
Somatic Reprogramming—Above and Beyond Pluripotency


Abstract
:
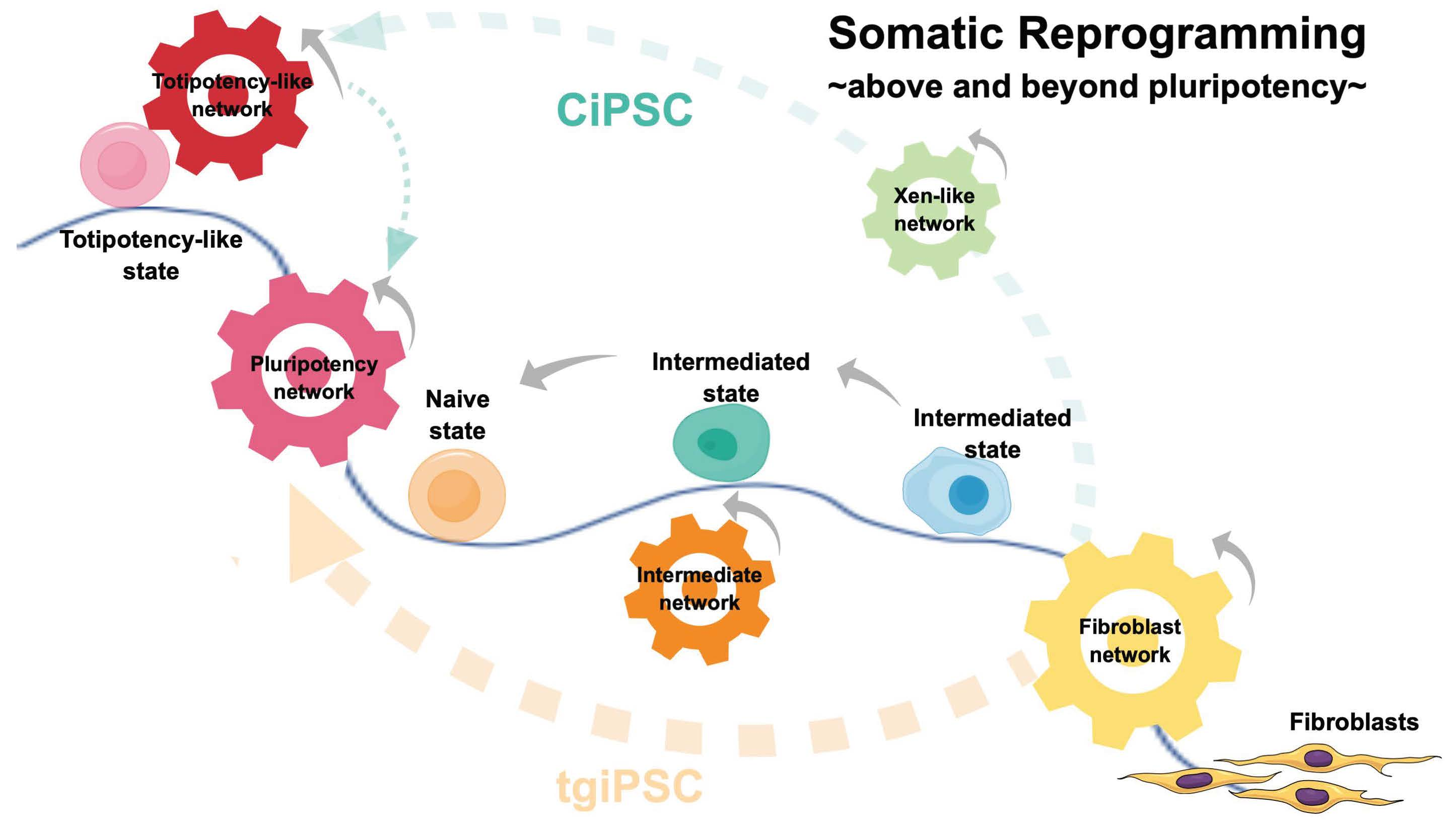{kind=link}
{kind=link}
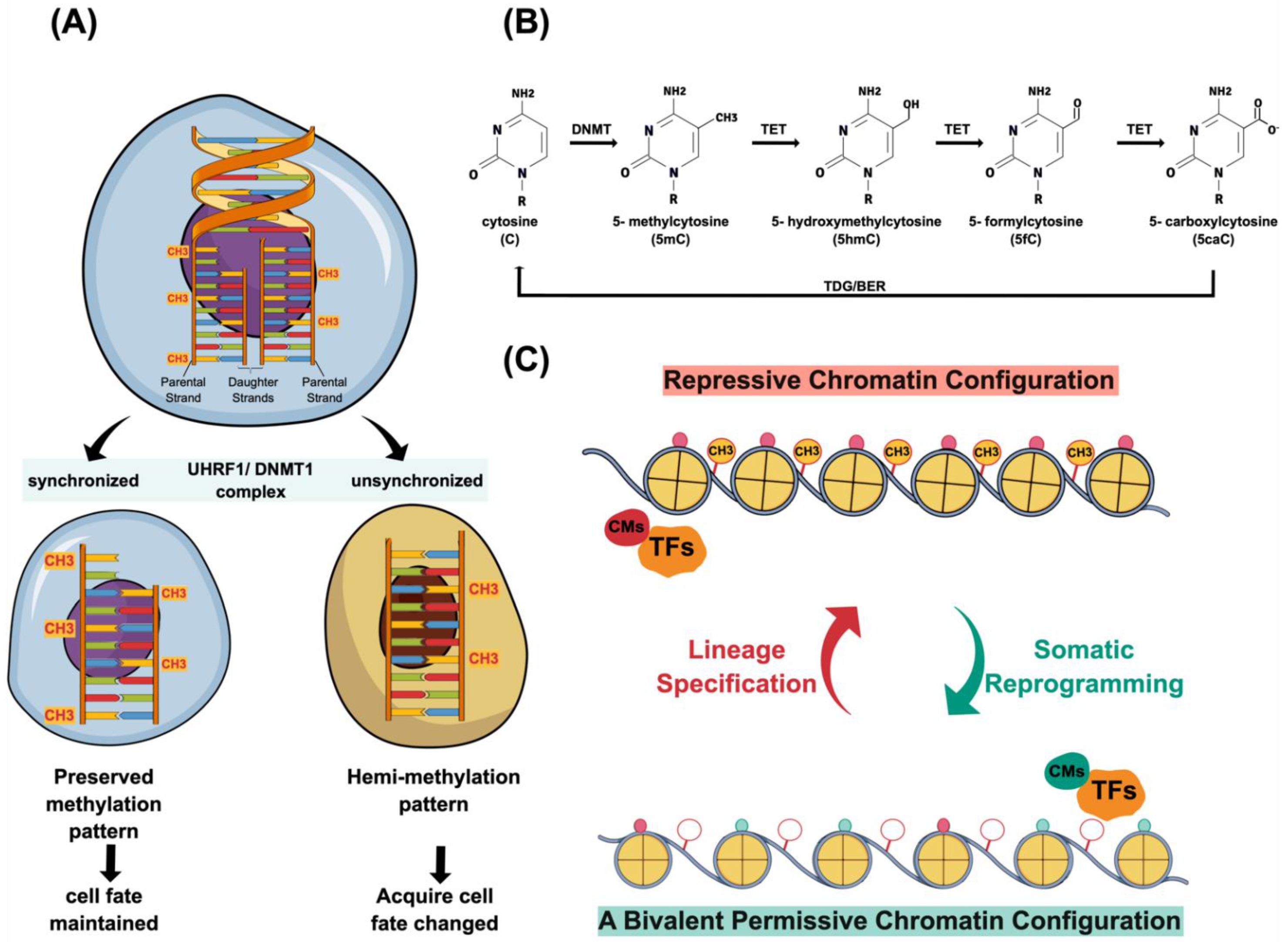{kind=link}
{kind=link}
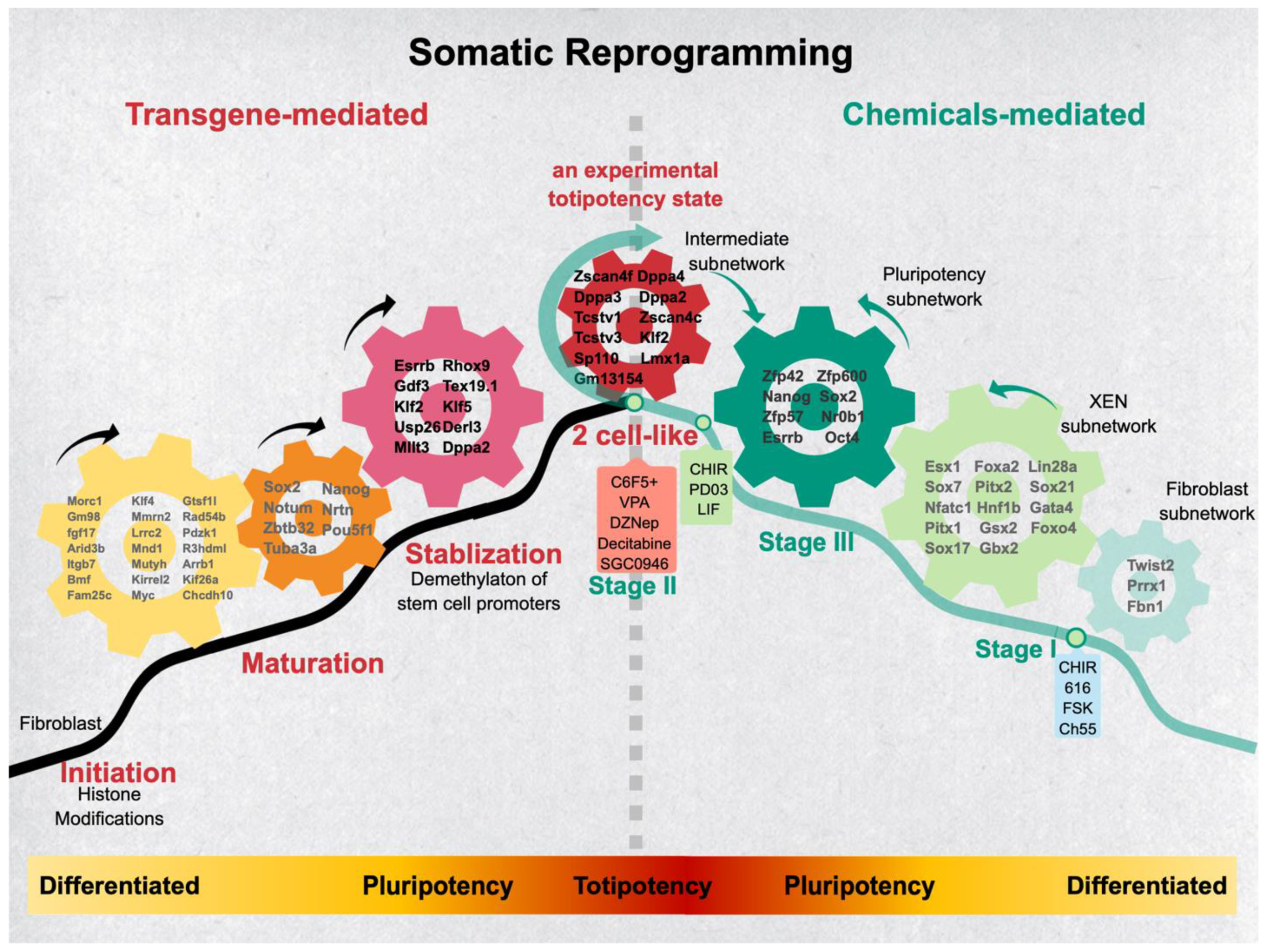{kind=link}
1. Introduction
2. Genome Plasticity Endows Cell Fate Change
2.1. DNA Methylation
2.2. Chromatin Modifications
2.3. Transcriptional Network Formation
2.4. A Two-Way Relationship between Transcription Factor and Chromatin Structure
2.5. Diverse Mechanisms Coordinating on Genome Plasticity
3. A Transgene-Based Pluripotency Acquisition
4. The Route Choice—Molecular Control of Induced Pluripotency Initiation
5. The Routes—Stepwise Phases of Reprogramming Mechanism
5.1. The Pros and Cons of Different Transgenesis Systems Used in iPSC Production
5.2. The Transgene-Based Somatic Reprogramming
5.3. The Initiation Phase
5.4. The Maturation Phase
5.5. The Stabilization Phase
5.6. The Chemical-Based Somatic Reprogramming
5.7. The Distinctive and Common Phases between tgiPSC and CiPSC
6. Reprogramming beyond Pluripotency
6.1. The 2C-like State
6.2. Expanded Potential Stem Cell—An In Vitro Captured 2C-like State
7. Reprogramming to Generate Germ Cell
8. The iPSC-Based Disease Modelling and iPSC Therapy in Clinical Trials
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cheloufi, S.; Elling, U.; Hopfgartner, B.; Jung, Y.L.; Murn, J.; Ninova, M.; Hubmann, M.; Badeaux, A.I.; Ang, C.E.; Tenen, D.; et al. The histone chaperone CAF-1 safeguards somatic cell identity. Nat. Cell Biol. 2015, 528, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Markoulaki, S.; Mitalipova, M.; Cheng, A.W.; Cassady, J.P.; Staerk, J.; Carey, B.W.; Lengner, C.; Foreman, R.; Love, J.; et al. Metastable Pluripotent States in NOD-Mouse-Derived ESCs. Cell Stem Cell 2009, 4, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtfeld, M.; Maherali, N.; Breault, D.T.; Hochedlinger, K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2008, 2, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Plath, K.; Lowry, W.E. Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 2011, 12, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Hansson, J.; Rafiee, M.-R.; Reiland, S.; Polo, J.M.; Gehring, J.; Okawa, S.; Huber, W.; Hochedlinger, K.; Krijgsveld, J. Highly Coordinated Proteome Dynamics during Reprogramming of Somatic Cells to Pluripotency. Cell Rep. 2012, 2, 1579–1592. [Google Scholar] [CrossRef] [Green Version]
- Gurdon, J.; Elsdale, T.R.; Fischberg, M. Sexually Mature Individuals of Xenopus laevis from the Transplantation of Single Somatic Nuclei. Nat. Cell Biol. 1958, 182, 64–65. [Google Scholar] [CrossRef]
- Gurdon, J.B. The Developmental Capacity of Nuclei Taken from Differentiating Endoderm Cells of Xenopus laevis. Development 1960, 8, 505–526. [Google Scholar] [CrossRef]
- Gurdon, J.B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 1962, 10, 622–640. [Google Scholar] [PubMed]
- Wilmut, I.; Schnieke, A.; McWhir, J.; Kind, A.J.; Campbell, K.H.S. Viable offspring derived from fetal and adult mammalian cells. Nat. Cell Biol. 1997, 385, 810–813. [Google Scholar] [CrossRef]
- Wakayama, T.; Perry, A.C.F.; Zuccotti, M.; Johnson, K.R.; Yanagimachi, R. Full-term development of mice form enucleated oocytes injected with cumulus cell nuclei. Nature 1998, 394, 369–374. [Google Scholar] [CrossRef]
- Zhou, Q.; Renard, J.-P.; Le Friec, G.; Brochard, V.; Beaujean, N.; Cherifi, Y.; Fraichard, A.; Cozzi, J. Generation of fertile cloned rats by regulating oocyte activation. Science 2003, 302, 1179. [Google Scholar] [CrossRef]
- Chiu, C.-P.; Blau, H.M. Reprogramming cell differentiation in the absence of DNA synthesis. Cell 1984, 37, 879–887. [Google Scholar] [CrossRef]
