
An Inducible System for Silencing Establishment Reveals a Stepwise Mechanism in Which Anchoring at the Nuclear Periphery Precedes Heterochromatin Formation

Abstract
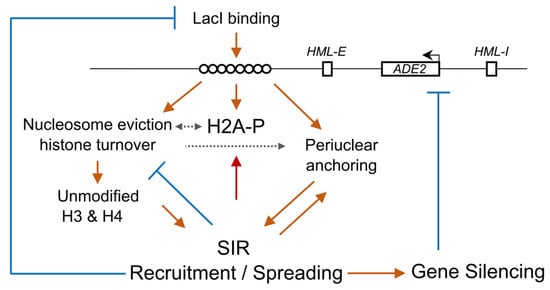
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Media and Growth Conditions
2.2. Strains
2.3. Plasmids
2.4. ChIP and Quantitative PCR Analysis
2.5. Quantitative Transcript Analysis
2.6. Microscopy
2.7. Image Processing and Analysis
2.8. Spot Localization
2.9. Intensity Quantifications
2.10. Statistical Analysis
3. Results
3.1. An Inducible and Reversible System to Establish Gene Silencing at an Ectopic Locus
3.2. Perinuclear Anchoring Occurs independently of the Cell Cycle and Precedes Gene Silencing Establishment
3.3. Sir3 and Sir4 Accumulate over Several Cell Cycles following LacI Induction
3.4. Perinuclear Anchoring Is Partly Sir3 Independent
3.5. LacI Binding Induces a Mec1 Dependent H2A-P
3.6. H2A-P Induced by LacI Binding Is Sir3 Independent but Is Stabilized by Sir3 Spreading
3.7. Sir3 Spreading Counteracts LacI-Induced Nucleosome Eviction and Limits LacI Binding
4. Discussion
4.1. LacI Binding Induces H2A Phosphorylation and SIR Recruitment Independently
4.2. Silencing Establishment Occurs over Several Cell Cycle
4.3. Anchoring at the Nuclear Periphery Precedes Silencing Establishment
4.4. Possible Mechanisms for Silencing Establishment
4.5. SIR Spreading Stabilizes Nucleosome and Limits the Access to DNA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meister, P.; Taddei, A. Building Silent Compartments at the Nuclear Periphery: A Recurrent Theme. Curr. Opin. Genet. Dev. 2013, 23, 96–103. [Google Scholar] [CrossRef]
- Bizhanova, A.; Kaufman, P.D. Close to the Edge: Heterochromatin at the Nucleolar and Nuclear Peripheries. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2021, 1864, 194666. [Google Scholar] [CrossRef] [PubMed]
- Gartenberg, M.R.; Smith, J.S. The Nuts and Bolts of Transcriptionally Silent Chromatin in Saccharomyces cerevisiae. Genetics 2016, 203, 1563–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maison, C.; Almouzni, G. HP1 and the Dynamics of Heterochromatin Maintenance. Nat. Rev. Mol. Cell. Biol. 2004, 5, 296–305. [Google Scholar] [CrossRef]
- Hocher, A.; Ruault, M.; Kaferle, P.; Descrimes, M.; Garnier, M.; Morillon, A.; Taddei, A. Expanding Heterochromatin Reveals Discrete Subtelomeric Domains Delimited by Chromatin Landscape Transitions. Genome Res. 2018, 28, 1867–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Chen, H.; Bai, L. Systematic Study of Nucleosome-Displacing Factors in Budding Yeast. Mol. Cell 2018, 71, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Straight, A.F.; Shou, W.; Dowd, G.J.; Turck, C.W.; Deshaies, R.J.; Johnson, A.D.; Moazed, D. Net1, a Sir2-Associated Nucleolar Protein Required for RDNA Silencing and Nucleolar Integrity. Cell 1999, 97, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Radman-Livaja, M.; Ruben, G.; Weiner, A.; Friedman, N.; Kamakaka, R.; Rando, O.J. Dynamics of Sir3 Spreading in Budding Yeast: Secondary Recruitment Sites and Euchromatic Localization: Dynamics of Sir3 Spreadingin Budding Yeast. EMBO J. 2011, 30, 1012–1026. [Google Scholar] [CrossRef] [Green Version]
- Ruault, M.; Scolari, V.F.; Lazar-Stefanita, L.; Hocher, A.; Loïodice, I.; Koszul, R.; Taddei, A. Sir3 Mediates Long-Range Chromosome Interactions in Budding Yeast. Genome Res. 2021, 31, 411–425. [Google Scholar] [CrossRef]
- Sperling, A.S.; Grunstein, M. Histone H3 N-Terminus Regulates Higher Order Structure of Yeast Heterochromatin. Proc. Natl. Acad. Sci. USA 2009, 106, 13153–13159. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.-H.; Schulze, J.M.; Jackson, J.; Hentrich, T.; Seidel, C.; Jaspersen, S.L.; Kobor, M.S.; Shilatifard, A. Dot1 and Histone H3K79 Methylation in Natural Telomeric and HM Silencing. Mol. Cell 2011, 42, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Mitsumori, R.; Ohashi, T.; Kugou, K.; Ichino, A.; Taniguchi, K.; Ohta, K.; Uchida, H.; Oki, M. Analysis of Novel Sir3 Binding Regions in Saccharomyces cerevisiae. J. Biochem. 2016, 160, 11–17. [Google Scholar] [CrossRef]
