
Alzheimer’s Disease, Sleep Disordered Breathing, and Microglia: Puzzling out a Common Link

 ,
, 
Abstract
:
1. Introduction
2. Neurodegeneration Is Common in Both SDB and AD
2.1. Sleep Disordered Breathing
2.2. Neurodegeneration in SDB
2.3. Experimental Models of SDB
2.4. Alzheimer’s Disease
2.5. Experimental Models of AD
3. Neuroinflammation in AD and SDB
3.1. Microglia and the Inflammasome in AD and SDB
3.2. Astrocytes in AD and SDB
3.3. Molecular Mechanisms of Neuroinflammation in AD and SDB
4. Common Mechanisms of SDB and AD
4.1. Bidirectional Effects of SDB and AD on One Another
4.2. Sleep Disturbances
4.3. Sex Hormones and Gender Differences
4.4. Common Risk Factors and Comorbidities for SDB and AD
4.5. Studying SDB in AD Animal Models
5. Summary and Closing Thoughts
6. Experimental Methods
6.1. Intermittent Hypoxia (IH) Exposure
6.2. Tissue Isolation and Fixation for Flow Cytometry
6.3. Staining for Flow Cytometry
6.4. Statistical ANALYSIS
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoffstein, V.; Mateika, S. Differences in Abdominal and Neck Circumferences in Patients with and without Obstructive Sleep Apnoea. Eur. Respir. J. 1992, 5, 377–381. [Google Scholar] [PubMed]
- Davies, R.J.O.; Ali, N.J.; Stradling, J.R. Neck Circumference and Other Clinical Features in the Diagnosis of the Obstructive Sleep Apnoea Syndrome. Thorax 1992, 47, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.K.; Kurian, S.; Malik, V.; Mohan, A.; Banga, A.; Pandey, R.M.; Handa, K.K.; Mukhopadhyay, S. A Stepped Approach for Prediction of Obstructive Sleep Apnea in Overtly Asymptomatic Obese Subjects: A Hospital Based Study. Sleep Med. 2004, 5, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Skatrud, J.; Peppard, P.E. Risk Factors for Obstructive Sleep Apnea in Adults. JAMA 2004, 291, 2013–2016. [Google Scholar] [CrossRef]
- Young, T.; Peppard, P.E.; Taheri, S. Excess Weight and Sleep-Disordered Breathing. J. Appl. Physiol. 2005, 99, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Palta, M.; Dempsey, J.; Skatrud, J.; Weber, S.; Badr, S. The Occurrence of Sleep-Disordered Breathing among Middle-Aged Adults. N. Engl. J. Med. 1993, 328, 1230–1235. [Google Scholar] [CrossRef] [Green Version]
- Peppard, P.E.; Young, T.; Barnet, J.H.; Palta, M.; Hagen, E.W.; Hla, K.M. Increased Prevalence of Sleep-Disordered Breathing in Adults. Am. J. Epidemiol. 2013, 177, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Snyder, B.; Cunningham, R.L. Sex Differences in Sleep Apnea and Comorbid Neurodegenerative Diseases. Steroids 2018, 133, 28–33. [Google Scholar] [CrossRef]
- Chang, H.-P.; Chen, Y.-F.; Du, J.-K. Obstructive Sleep Apnea Treatment in Adults. Kaohsiung J. Med. Sci. 2020, 36, 7–12. [Google Scholar] [CrossRef]
- Dempsey, J.A.; Smith, C.A. Pathophysiology of Human Ventilatory Control. Eur. Respir. J. 2014, 44, 495–512. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, J.A.; Xie, A.; Patz, D.S.; Wang, D. Physiology in Medicine: Obstructive Sleep Apnea Pathogenesis and Treatment—Considerations beyond Airway Anatomy. J. Appl. Physiol. 2014, 116, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, J.A.; Veasey, S.C.; Morgan, B.J.; O’donnell, C.P. Pathophysiology of Sleep Apnea. Physiol. Rev. 2010, 90, 47–112. [Google Scholar] [CrossRef]
- Zhang, S.X.L.; Wang, Y.; Gozal, D. Pathological Consequences of Intermittent Hypoxia in the Central Nervous System. Compr. Physiol. 2012, 2, 1767–1777. [Google Scholar] [CrossRef]
- Jackson, M.L.; Howard, M.E.; Barnes, M. Cognition and Daytime Functioning in Sleep-Related Breathing Disorders. Prog. Brain Res. 2011, 190, 53–68. [Google Scholar] [CrossRef]
- Zimmerman, M.E.; Aloia, M.S. A Review of Neuroimaging in Obstructive Sleep Apnea. J. Clin. Sleep Med. 2006, 2, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Celle, S.; Peyron, R.; Faillenot, I.; Pichot, V.; Alabdullah, M.; Gaspoz, J.-M.; Laurent, B.; Barthélémy, J.-C.; Roche, F. Undiagnosed Sleep-Related Breathing Disorders Are Associated with Focal Brainstem Atrophy in the Elderly. Hum. Brain Mapp. 2009, 30, 2090–2097. [Google Scholar] [CrossRef]
- Macey, P.M.; Henderson, L.A.; Macey, K.E.; Alger, J.R.; Frysinger, R.C.; Woo, M.A.; Harper, R.K.; Yan-Go, F.L.; Harper, R.M. Brain Morphology Associated with Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2002, 166, 1382–1387. [Google Scholar] [CrossRef]