- Chiu, C.-P.; Blau, H.M. 5-Azacytidine permits gene activation in a previously noninducible cell type. Cell 1985, 40, 417–424. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells for mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, S.; Blau, H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nat. Cell Biol. 2010, 465, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, G.C.; Rajagopal, N.; Shen, Y.; McCleary, D.F.; Yue, F.; Dang, M.D.; Ren, B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 2013, 45, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nat. Cell Biol. 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Active DNA Demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef] [Green Version]
- Rai, K.; Huggins, I.J.; James, S.R.; Karpf, A.; Jones, D.A.; Cairns, B.R. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and Gadd45. Cell 2008, 135, 1201–1212. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Chen, J.; Li, K.; Wu, T.; Huang, B.; Liu, W.; Kou, X.; Zhang, Y.; Huang, H.; Jiang, Y.; et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell 2013, 12, 453–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutani, N.; Brady, J.J.; Damian, M.; Sacco, A.; Corbel, S.Y.; Blau, H.M. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nat. Cell Biol. 2010, 463, 1042–1047. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, N.; Decker, M.N.; Brady, J.J.; Bussat, R.T.; Burns, D.M.; Corbel, S.Y.; Blau, H.M. A critical role for AID in the initiation of reprogramming to induced pluripotent stem cells. FASEB J. 2013, 27, 1107–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.-L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Adli, M.; Zou, J.Y.; Verstappen, G.; Coyne, M.; Zhang, X.; Durham, T.; Miri, M.; Deshpande, V.; De Jager, P.L.; et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 2013, 152, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef]
- Sridharan, R.; Gonzales-Cope, M.; Chronis, C.; Bonora, G.; McKee, R.; Huang, C.; Patel, S.; Lopez, D.; Mishra, N.; Pellegrini, M.; et al. Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Mancarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar] [CrossRef]
- Yeap, L.-S.; Hayashi, K.; Surani, M.A. ERG-associated protein with SET domain (ESET)-Oct4 interaction regulates pluripotency and represses the trophectoderm lineage. Epigenetics Chromatin 2009, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Han, J.; Guo, G.; Orlov, Y.; Huss, M.; Loh, Y.-H.; Yaw, L.-P.; Robson, P.; Lim, B.; Ng, H.H. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 2009, 23, 2507–2520. [Google Scholar] [CrossRef] [Green Version]
- Koche, R.P.; Smith, Z.D.; Adli, M.; Gu, H.; Ku, M.; Gnirke, A.; Bernstein, B.E.; Meissner, A. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell 2011, 8, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Hussein, S.M.I.; Puri, M.C.; Tonge, P.D.; Benevento, M.; Corso, A.J.; Clancy, J.L.; Mosbergen, R.; Li, M.; Lee, D.-S.; Cloonan, N.; et al. Genome-wide characterization of the routes to pluripotency. Nature 2014, 516, 198–206. [Google Scholar] [CrossRef]
