A.; Turner, M.; Kioussis, D.; Tybulewicz, V.L.J. Rac Gtpases Play Critical Roles in Early T-Cell Development. Blood 2009, 113, 3990–3998. [Google Scholar] [CrossRef]
- Khosravi-Far, R.; Solski, A.P.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of Rac1, Rhoa, and Mitogen-Activated Protein Kinases Is Required for Ras Transformation. Mol. Cell. Biol. 1995, 15, 6443–6453. [Google Scholar] [CrossRef] [Green Version]
- Kissil, A.L.; Walmsley, M.J.; Hanlon, L.; Haigis, K.M.; Kim, C.F.; Sweet-Cordero, A.; Eckman, M.S.; Tuveson, D.A.; Capobianco, A.J.; Tybulewicz, V.L.J.; et al. Requirement for Rac1 in a K-Ras Induced Lung Cancer in the Mouse. Cancer Res. 2007, 67, 8089–8094. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Pedersen, E.; Basse, A.; Lefever, T.W.; Peyrollier, K.; Kapoor, S.; Mei, Q.B.; Karlsson, R.; Chrostekgrashoff, A.; Brakebusch, C. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene 2010, 29, 3362–3373. [Google Scholar] [CrossRef] [Green Version]
- Kotelevets, L.; Chastre, E. Rac1 Signaling: From Intestinal Homeostasis to Colorectal Cancer Metastasis. Cancers 2020, 12, 665. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.M.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Sheth, K.R.; Clary, B.M. Management of Hepatic Metastases from Colorectal Cancer. Clin. Colon Rectal Surg. 2005, 18, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Guo, H.; Lu, Y.; Feng, W.; Sun, X.; Tang, C.; Wang, X.; Shen, M. Blocking hepatic metastases of colon cancer cells using an shRNA against Rac1 delivered by activatable cell-penetrating peptide. Oncotarget 2016, 7, 77183–77195. [Google Scholar] [CrossRef]
- Lou, S.; Wang, P.; Yang, J.; Ma, J.; Liu, C.; Zhou, M. Prognostic and Clinicopathological Value of Rac1 in Cancer Survival: Evidence from a Meta-Analysis. J. Cancer 2018, 9, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Lin, J.; Su, J.; Oyang, L.; Wang, H.; Tan, S.; Tang, Y.; Chen, X.; Liu, W.; Luo, X.; et al. Diallyl Disulfide Inhibits Colon Cancer Metastasis by Suppressing Rac1-Mediated Epithelial-Mesenchymal Transition. OncoTargets Ther. 2019, 12, 5713–5728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. Ros Production and Nf-Kappab Activation Triggered by Rac1 Facilitate Wnt-Driven Intestinal Stem Cell Proliferation and Colorectal Cancer Initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Goka, E.T.; Chaturvedi, P.; Mesa-Lopez, D.T.; de la Garza, A.; Lippman, M.E. Rac1b Overexpression Confers Resistance to Chemotherapy Treatment in Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotelevets, L.; Walker, F.; Mamadou, G.; Lehy, T.; Jordan, P.; Chastre, E. The Rac1 Splice Form Rac1b Favors Mouse Colonic Mucosa Regeneration and Contributes to Intestinal Cancer Progression. Oncogene 2018, 37, 6054–6068. [Google Scholar] [CrossRef]