- Buck, S.W.; Shore, D. Action of a RAP1 Carboxy-Terminal Silencing Domain Reveals an Underlying Competition between HMR and Telomeres in Yeast. Genes Dev. 1995, 9, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.S.; Brachmann, C.B.; Pillus, L.; Boeke, J.D. Distribution of a Limited Sir2 Protein Pool Regulates the Strength of Yeast RDNA Silencing and Is Modulated by Sir4p. Genetics 1998, 149, 1205–1219. [Google Scholar] [CrossRef]
- Michel, A.H. Spontaneous RDNA Copy Number Variation Modulates Sir2 Levels and Epigenetic Gene Silencing. Genes Dev. 2005, 19, 1199–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, A.; Gasser, S.M. Structure and Function in the Budding Yeast Nucleus. Genetics 2012, 192, 107–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubarry, M.; Loiodice, I.; Chen, C.L.; Thermes, C.; Taddei, A. Tight Protein-DNA Interactions Favor Gene Silencing. Genes Dev. 2011, 25, 1365–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillet, L.; Gaden, F.; Brevet, V.; Fourel, G.; Martin, S.G.; Dubrana, K.; Gasser, S.M.; Gilson, E. Ku-deficient Yeast Strains Exhibit Alternative States of Silencing Competence. EMBO Rep. 2001, 2, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Marcand, S.; Buck, S.W.; Moretti, P.; Gilson, E.; Shore, D. Silencing of Genes at Nontelomeric Sites in Yeast Is Controlled by Sequestration of Silencing Factors at Telomeres by Rap 1 Protein. Genes Dev. 1996, 10, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Maillet, L.; Boscheron, C.; Gotta, M.; Marcand, S.; Gilson, E.; Gasser, S.M. Evidence for Silencing Compartments within the Yeast Nucleus: A Role for Telomere Proximity and Sir Protein Concentration in Silencer-Mediated Repression. Genes Dev. 1996, 10, 1796–1811. [Google Scholar] [CrossRef] [Green Version]
- Taddei, A.; Van Houwe, G.; Nagai, S.; Erb, I.; van Nimwegen, E.; Gasser, S.M. The Functional Importance of Telomere Clustering: Global Changes in Gene Expression Result from SIR Factor Dispersion. Genome Res. 2009, 19, 611–625. [Google Scholar] [CrossRef] [Green Version]
- Andrulis, E.D.; Neiman, A.M.; Zappulla, D.C.; Sternglanz, R. Perinuclear Localization of Chromatin Facilitates Transcriptional Silencing. Nature 1998, 394, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Katan-Khaykovich, Y.; Struhl, K. Heterochromatin Formation Involves Changes in Histone Modifications over Multiple Cell Generations. EMBO J. 2005, 24, 2138–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, E.A.; Dudoit, S.; Rine, J. The Establishment of Gene Silencing at Single-Cell Resolution. Nat. Genet. 2009, 41, 800–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, E.A.; Hiraoka, Y.; Rine, J. Symmetry, Asymmetry, and Kinetics of Silencing Establishment in Saccharomyces cerevisiae Revealed by Single-Cell Optical Assays. Proc. Natl. Acad. Sci. USA 2011, 108, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.M.; Nasmyth, K.A. Role of DNA Replication in the Repression of Silent Mating Type Loci in Yeast. Nature 1984, 312, 247–251. [Google Scholar] [CrossRef]
- Lau, A. Cell-Cycle Control of the Establishment of Mating-Type Silencing in S. cerevisiae. Genes Dev. 2002, 16, 2935–2945. [Google Scholar] [CrossRef] [Green Version]
- Martins-Taylor, K.; Sharma, U.; Rozario, T.; Holmes, S.G. H2A.Z (Htz1) Controls the Cell-Cycle-Dependent Establishment of Transcriptional Silencing at Saccharomyces cerevisiae Telomeres. Genetics 2011, 187, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Martins-Taylor, K.; Dula, M.L.; Holmes, S.G. Heterochromatin Spreading at Yeast Telomeres Occurs in M Phase. Genetics 2004, 168, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wang, C.-L.; Sternglanz, R. Promoter Strength Influences the S Phase Requirement for Establishment of Silencing at the Saccharomyces cerevisiae Silent Mating Type Loci. Genetics 2010, 186, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, A.G.; Holmes, S.G. A Cis -Acting TRNA Gene Imposes the Cell Cycle Progression Requirement for Establishing Silencing at the HMR Locus in Yeast. Genetics 2011, 187, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Goodnight, D.; Rine, J. S-Phase-Independent Silencing Establishment in Saccharomyces cerevisiae. eLife 2020, 9, e58910. [Google Scholar] [CrossRef]