- Tonge, P.D.; Corso, A.J.; Monetti, C.; Hussein, S.M.I.; Puri, M.C.; Michael, I.; Li, M.; Lee, D.-S.; Mar, J.; Cloonan, N.; et al. Divergent reprogramming routes lead to alternative stem-cell states. Nat. Cell Biol. 2014, 516, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gafni, O.; Weinberger, L.; Zviran, A.; Ayyash, M.; Rais, Y.; Krupalnik, V.; Zerbib, M.; Amann-Zalcenstein, D.; Maza, I.; et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nat. Cell Biol. 2012, 488, 409–413. [Google Scholar] [CrossRef]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Xie, H.; Ye, M.; Feng, R.; Graf, T. Stepwise reprogramming of B cells into macrophages. Cell 2004, 117, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Graf, T.; Enver, T. Forcing cells to change lineages. Nature 2009, 462, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.M.; Lim, B. A Precarious balance: Pluripotency factors as lineage specifiers. Cell Stem Cell 2011, 8, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeineddine, D.; Papadimou, E.; Chebli, K.; Gineste, M.; Liu, J.; Grey, C.; Thurig, S.; Behfar, A.; Wallace, V.A.; Skerjanc, I.S.; et al. Oct-3/4 dose dependently regulates specification of embryonic stem cells toward a cardiac lineage and early heart development. Dev. Cell 2006, 11, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Teo, A.K.K.; Arnold, S.J.; Trotter, M.W.B.; Brown, S.; Ang, L.T.; Chng, Z.; Robertson, E.J.; Dunn, N.R.; Vallier, L. Pluripotency factors regulate definitive endoderm specification through eomesodermin. Genes Dev. 2011, 25, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivánek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nat. Cell Biol. 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Feldmann, A.; Ivánek, R.; Murr, R.; Gaidatzis, D.; Burger, L.; Schübeler, D. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 2013, 9, e1003994. [Google Scholar] [CrossRef]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, T.; Guan, J.; Zhang, X.; Fu, Y.; Ye, J.; Zhu, J.; Meng, G.; Ge, J.; Yang, S.; et al. A XEN-like state bridges somatic cells to pluripotency during chemical reprogramming. Cell 2015, 163, 1678–1691. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Li, Y.; Deng, H. Cell fate conversion—From the viewpoint of small molecules and lineage specifiers. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Fu, Y.; Zhu, J.; Liu, Y.; Zhang, Q.; Yi, Z.; Chen, S.; Jiao, Z.; Xu, X.; Xu, J.; et al. Single-Cell RNA-seq reveals dynamic early embryonic-like programs during chemical reprogramming. Cell Stem Cell 2018, 23, 31–45.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the methylome: Erasing memory and creating diversity. Cell Stem Cell. 2014, 14, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Matoba, S.; Zhang, Y. Somatic cell nuclear transfer reprogramming: Mechanisms and applications. Cell Stem Cell 2018, 23, 471–485. [Google Scholar] [CrossRef] [Green Version]
- Marión, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nat. Cell Biol. 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nat. Cell Biol. 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nat. Cell Biol. 2009, 460, 1145–1148. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nat. Cell Biol. 2009, 460, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nat. Cell Biol. 2011, 474, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Buganim, Y.; Faddah, D.A.; Cheng, A.W.; Itskovich, E.; Markoulaki, S.; Ganz, K.; Klemm, S.L.; van Oudenaarden, A.; Jaenisch, R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012, 150, 1209–1222. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S. Elite and stochastic models for induced pluripotent stem cell generation. Nature 2009, 460, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.; Jaenisch, R. Molecular control of induced pluripotency. Cell Stem Cell 2014, 14, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Hufton, P.G.; Lee, E.J.; Potoyan, D.A. A stochastic and dynamical view of pluripotency in mouse embryonic stem cells. PLoS Comput Biol. 2018, 14, e1006000. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, B.; Sardina, J.L.; van Oevelen, C.; Collombet, S.; Kallin, E.M.; Vicent, G.P.; Lu, J.; Thieffry, D.; Beato, M.; Graf, T. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature 2013, 506, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Zi, X.; Schulz, V.; Cheng, J.; Zhong, M.; Koochaki, S.H.; Megyola, C.M.; Pan, X.; Heydari, K.; Weissman, S.M.; et al. Nonstochastic Reprogramming from a Privileged Somatic Cell State. Cell 2014, 156, 649–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-H.; Chen, C.-C.; Hsueh, Y.-J.; Hung, L.-M.; Ma, H.-K.D.; Chen, H.-C.; Len, W.-B.; Meir, Y.-J.J. Extraneous E-Cadherin Engages the Deterministic Process of Somatic Reprogramming through Modulating STAT3 and Erk1/2 Activity. Cells 2021, 10, 284. [Google Scholar] [CrossRef] [PubMed]
- Rais, Y.; Zviran, A.; Geula, S.; Gafni, O.; Chomsky, E.; Viukov, S.; Mansour, A.A.; Caspi, I.; Krupalnik, V.; Zerbib, M.; et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nat. Cell Biol. 2013, 502, 65–70. [Google Scholar] [CrossRef]
- Yang, J.; van Oosten, A.L.; Theunissen, T.; Guo, G.; Silva, J.C.R.; Smith, A. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell Stem Cell 2010, 7, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and Cellular Reprogramming: Facts, Hypotheses, Unresolved Issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oosten, A.L.; Costa, Y.; Smith, A.; Silva, J.C. JAK/STAT3 signalling is sufficient and dominant over antagonistic cues for the establishment of naive pluripotency. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shakiba, N.; Fahmy, A.; Jayakumaran, G.; McGibbon, S.; David, L.; Trcka, D.; Elbaz, J.; Puri, M.C.; Nagy, A.; van der Kooy, D.; et al. Cell competition during reprogramming gives rise to dominant clones. Science 2019, 364, eaan0925. [Google Scholar] [CrossRef]
- Chung, K.M.; Kolling, F.W., 4th; Gajdosik, M.D.; Burger, S.; Russell, A.C.; Nelson, C.E. Single cell analysis reveals the stochastic phase of reprogramming to pluripotency is an ordered probabilistic process. PLoS ONE 2014, 9, e95304. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Maherali, N.; Borkent, M.; Hochedlinger, K. A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat. Methods 2009, 7, 53–55. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Ng, J.-H.; Heng, J.-C.D.; Ng, H.H. Molecules that promote or enhance reprogramming of somatic cells to induced pluripotent stem cells. Cell Stem Cell 2009, 4, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, A.; Akagi, K.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nat. Cell Biol. 2005, 436, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Collier, L.S.; Carlson, C.M.; Ravimohan, S.; Dupuy, A.; Largaespada, D.A. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nat. Cell Biol. 2005, 436, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Keng, V.; Villanueva, A.; Chiang, D.; Dupuy, A.; Ryan, B.J.; Matise, I.; Silverstein, K.A.; Sarver, A.L.; Starr, T.; Akagi, K.; et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat. Biotechnol. 2009, 27, 264–274. [Google Scholar] [CrossRef]
- Starr, T.K.; Allaei, R.; Silverstein, K.A.T.; Staggs, R.A.; Sarver, A.L.; Bergemann, T.L.; Gupta, M.; O’Sullivan, M.G.; Matise, I.; Dupuy, A.J.; et al. A Transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009, 323, 1747–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-Y.S.; Meir, Y.-J.J.; Coates, C.J.; Handler, A.M.; Moisyadi, S.; Pelczar, P.; Kaminski, J.M. piggyBac is a flexible and highly active transposon as compared to Sleeping Beauty, Tol2, and Mos1 in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15008–15013. [Google Scholar] [CrossRef] [Green Version]