- Mizukawa, B.; Wei, J.; Shrestha, M.; Wunderlich, M.; Chou, F.-S.; Griesinger, A.; Harris, C.E.; Kumar, A.R.; Zheng, Y.; Williams, D.A.; et al. Inhibition of Rac Gtpase Signaling and Downstream Prosurvival Bcl-2 Proteins as Combination Targeted Therapy in Mll-Af9 Leukemia. Blood 2011, 118, 5235–5245. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Singh, A.R.; Zulcic, M.; Bao, L.; Messer, K.; Ideker, T.; Dutkowski, J.; Durden, D.L. Rac2 Controls Tumor Growth, Metastasis and M1-M2 Macrophage Differentiation in Vivo. PLoS ONE 2014, 9, e95893. [Google Scholar] [CrossRef] [Green Version]
- Yusung, S.; McGovern, D.; Lin, L.; Hommes, D.; Lagishetty, V.; Braun, J. Nk Cells Are Biologic and Biochemical Targets of 6-Mercaptopurine in Crohn’s Disease Patients. Clin. Immunol. 2017, 175, 82–90. [Google Scholar] [CrossRef]
- Zheng, X.-B.; Liu, H.-S.; Zhang, L.-J.; Liu, X.-H.; Zhong, X.-L.; Zhou, C.; Hu, T.; Wu, X.-R.; Hu, J.-C.; Lian, L.; et al. Engulfment and Cell Motility Protein 1 Protects against Dss-Induced Colonic Injury in Mice Via Rac1 Activation. J. Crohns Coliti 2018, 13, 100–114. [Google Scholar] [CrossRef]
- Birkl, D.; Quiros, M.; Garcia-Hernandez, V.; Zhou, D.W.; Brazil, J.C.; Hilgarth, R.; Keeney, J.; Yulis, M.; Bruewer, M.; Garcia, A.J.; et al. Tnfalpha Promotes Mucosal Wound Repair through Enhanced Platelet Activating Factor Receptor Signaling in the Epithelium. Mucosal Immunol. 2019, 12, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.H.; Giri, R.; Davies, J.; Schreiber, V.; Alabbas, S.; Movva, R.; He, Y.; Wu, A.; Hooper, J.; McWhinney, B.; et al. A Nucleotide Analog Prevents Colitis-Associated Cancer Via Beta-Catenin Independently of Inflammation and Autophagy. Cell Mol. Gastroenterol. Hepatol. 2020, 11, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, X.; Duan, S.; Liu, X.; Chen, J.; Lan, J.; Liu, X.; Huang, W.; Geng, J.; Zhou, J. Csrp2 Suppresses Colorectal Cancer Progression Via P130cas/Rac1 Axis-Meditated Erk, Pak, and Hippo Signaling Pathways. Theranostics 2020, 10, 11063–11079. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, W.; Lu, T.; Zhou, J.; Ge, X.; Hua, D. Microrna-142-3p Promotes Cellular Invasion of Colorectal Cancer Cells by Activation of Rac1. Technol. Cancer Res. Treat. 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.H.; Lu, Y.X.; Zhang, Z.Y.; Zhang, J.M.; Zhang, W.J.; Zheng, L.; Lin, W.H.; Zhang, W.; Li, X.N. Ssh3 Facilitates Colorectal Cancer Cell Invasion and Metastasis by Affecting Signaling Cascades Involving Limk1/Rac1. Am. J. Cancer Res. 2019, 9, 1061–1073. [Google Scholar]
- Zhang, T.; Wang, Z.; Liu, Y.; Huo, Y.; Liu, H.; Xu, C.; Mao, R.; Zhu, Y.; Liu, L.; Wei, D.; et al. Plastin 1 Drives Metastasis of Colorectal Cancer through the Iqgap1/Rac1/Erk Pathway. Cancer Sci. 2020, 111, 2861–2871. [Google Scholar] [CrossRef]
- Ye, Y.P.; Jiao, H.L.; Wang, S.Y.; Xiao, Z.Y.; Zhang, D.; Qiu, J.F.; Zhang, L.J.; Zhao, Y.L.; Li, T.T.; Li, L.; et al. Hypermethylation of Dmtn Promotes the Metastasis of Colorectal Cancer Cells by Regulating the Actin Cytoskeleton through Rac1 Signaling Activation. J. Exp. Clin. Cancer Res. 2018, 37, 299. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Zhang, Z.; Chen, Q.; Lu, Y.; Zhang, J.; Lin, C.; Zhang, F.; Zhang, W.; Li, X.; Zhang, W.; et al. Irf1 Inhibits the Proliferation and Metastasis of Colorectal Cancer by Suppressing the Ras-Rac1 Pathway. Cancer Manag. Res. 2019, 11, 369–378. [Google Scholar] [CrossRef] [Green Version]