- Thomas, B.J.; Rothstein, R. Elevated Recombination Rates in Transcriptionally Active DNA. Cell 1989, 56, 619–630. [Google Scholar] [CrossRef]
- Winston, F.; Dollard, C.; Ricupero-Hovasse, S.L. Construction of a Set of Convenient Saccharomyces cerevisiae Strains That Are Isogenic to S288C. Yeast 1995, 11, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Longtine, M.S.; Iii, A.M.; Demarini, D.J.; Shah, N.G.; Wach, A.; Brachat, A.; Philippsen, P.; Pringle, J.R. Additional Modules for Versatile and Economical PCR-Based Gene Deletion and Modification in Saccharomyces cerevisiae. Yeast 1998, 14, 953–961. [Google Scholar] [CrossRef]
- Janke, C.; Magiera, M.M.; Rathfelder, N.; Taxis, C.; Reber, S.; Maekawa, H.; Moreno-Borchart, A.; Doenges, G.; Schwob, E.; Schiebel, E.; et al. A Versatile Toolbox for PCR-Based Tagging of Yeast Genes: New Fluorescent Proteins, More Markers and Promoter Substitution Cassettes. Yeast 2004, 21, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Nistér, D.; Stewénius, H. Linear Time Maximally Stable Extremal Regions. In Proceedings of the Computer Vision—ECCV 2008, Marseille, France, 12–18, October 2008; Forsyth, D., Torr, P., Zisserman, A., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2008; Volume 5303, pp. 183–196. ISBN 978-3-540-88685-3. [Google Scholar]
- Kruskal, W.H. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 40. [Google Scholar] [CrossRef]
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, E.; Blow, J.J.; Tanaka, T.U. Live-Cell Imaging Reveals Replication of Individual Replicons in Eukaryotic Replication Factories. Cell 2006, 125, 1297–1308. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, C.L.; Storey, J.D.; Tibshirani, R.J.; Herschlag, D.; Brown, P.O. Precision and Functional Specificity in MRNA Decay. Proc. Natl. Acad. Sci. USA 2002, 99, 5860–5865. [Google Scholar] [CrossRef] [Green Version]
- Grigull, J.; Mnaimneh, S.; Pootoolal, J.; Robinson, M.D.; Hughes, T.R. Genome-Wide Analysis of MRNA Stability Using Transcription Inhibitors and Microarrays Reveals Posttranscriptional Control of Ribosome Biogenesis Factors. Mol. Cell. Biol. 2004, 24, 5534–5547. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.; Schwalb, B.; Maier, K.; Schulz, D.; Dümcke, S.; Zacher, B.; Mayer, A.; Sydow, J.; Marcinowski, L.; Dölken, L.; et al. Dynamic Transcriptome Analysis Measures Rates of MRNA Synthesis and Decay in Yeast. Mol. Syst. Biol. 2011, 7, 458. [Google Scholar] [CrossRef] [PubMed]
- Geisberg, J.V.; Moqtaderi, Z.; Fan, X.; Ozsolak, F.; Struhl, K. Global Analysis of MRNA Isoform Half-Lives Reveals Stabilizing and Destabilizing Elements in Yeast. Cell 2014, 156, 812–824. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Schleker, T.; Sperling, A.S.; Xie, W.; Gasser, S.M.; Grunstein, M. ΓH2A Is a Component of Yeast Heterochromatin Required for Telomere Elongation. Cell. Cycle 2011, 10, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Szilard, R.K.; Jacques, P.-É.; Laramée, L.; Cheng, B.; Galicia, S.; Bataille, A.R.; Yeung, M.; Mendez, M.; Bergeron, M.; Robert, F.; et al. Systematic Identification of Fragile Sites via Genome-Wide Location Analysis of γ-H2AX. Nat. Struct. Mol. Biol. 2010, 17, 299–305. [Google Scholar] [CrossRef]