- Meir, Y.-J.J.; Lin, A.; Huang, M.-F.; Weirauch, M.T.; Chou, H.-C.; Lin, S.-J.; Wu, C.-Y. A Versatile, highly efficient, and potentially safer piggyBac transposon system for mammalian genome manipulations. FASEB J. 2013, 27, 4429–4443. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nat. Cell Biol. 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef]
- Yusa, K.; Rashid, S.T.; Strick-Marchand, H.; Varela, I.; Liu, P.-Q.; Paschon, D.E.; Miranda, E.; Ordóñez, A.; Hannan, N.; Rouhani, F.J.; et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nat. Cell Biol. 2011, 478, 391–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki-Takahashi, Y.; Shikina, S.; Watanabe, M.; Banba, A.; Yagisawa, M.; Takahashi, K.; Fujihara, R.; Okabe, T.; Valdez, D.M., Jr.; Yamauchi, A.; et al. Production of functional eggs and sperm from in vitro-expanded type A spermatogonia in rainbow trout. Commun. Biol. 2020, 15, 308. [Google Scholar] [CrossRef]
- Nakanishi, M.; Otsu, M. Development of Sendai virus vectors and their potential applications gene therapy and regenerative medicine. Curr. Gene. Ther. 2012, 12, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Okumura, T.; Horie, Y.; Lai, C.-Y.; Lin, H.-T.; Shoda, H.; Natsumoto, B.; Fujio, K.; Kumaki, E.; Okano, T.; Ono, S.; et al. Roust and highly efficient hiPSC generation form patient non-mobilized peripheral blood-derived CD34+ cells using the auto-erasable Sendai virus vector. Stem Cell Res. Ther. 2019, 10, 185. [Google Scholar] [CrossRef]
- Buganim, Y.; Markoulaki, S.; van Wietmarschen, N.; Hoke, H.; Wu, T.; Ganz, K.; Akhtar-Zaidi, B.; He, Y.; Abraham, B.J.; Porubsky, D.; et al. The developmental potential of iPSCs is greatly influenced by reprogramming factor selection. Cell Stem Cell 2014, 15, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Churko, J.M.; Lee, J.; Ameen, M.; Gu, M.; Venkatasubramanian, M.; Diecke, S.; Sallam, K.; Im, H.; Wang, G.; Gold, J.D.; et al. Transcriptomic and epigenomic differences in human induced pluripotent stem cells generated from six reprogramming methods. Nat. Biomed. Eng. 2017, 1, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Roost, M.S.; Slieker, R.C.; Bialecka, M.; Van Iperen, L.; Fernandes, M.M.G.; He, N.; Suchiman, H.E.D.; Szuhai, K.; Carlotti, F.; De Koning, E.J.P.; et al. DNA methylation and transcriptional trajectories during human development and reprogramming of isogenic pluripotent stem cells. Nat. Commun. 2017, 8, 908. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Markoulaki, S.; Hanna, J.H.; Faddah, D.A.; Buganim, Y.; Kim, J.; Ganz, K.; Steine, E.J.; Cassady, J.P.; Creyghton, M.P.; et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell 2011, 9, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Bar, S.; Benvenisty, N. Epigenetic aberrations in human pluripotent stem cells. EMBO J. 2019, 38, 101033. [Google Scholar] [CrossRef]
- Maherali, N.; Sridharan, R.; Xei, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchiew, J.; Jaenisch, R.; et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, J.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Y.; Li, W.; Lv, Z.; Liu, L.; Tong, M.; Hai, T.; Hao, J.; Guo, C.-L.; Ma, Q.-W.; Wang, L.; et al. iPS cells produce viable mice through tetraploid complementation. Nat. Cell Biol. 2009, 461, 86–90. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Apostolou, E.; Akutsu, H.; Fukuda, A.; Follett, P.; Natesan, S.; Kono, T.; Shioda, T.; Hochedlinger, K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nat. Cell Biol. 2010, 465, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundry, R.L.; Riordon, D.R.; Tarasova, Y.; Chuppa, S.; Bhattacharya, S.; Juhasz, O.; Wiedemeier, O.; Milanovich, S.; Noto, F.K.; Tchernyshyov, I.; et al. A cell surfaceome map for immunophenotyping and sorting pluripotent stem cells. Mol. Cell. Proteom. 