- An, N.; Liu, T.; Zhu, B.; Yang, Y.; Yan, X.; Cao, M.; Chen, Y.; Liu, R.; Xia, P.; Liu, C.; et al. A Bidirectional Effect of Rac1 Inhibition-Protects Radiation-Induced Intestinal Injury While Inhibits Tumor. Life Sci. 2020, 240, 117105. [Google Scholar] [CrossRef]
- Elias, B.C.; Elias, B.C.; Das, A.; Parekh, D.V.; Mernaugh, G.; Adams, R.; Yang, Z.; Brakebusch, C.; Pozzi, A.; Marciano, D.K.; et al. Cdc42 Regulates Epithelial Cell Polarity and Cytoskeletal Function During Kidney Tubule Development. J. Cell Sci. 2015, 128, 4293–4305. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Rohatgi, R.; Kirschner, M.W. The Arp2/3 Complex Mediates Actin Polymerization Induced by the Small Gtp-Binding Protein Cdc42. Proc. Natl. Acad. Sci. USA 1998, 95, 15362–15367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozma, R.; Ahmed, S.; Best, A.; Lim, L. The Ras-Related Protein Cdc42hs and Bradykinin Promote Formation of Peripheral Actin Microspikes and Filopodia in Swiss 3t3 Fibroblasts. Mol. Cell. Biol. 1995, 15, 1942–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, A.B.; Kaji, N.; Durgan, J.; Hall, A. Cdc42 Controls Spindle Orientation to Position the Apical Surface During Epithelial Morphogenesis. J. Cell Biol. 2008, 183, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.J.; Peng, K.Y.; Tang, Z.F.; Wang, Y.H.; Xue, A.J.; Huang, Y. Microrna-15a—Cell Division Cycle 42 Signaling Pathway in Pathogenesis of Pediatric Inflammatory Bowel Disease. World J. Gastroenterol. 2018, 24, 5234–5245. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Gu, H.; Zhao, X.; Huang, L.; Zhou, S.; Zhi, F. Involvement of Activated Cdc42 Kinase1 in Colitis and Colorectal Neoplasms. Med Sci. Monit. 2016, 22, 4794–4802. [Google Scholar] [CrossRef] [Green Version]
- Sakamori, R.; Das, S.; Yu, S.; Feng, S.; Stypulkowski, E.; Guan, Y.; Douard, Y.; Tang, W.; Ferraris, R.P.; Harada, A.; et al. Cdc42 and Rab8a Are Critical for Intestinal Stem Cell Division, Survival, and Differentiation in Mice. J. Clin. Investig. 2012, 122, 1052–1065. [Google Scholar] [CrossRef]
- Melendez, J.; Liu, M.; Sampson, L.; Akunuru, S.; Han, X.; Vallance, J.; Witte, D.; Shroyer, N.; Zheng, Y. Cdc42 Coordinates Proliferation, Polarity, Migration, and Differentiation of Small Intestinal Epithelial Cells in Mice. Gastroenterology 2013, 145, 808–819. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.M.; Chen, X.-W.; He, X.-X.; Jiang, X.-P.; Wu, F. Cell Division Cycle Protein 42 Regulates the Inflammatory Response in Mice Bearing Inflammatory Bowel Disease. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Chemin, K.; Bohineust, A.; Dogniaux, S.; Tourret, M.; Guégan, S.; Miró-Mur, F.; Hivroz, C. Cytokine Secretion by Cd4+ T Cells at the Immunological Synapse Requires Cdc42-Dependent Local Actin Remodeling but Not Microtubule Organizing Center Polarity. J. Immunol. 2012, 189, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Zhang, S.; Tripathi, P.; Mattner, J.; Phelan, J.; Sproles, A.; Mo, J.; Wills-Karp, M.; Grimes, H.L.; Hildeman, D.; et al. Distinct Roles of Cdc42 in Thymopoiesis and Effector and Memory T Cell Differentiation. LOS ONE 2011, 6, e18002. [Google Scholar] [CrossRef] [Green Version]
- Kalim, K.W.; Yang, J.Q.; Li, Y.; Meng, Y.; Zheng, Y.; Guo, F. Reciprocal Regulation of Glycolysis-Driven Th17 Pathogenicity and Regulatory T Cell Stability by Cdc42. J. Immunol. 2018, 200, 2313–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Bagrodia, S.; Cerione, R.; Manor, D. A Novel Cdc42hs Mutant Induces Cellular Transformation. Curr. Biol. 1997, 7, 794–797. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Bai, L.; Nan, Q.Z. Activation of Rho Gtpase Cdc42 Promotes Adhesion and Invasion in Colorectal Cancer Cells. Med. Sci. Monit. Basic Res. 2013, 19, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez Del Pulgar, T.; Valdés-Mora, F.; Bandrés, E.; Pérez-Palacios, R.; Espina, C.; Cejas, P.; García-Cabezas, M.A.; Nistal, M.; Casado, E.; González-Barón, M.; et al. Cdc42 Is Highly Expressed in Colorectal Adenocarcinoma and Downregulates Id4 through an Epigenetic Mechanism. Int. J. Oncol. 2018, 33, 185–193. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, Y.; Wang, C.; Li, X.; Cui, X.; Tu, S.; You, L.; Fu, J.; Chen, Z.; Hu, W.; et al. Pak5 Facilitates the Proliferation, Invasion and Migration in Colorectal Cancer Cells. Cancer Med. 2020, 9, 4777–4790. [Google Scholar] [CrossRef]
- Hamann, M.J.; Lubking, C.M.; Luchini, D.N.; Billadeau, D.D. Asef2 Functions as a Cdc42 Exchange Factor and Is Stimulated by the Release of an Autoinhibitory Module from a Concealed C-Terminal Activation Element. Mol. Cell Biol. 2007, 27, 1380–1393. [Google Scholar] [CrossRef] [Green Version]
- Mitin, N.; Betts, L.; Yohe, M.E.; Der, C.J.; Sondek, J.; Rossman, K.L. Release of Autoinhibition of Asef by Apc Leads to Cdc42 Activation and Tumor Suppression. Nat. Struct. Mol. Biol. 2007, 14, 814–823. [Google Scholar] [CrossRef]
- Sakamori, R.; Yu, S.; Zhang, X.; Hoffman, A.; Sun, J.; Das, S.; Vedula, P.; Li, G.; Fu, J.; Walker, F.; et al. Cdc42 Inhibition Suppresses Progression of Incipient Intestinal Tumors. Cancer Res. 2014, 74, 5480–5492. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ranftl, R.; Hooper, S.; Farrugia, A.J.; Moeendarbary, E.; Bruckbauer, A.; Batista, F.; Charras, G.; Sahai, E. Cdc42ep3/Borg2 and Septin Network Enables Mechano-Transduction and the Emergence of Cancer-Associated Fibroblasts. Cell Rep 2015, 13, 2699–2714. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, A.G.; Evans, R.; Kiaii, S.; Svensson, L.; Hogg, N.; Gribben, J.G. Chronic Lymphocytic Leukemia Cells Induce Defective Lfa-1-Directed T-Cell Motility by Altering Rho Gtpase Signaling That Is Reversible with Lenalidomide. Blood 2013, 121, 2704–2714. [Google Scholar] [CrossRef]