- Colombo, C.V.; Gnugnoli, M.; Gobbini, E.; Longhese, M.P. How Do Cells Sense DNA Lesions? Biochem. Soc. Trans. 2020, 48, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, A.; Weiss, K.; Simpson, R.T. High-Resolution Structural Analysis of Chromatin at Specific Loci: Saccharomyces cerevisiae Silent Mating-Type Locus HMRa. Mol. Cell. Biol. 1999, 19, 7944–7950. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Simpson, R.T. High-Resolution Structural Analysis of Chromatin at Specific Loci: Saccharomyces cerevisiae Silent Mating Type Locus HMLα. Mol. Cell. Biol. 1998, 18, 5392–5403. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Yu, Q.; Bi, X. Asymmetric Positioning of Nucleosomes and Directional Establishment of Transcriptionally Silent Chromatin by Saccharomyces cerevisiae Silencers. Mol. Cell. Biol. 2006, 26, 7806–7819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.G.; Peterson, M.R.; Still, C.D.; Brueggeman, L.; Dhillon, N.; Kamakaka, R.T. Heterochromatin Formation via Recruitment of DNA Repair Proteins. MBoC 2015, 26, 1395–1410. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Kuryan, B.G.; Tran, N.N.H.; Song, C.; Xue, Y.; Carey, M.; Grunstein, M. Mechanism for Epigenetic Variegation of Gene Expression at Yeast Telomeric Heterochromatin. Genes Dev. 2012, 26, 2443–2455. [Google Scholar] [CrossRef] [Green Version]
- Saxton, D.S.; Rine, J. Nucleosome Positioning Regulates the Establishment, Stability, and Inheritance of Heterochromatin in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2020, 117, 27493–27501. [Google Scholar] [CrossRef]
- Garcia, J.F.; Dumesic, P.A.; Hartley, P.D.; El-Samad, H.; Madhani, H.D. Combinatorial, Site-Specific Requirement for Heterochromatic Silencing Factors in the Elimination of Nucleosome-Free Regions. Genes Dev. 2010, 24, 1758–1771. [Google Scholar] [CrossRef] [Green Version]
- Emden, T.S.; Forn, M.; Forné, I.; Sarkadi, Z.; Capella, M.; Martín Caballero, L.; Fischer-Burkart, S.; Brönner, C.; Simonetta, M.; Toczyski, D.; et al. Shelterin and Subtelomeric DNA Sequences Control Nucleosome Maintenance and Genome Stability. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Swygert, S.G.; Senapati, S.; Bolukbasi, M.F.; Wolfe, S.A.; Lindsay, S.; Peterson, C.L. SIR Proteins Create Compact Heterochromatin Fibers. Proc. Natl. Acad. Sci. USA 2018, 115, 12447–12452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kueng, S.; Oppikofer, M.; Gasser, S.M. SIR Proteins and the Assembly of Silent Chromatin in Budding Yeast. Annu. Rev. Genet. 2013, 47, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Ruault, M.; De Meyer, A.; Loïodice, I.; Taddei, A. Clustering heterochromatin: Sir3 promotes telomere clustering independently of silencing in yeast. J. Cell Biol. 2011, 192, 417–431. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, H.; Hawke, D.; Kobayashi, R.; Verreault, A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 2005, 436, 294–298. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loïodice, I.; Garnier, M.; Nikolov, I.; Taddei, A. An Inducible System for Silencing Establishment Reveals a Stepwise Mechanism in Which Anchoring at the Nuclear Periphery Precedes Heterochromatin Formation. Cells 2021, 10, 2810. https://doi.org/10.3390/cells10112810
Loïodice I, Garnier M, Nikolov I, Taddei A. An Inducible System for Silencing Establishment Reveals a Stepwise Mechanism in Which Anchoring at the Nuclear Periphery Precedes Heterochromatin Formation. Cells. 2021; 10(11):2810. https://doi.org/10.3390/cells10112810
Chicago/Turabian StyleLoïodice, Isabelle, Mickael Garnier, Ivaylo Nikolov, and Angela Taddei. 2021. "An Inducible System for Silencing Establishment Reveals a Stepwise Mechanism in Which Anchoring at the Nuclear Periphery Precedes Heterochromatin Formation" Cells 10, no. 11: 2810. https://doi.org/10.3390/cells10112810
APA StyleLoïodice, I., Garnier, M., Nikolov, I., & Taddei, A. (2021). An Inducible System for Silencing Establishment Reveals a Stepwise Mechanism in Which Anchoring at the Nuclear Periphery Precedes Heterochromatin Formation. Cells, 10(11), 2810. https://doi.org/10.3390/cells10112810