2012, 11, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Stadtfeld, M.; Apostolou, E.; Ferrari, F.; Choi, J.; Walsh, R.M.; Chen, T.; Ooi, S.S.; Kim, S.Y.; Bestor, T.H.; Shioda, T.; et al. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nat. Genet. 2012, 44, 398–405. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Blaschke, K.; Ebata, K.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nat. Cell Biol. 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [Green Version]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-Mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef] [PubMed]
- Markoulaki, S.; Hanna, J.H.; Beard, C.; Carey, B.W.; Cheng, A.; Lengner, C.; Dausman, J.A.; Fu, D.; Gao, Q.; Wu, S.; et al. Transgenic mice with defined combinations of drug-inducible reprogramming factors. Nat. Biotechnol. 2009, 27, 169–171. [Google Scholar] [CrossRef]
- Carey, B.W.; Markoulaki, S.; Beard, C.; Hanna, J.; Jaenisch, R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods 2009, 7, 56–59. [Google Scholar] [CrossRef]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.-K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golipour, A.; David, L.; Liu, Y.; Jayakumaran, G.; Hirsch, C.; Trcka, D.; Wrana, J.L. A late transition in somatic cell reprogramming requires regulators distinct from the pluripotency network. Cell Stem Cell 2012, 11, 769–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.; Polo, J.M. Phases of reprogramming. Stem Cell Res. 2014, 12, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Friedli, M.; Turelli, P.; Kapopoulou, A.; Rauwel, B.; Castro-Díaz, N.; Rowe, H.; Ecco, G.; Unzu, C.; Planet, E.; Lombardo, A.L.; et al. Loss of transcriptional control over endogenous retroelements during reprogramming to pluripotency. Genome Res. 2014, 24, 1251–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Eckersley-Maslin, M.A.; Svensson, V.; Krueger, C.; Stubbs, T.M.; Giehr, P.; Krueger, F.; Miragaia, R.J.; Kyriakopoulos, C.; Berrens, R.V.; Milagre, I.; et al. MERVL/Zscan4 network activation results in transient genome-wide DNA demethylation of mESCs. Cell Rep. 2016, 17, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.F.; Chuang, C.Y.; Lee, W.C.; Huang, H.P.; Wu, H.C.; Ho, H.N.; Chen, Y.J.; Kuo, H.C. Surface marker epithelial cell adhesion molecule and E-cadherin facilitate the identification and selection of induced pluripotent stem cells. Stem Cell Rev. Rep. 2011, 7, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Zhang, L.; Mao, S.-Q.; Li, Z.; Chen, J.; Zhang, R.-R.; Wu, H.-P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Ling, T.; Xie, W.; Sun, H.; Zhou, Y.; Zhu, Q.; Shen, M.; Zong, L.; Lyu, G.; Zhao, Y.; et al. NuRD blocks reprogramming of mouse somatic cells into pluripotent stem cells. Stem Cells 2013, 31, 1278–1286. [Google Scholar] [CrossRef]
- Tobias, B.; Foreman, R.; Welstead, G.G.; Lengner, C.J.; Wernig, M.; Suh, H.; Jaenisch, R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2008, 2, 151–159. [Google Scholar]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nat. Cell Biol. 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In Vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nat. Cell Biol. 2007, 448, 318–324. [Google Scholar] [CrossRef]
- Papp, B.; Plath, K. Epigenetics of reprogramming to induced pluripotency. Cell 2013, 152, 1324–1343. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, D.; Ma, Y.; Du, X.; Jing, J.; Wang, L.; Xie, B.; Sun, D.; Sun, S.; Jin, X.; et al. Direct reprogramming of fibroblasts via a chemically induced XEN-like state. Cell Stem Cell 2017, 21, 264–273.e7. [Google Scholar] [CrossRef]