- Liu, L.; Zhuang, R.; Xiao, L.; Chung, H.K.; Luo, J.; Turner, D.J.; Rao, J.N.; Gorospe, M.; Wang, J.Y. Hur Enhances Early Restitution of the Intestinal Epithelium by Increasing Cdc42 Translation. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdés-Mora, F.; Locke, W.; Bandrés, E.; Gallego-Ortega, D.; Cejas, P.; García-Cabezas, M.Á.; Colino-Sanguino, Y.; Feliu, J.; Del Pulgar, T.G.; Lacal, J.C. Clinical Relevance of the Transcriptional Signature Regulated by Cdc42 in Colorectal Cancer. Oncotarget 2017, 8, 26755–26770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Chen, J.; Peng, M.; Duan, S.; Hu, Y.; Guo, D.; Geng, J.; Zhou, J. Potee Promotes Colorectal Carcinoma Progression Via Activating the Rac1/Cdc42 Pathway. Exp. Cell Res. 2020, 390, 111933. [Google Scholar] [CrossRef]
- Zhu, G.-F.; Xu, Y.-W.; Li, J.; Niu, H.-L.; Ma, W.-X.; Xu, J.; Zhou, P.-R.; Liu, X.; Ye, D.-L.; Liu, X.-R.; et al. Mir20a/106a-Wtx Axis Regulates Rhogdia/Cdc42 Signaling and Colon Cancer Progression. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Chen, Y.-R.; Liu, S.-S.; Ye, Y.-P.; Jiao, H.-L.; Wang, S.-Y.; Xiao, Z.-Y.; Wei, W.-T.; Qiu, J.-F.; Liang, L.; et al. Mir-384 Inhibits Human Colorectal Cancer Metastasis by Targeting Kras and Cdc42. Oncotarget 2016, 7, 84826–84838. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.L.; Guo, L.L.; Luo, Y.; Liu, G.L.; Lei, Y.; Jing, F.Y.; Zhang, Y.L.; Tong, G.H.; Jing, Z.L.; Shen, L.; et al. Cdc42 Subcellular Relocation in Response to Vegf/Nrp1 Engagement Is Associated with the Poor Prognosis of Colorectal Cancer. Cell Death Dis. 2020, 11, 171. [Google Scholar] [CrossRef]
- Loebel, D.A.; Studdert, J.B.; Power, M.; Radziewic, T.; Jones, V.; Coultas, L.; Jackson, Y.; Rao, R.S.; Steiner, K.; Fossat, N.; et al. Rhou Maintains the Epithelial Architecture and Facilitates Differentiation of the Foregut Endoderm. Development 2011, 138, 4511–4522. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. Microrna-21 Knockout Improve the Survival Rate in Dss Induced Fatal Colitis through Protecting against Inflammation and Tissue Injury. PLoS ONE 2013, 8, e66814. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ma, Y.; Shi, C.; Chen, H.; Zhang, H.; Chen, N.; Zhang, P.; Wang, F.; Yang, J.; Yang, J.; et al. Overexpression of Mir-21 in Patients with Ulcerative Colitis Impairs Intestinal Epithelial Barrier Function through Targeting the Rho Gtpase Rhob. Biochem. Biophys. Res. Commun. 2013, 434, 746–752. [Google Scholar] [CrossRef]
- Huang, M.; Prendergast, G.C. Rhob in Cancer Suppression. Histol. Histopathol. 2006, 21, 213–218. [Google Scholar]
- Liu, M.; Tang, Q.; Qiu, M.; Lang, N.; Li, M.; Zheng, Y.; Bi, F. Mir-21 Targets the Tumor Suppressor Rhob and Regulates Proliferation, Invasion and Apoptosis in Colorectal Cancer Cells. FEBS Lett. 2011, 585, 2998–3005. [Google Scholar] [CrossRef] [PubMed]