- Cao, S.; Yu, S.; Li, D.; Ye, J.; Yang, X.; Li, C.; Wang, X.; Mai, Y.; Qin, Y.; Wu, J.; et al. Chromatin accessibility dynamics during chemical induction of pluripotency. Cell Stem Cell 2018, 22, 529–542.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, W.; Hu, J.; Hu, G.; Song, Y.; Xia, Q.; Yao, M.; Gong, S.; Jiang, C.; Yao, H. Genome-wide DNA methylation analysis reveals that mouse chemical iPSCs have closer epigenetic features to mESCs than OSKM-integrated iPSCs. Cell Death Dis. 2018, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- Zalzman, M.; Falco, G.; Sharova, L.V.; Nishiyama, A.; Thomas, M.; Lee, S.L.; Stagg, C.A.; Hoang, H.G.; Yang, H.-T.; Indig, F.E.; et al. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 2010, 464, 858–863. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.S.C.; Wong, R.C.B.; Sharov, A.A.; Nakatake, Y.; Yu, H.; Ko, M.S.H. Repression of global protein synthesis by Eif1a-Like genes that are expressed specifically in the two-cell embryos and the transient Zscan4-positive state of embryonic stem cells. DNA Res. 2013, 20, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Wu, X.; Djekidel, M.N.; Zhang, Y. Myc and Dnmt1 impede the pluripotent to totipotent state transition in embryonic stem cells. Nat. Cell Biol. 2019, 21, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Macfarlan, T.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nat. Cell Biol. 2012, 487, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgani, S.M.; Canham, M.; Nichols, J.; Sharov, A.A.; Migueles, R.P.; Ko, M.S.; Brickman, J.M. Totipotent embryonic stem cells arise in ground-state culture conditions. Cell Rep. 2013, 3, 1945–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Cañamero, M.; Rayon, T.; Ors, I.; Graña, O.; Megías, D.; Domínguez, O.; Martínez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ryan, D.J.; Wang, W.; Tsang, J.C.-H.; Lan, G.; Masaki, H.; Gao, X.; Antunes, L.; Yu, Y.; Zhu, Z.; et al. Establishment of mouse expanded potential stem cells. Nat. Cell Biol. 2017, 550, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, B.; Xu, J.; Wang, J.; Wu, J.; Shi, C.; Xu, Y.; Dong, J.; Wang, C.; Lai, W.; et al. Derivation of pluripotent stem cells with in vivo embryonic and extraembryonic potency. Cell 2017, 169, 243–257.e25. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, A.A.; Kim, J.K.; Tsang, J.C.; Ilicic, T.; Henriksson, J.; Natarajan, K.N.; Tuck, A.C.; Gao, X.; Bühler, M.; Liu, P.; et al. Single cell RNA sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 2015, 17, 471–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genet, M.; Torres-Padilla, M.-E. The molecular and cellular features of 2-cell-like cells: A reference guide. Development 2020, 147, 189688. [Google Scholar] [CrossRef]
- Redo Riveiro, A.; Brickman, J.M. From pluripotency to totipotency: An experimentalist’s guide to cellular potency. Development 2020, 147, 189845. [Google Scholar] [CrossRef]
- Wobus, A.M.; Boheler, K.R. Embryonic stem cells: Prospects for developmental biology and cell therapy. Physiol. Rev. 2005, 85, 635–678. [Google Scholar] [CrossRef]
- Kobayashi, T.; Surani, M.A. On the origin of the human germline. Development 2018, 145, dev150433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, M. Mammalian germ cell development: From mechanism to in vitro reconstitution. Stem Cell Rep. 2021, 16, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Ohta, H.; Kurimoto, K.; Aramaki, S.; Saitou, M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 2011, 146, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Ogushi, S.; Kurimoto, K.; Shimamoto, S.; Ohta, H.; Saitou, M. Offspring from oocytes derived from in vitro primordial germ cell–like cells in mice. Science 2012, 338, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Hikabe, O.; Hamazaki, N.; Nagamatsu, G.; Obata, Y.; Hirao, Y.; Hamada, N.; Shimamoto, S.; Imamura, T.; Nakashima, K.; Saitou, M.; et al. Reconstitution in vitro of the entire cycle of the mouse female germ line. Nat. Cell Biol. 2016, 539, 299–303. [Google Scholar] [CrossRef]
- Ishikura, Y.; Yabuta, Y.; Ohta, H.; Hayashi, K.; Nakamura, T.; Okamoto, I.; Yamamoto, T.; Kurimoto, K.; Shirane, K.; Sasaki, H.; et al. In vitro derivation and propagation of spermatogonial stem cell activity from mouse pluripotent stem cells. Cell Rep. 2016, 17, 2789–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashiro, C.; Sasaki, K.; Yabuta, Y.; Kojima, Y.; Nakamura, T.; Okamoto, I.; Yokobayashi, S.; Murase, Y.; Ishikura, Y.; Shirane, K.; et al. Generation of human oogonia from induced pluripotent stem cells in vitro. Science 2018, 362, 356–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.S.; Suzuki, S.; Seita, Y.; Ito, J.; Sakata, Y.; Aso, H.; Sato, K.; Hermann, B.P.; Sasaki, K. Reconstitution of prospermatogonial specification in vitro from human induced pluripotent stem cells. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.; Casini, S.; Davis, R.; D’Aniello, C.; Haas, J.; Oostwaard, D.W.-V.; Tertoolen, L.G.J.; Jung, C.B.; Elliott, D.; Welling, A.; et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013, 32, 3161–3175. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flugel, L.; Dorn, T.; Goedel, A.; Hohnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cellmodels for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [Green Version]
- Jacquet, L.; Stephenson, E.; Collins, R.; Patel, H.; Trussler, J.; Al-Bedaery, R.; Renwick, P.; Ogilvie, C.; Vaughan, R.; Ilic, D. Strategy for the creation of clinical grade hESC line banks that HLA-match a target population. EMBO Mol. Med. 2013, 5, 10–17. [Google Scholar] [CrossRef]
- Bravery, C.A. Do human leukocyte antigen-typed cellular therapeutics based on induced pluripotent stem cells make commercial sense? Stem Cells Dev. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Palomo, B.; García-Martinez, I.; Gayoso, J.; Raya, A.; Veiga, A.; Abad, M.L.; Eiras, A.; Guzmán-Fulgencio, M.; Luis-Hidalgo, M.; Eguizabal, C.; et al. Evaluation of the Spanish population coverage of a prospective HLA haplobank of induced pluripotent stem cells. Stem Cell Res. Ther. 2021, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Huh, J.Y.; Turner, D.M.; Lee, S.; Robinson, J.; Stein, J.E.; Shim, S.H.; Hong, C.P.; Kang, M.S.; Nakagawa, M.; et al. Repurposing the cord blood bank for haplobanking of HLA-homozygous iPSCs and their usefulness to multiple populations. Stem Cells 2018, 36, 1552–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourraud, P.-A.; Gilson, L.; Girard, M.; Peschanski, M. The role of human leukocyte antigen matching in the development of multiethnic “Haplobank” of induced pluripotent stem cell lines. Stem Cells 2012, 30, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meir, Y.-J.J.; Li, G. Somatic Reprogramming—Above and Beyond Pluripotency. Cells 2021, 10, 2888. https://doi.org/10.3390/cells10112888
Meir Y-JJ, Li G. Somatic Reprogramming—Above and Beyond Pluripotency. Cells. 2021; 10(11):2888. https://doi.org/10.3390/cells10112888
Chicago/Turabian StyleMeir, Yaa-Jyuhn James, and Guigang Li. 2021. "Somatic Reprogramming—Above and Beyond Pluripotency" Cells 10, no. 11: 2888. https://doi.org/10.3390/cells10112888
APA StyleMeir, Y. -J. J., & Li, G. (2021). Somatic Reprogramming—Above and Beyond Pluripotency. Cells, 10(11), 2888. https://doi.org/10.3390/cells10112888