- Adnane, J.; Muro-Cacho, C.; Mathews, L.; Sebti, S.M.; Muñoz-Antonia, T. Suppression of rho B expression in invasive carcinoma from head and neck cancer patients. Clin. Cancer Res. 2002, 8, 2225–2232. [Google Scholar] [PubMed]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic Analysis of Metastasis Reveals an Essential Role for Rhoc. Nat. Cell Biol. 2000, 406, 532–535. [Google Scholar] [CrossRef] [PubMed]
- van Golen, K.L.; Wu, Z.F.; Qiao, X.T.; Bao, L.; Merajver, S.D. Rhoc Gtpase Overexpression Modulates Induction of Angiogenic Factors in Breast Cells. Neoplasia 2000, 2, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Chen, S.; Zhao, Y. The Role of Rhoc in Malignant Tumor Invasion, Metastasis and Targeted Therapy. Histol. Histopathol. 2018, 33, 255–260. [Google Scholar]
- Ha, Y.J.; Tak, K.H.; Kim, S.K.; Kim, C.W.; Lee, J.L.; Roh, S.A.; Cho, D.H.; Kim, S.Y.; Kim, Y.S.; Kim, J.C. Biological Characteristics and Clinical Significance of Itgb1 and Rhoc in Patients with Recurrent Colorectal Cancer. Anticancer Res. 2019, 39, 4853–4864. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Liu, X.P.; Liang, J.; Yang, K.; Sui, A.H.; Liu, Y.J. Expression of Rhoa and Rhoc in Colorectal Carcinoma and Its Relations with Clinicopathological Parameters. Clin. Chem. Lab. Med. 2009, 47, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Hong, D.; Lou, Z.; Tu, X.; Jin, L. Lupeol Inhibits Migration and Invasion of Colorectal Cancer Cells by Suppressing Rhoa-Rock1 Signaling Pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 2185–2196. [Google Scholar] [CrossRef]
- Zeng, Y.F.; Xiao, Y.S.; Liu, Y.; Luo, X.J.; Wen, L.D.; Liu, Q.; Chen, M. Formin-Like 3 Regulates Rhoc/Fak Pathway and Actin Assembly to Promote Cell Invasion in Colorectal Carcinoma. World J. Gastroenterol. 2018, 24, 3884–3897. [Google Scholar] [CrossRef]
- Bellovin, D.I.; Simpson, K.J.; Danilov, T.; Maynard, E.; Rimm, D.L.; Oettgen, P.; Mercurio, A.M. Reciprocal Regulation of Rhoa and Rhoc Characterizes the Emt and Identifies Rhoc as a Prognostic Marker of Colon Carcinoma. Oncogene 2006, 25, 6959–6967. [Google Scholar] [CrossRef] [Green Version]
- Han, S.W.; Kim, H.P.; Shin, J.Y.; Jeong, E.G.; Lee, W.C.; Kim, K.Y.; Park, S.Y.; Lee, D.W.; Won, J.K.; Jeong, S.Y.; et al. Rna Editing in Rhoq Promotes Invasion Potential in Colorectal Cancer. J. Exp. Med. 2014, 211, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.H.; Chang, S.C.; Wang, H.H.; Yang, S.H.; Lai, K.C.; Lee, T.C. Prognostic Values of Epdr1 Hypermethylation and Its Inhibitory Function on Tumor Invasion in Colorectal Cancer. Cancers 2018, 10, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaymi, C.; Vignal, E.; Crès, G.; Roux, P.; Blangy, A.; Raynaud, P.; Fort, P. The Atypical Rhou/Wrch1 Rho Gtpase Controls Cell Proliferation and Apoptosis in the Gut Epithelium. Biol. Cell 2019, 111, 121–141. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cluster | Subfamily Name | Name | Approved Name | Alternative Name(s)/Symbol(s) | Original Reference | |
|---|---|---|---|---|---|---|
| Rho Family | I | Rnd | RND1 | Rho family GTPase 1 | Rho6, ARHS, RHOS | Nobes, C.D. et al., 1998 [37] |
| RND2 | Rho family GTPase 2 | ARHN, Rho7, RhoN | ||||
| RND3 | Rho family GTPase 3 | ARHE, RhoE, Rho8 | Foster, R.K. et al., 1996 [38] | |||
| Rho | RHOA | ras homolog family member A | ARHA, ARH12, Rho12, RhoH12 | Madaule, P. et al., 1985 [39] | ||
| RHOB | ras homolog family member B | ARH6, ARHB, RhoH6, MST081 | Cannizzaro, L.A. et al., 1990 [40] | |||
| RHOC | ras homolog family member C | ARH9, ARHC, Rho9 | ||||
| RhoD/RhoF | RHOD | ras homolog family member D | RhoHP1,Rho, ARHD | Mrphy, C. et al., 1996 [41] | ||
| RHOF | ras homolog family member F, filopodia associated | ARHF, Rif, FLJ20247 | Ellis, S. et al., 2000 [42] | |||
| II | Rac1/RhoG | RAC1 | Rac family small GTPase 1 | TC-25, p21-Rac1, Rac-1 | Didsbury, J. et al., 1989 [43] | |
| RAC2 | Rac family small GTPase 2 | EN-7 | ||||
| RAC3 | Rac family small GTPase 3 | - | Haataja, L. et al., 1997 [44] | |||
| RHOG | ras homolog family member G | ARHG, MGC125835, MGC125836 | Vincent, S. et al., 1992 [45] | |||
| Cdc42/RhoJ/ RhoQ | CDC42 | cell division cycle 42 | G25K, CDC42Hs | Polakis, P.G. et al., 1989 [46] | ||
| RHOJ | ras homolog family member J | RASL7B, ARHJ, FLJ14445, TCL | Vignal, E. et al., 2000 [47] | |||
| RHOQ | ras homolog family member Q | RASL7A, ARHQ, TC10 | Neudauer, C.L. et al., 1998 [48] | |||
| RhoU/ RhoV | RHOU | ras homolog family member U | ARHU, WRCH-1, DJ646B12.2, FLJ10616, WRCH1, CDC42L1, hG28K, fJ646B12.2 | Tao, W. et al., 2001 [49] | ||
| RHOV | ras homolog family member V | ARHV, Chp, WRCH2 | Aronheim, A. et al., 1998 [50] | |||
| III | RhoH | RHOH | ras homolog family member H | ARHH, TTF | Dallery E. et al., 1995 [51] | |
| IV | RhoBTB | RHOBTB1 | Rho related BTB domain containing 1 | KIAA0740 | Rivero F. et al., 2001 [52] | |
| RHOBTB2 | Rho related BTB domain containing 2 | KIAA0717, DBC-2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pradhan, R.; Ngo, P.A.; Martínez-Sánchez, L.d.C.; Neurath, M.F.; López-Posadas, R. Rho GTPases as Key Molecular Players within Intestinal Mucosa and GI Diseases. Cells 2021, 10, 66. https://doi.org/10.3390/cells10010066
Pradhan R, Ngo PA, Martínez-Sánchez LdC, Neurath MF, López-Posadas R. Rho GTPases as Key Molecular Players within Intestinal Mucosa and GI Diseases. Cells. 2021; 10(1):66. https://doi.org/10.3390/cells10010066
Chicago/Turabian StylePradhan, Rashmita, Phuong A. Ngo, Luz d. C. Martínez-Sánchez, Markus F. Neurath, and Rocío López-Posadas. 2021. "Rho GTPases as Key Molecular Players within Intestinal Mucosa and GI Diseases" Cells 10, no. 1: 66. https://doi.org/10.3390/cells10010066
APA StylePradhan, R., Ngo, P. A., Martínez-Sánchez, L. d. C., Neurath, M. F., & López-Posadas, R. (2021). Rho GTPases as Key Molecular Players within Intestinal Mucosa and GI Diseases. Cells, 10(1), 66. https://doi.org/10.3390/cells10010066