- Gozal, D.; Daniel, J.M.; Dohanich, G.P. Behavioral and Anatomical Correlates of Chronic Episodic Hypoxia during Sleep in the Rat. J. Neurosci. 2001, 21, 2442–2450. [Google Scholar] [CrossRef]
- Xu, W.; Chi, L.; Row, B.W.; Xu, R.; Ke, Y.; Xu, B.; Luo, C.; Kheirandish, L.; Gozal, D.; Liu, R. Increased Oxidative Stress Is Associated with Chronic Intermittent Hypoxia-Mediated Brain Cortical Neuronal Cell Apoptosis in a Mouse Model of Sleep Apnea. Neuroscience 2004, 126, 313–323. [Google Scholar] [CrossRef]
- Machaalani, R.; Waters, K.A. Postnatal Nicotine and/or Intermittent Hypercapnic Hypoxia Effects on Apoptotic Markers in the Developing Piglet Brainstem Medulla. Neuroscience 2006, 142, 107–117. [Google Scholar] [CrossRef]
- Nanduri, J.; Nanduri, R.P. Cellular Mechanisms Associated with Intermittent Hypoxia. Essays Biochem. 2007, 43, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Douglas, R.M.; Ryu, J.; Kanaan, A.; del Carmen Rivero, M.; Dugan, L.L.; Haddad, G.G.; Ali, S.S. Neuronal Death during Combined Intermittent Hypoxia/Hypercapnia Is Due to Mitochondrial Dysfunction. Am. J. Physiol. Cell Physiol. 2010, 298, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Daulatzai, M.A. Quintessential Risk Factors: Their Role in Promoting Cognitive Dysfunction and Alzheimer’s Disease. Neurochem. Res. 2012, 37, 2627–2658. [Google Scholar] [CrossRef]
- Chou, Y.-T.; Zhan, G.; Zhu, Y.; Fenik, P.; Panossian, L.; Li, Y.; Zhang, J.; Veasey, S. C/EBP Homologous Binding Protein (CHOP) Underlies Neural Injury in Sleep Apnea Model. Sleep 2013, 36, 481–492. [Google Scholar] [CrossRef]
- Yuan, X.; Guo, X.; Deng, Y.; Zhu, D.; Shang, J.; Liu, H. Chronic Intermittent Hypoxia-Induced Neuronal Apoptosis in the Hippocampus Is Attenuated by Telmisartan through Suppression of INOS/NO and Inhibition of Lipid Peroxidation and Inflammatory Responses. Brain Res. 2015, 1596, 48–57. [Google Scholar] [CrossRef]
- Bliwise, D.L. Sleep Disorders in Alzheimer’s Disease and Other Dementias. Clin. Cornerstone 2004, 6, S16–S28. [Google Scholar] [CrossRef]
- Ancoli-Israel, S.; Palmer, B.W.; Cooke, J.R.; Corey-Bloom, J.; Fiorentino, L.; Natarajan, L.; Liu, L.; Ayalon, L.; He, F.; Loredo, J.S. Cognitive Effects of Treating Obstructive Sleep Apnea in Alzheimer’s Disease: A Randomized Controlled Study. J. Am. Geriatr. Soc. 2008, 56, 2076–2081. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.M.; Lau, C.F.; Fung, M.L. Melatonin Reduces Hippocampal β-Amyloid Generation in Rats Exposed to Chronic Intermittent Hypoxia. Brain Res. 2010, 1354, 163–171. [Google Scholar] [CrossRef]
- Shiota, S.; Takekawa, H.; Matsumoto, S.-E.; Takeda, K.; Nurwidya, F.; Yoshioka, Y.; Takahashi, F.; Hattori, N.; Tabira, T.; Mochizuki, H.; et al. Chronic Intermittent Hypoxia/Reoxygenation Facilitate Amyloid-Generation in Mice. J. Alzheimer’s Dis. 2013, 37, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.E.; Benediktsdottir, B.; Cook, E.; Olafsson, I.; Gislason, T.; Robinson, S.R. Alzheimer’s Disease Neuropathology in the Hippocampus and Brainstem of People with Obstructive Sleep Apnea. Sleep 2021, 44, zsaa195. [Google Scholar] [CrossRef]
- Fang, H.; Zhang, L.F.; Meng, F.T.; Du, X.; Zhou, J.N. Acute Hypoxia Promote the Phosphorylation of Tau via ERK Pathway. Neurosci. Lett. 2010, 474, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-E.; Yang, X.; Li, L.; Sui, X.; Tian, Q.; Wei, W.; Wang, J.; Liu, G. Hypoxia-Induced Tau Phosphorylation and Memory Deficit in Rats. Neurodegener. Dis. 2014, 14, 107–116. [Google Scholar] [CrossRef]
- Fletcher, E.C.; Lesske, J.; Behm, R.; Miller, C.C., 3rd; Stauss, H.; Unger, T. Carotid Chemoreceptors, Systemic Blood Pressure, and Chronic Episodic Hypoxia Mimicking Sleep Apnea. J. Appl. Physiol. 1992, 72, 1978–1984. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.C.; Lesske, J.; Qian, W.; Miller, C.C., 3rd; Unger, T. Repetitive, Episodic Hypoxia Causes Diurnal Elevation of Blood Pressure in Rats. Hypertension 1992, 19, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.C.; Brady, D.C.; Po, P.; Chuang, L.P.; Marcondes, L.; Kim, E.Y.; Keenan, B.T.; Guo, X.; Maislin, G.; Galante, R.J.; et al. Simulating Obstructive Sleep Apnea Patients’ Oxygenation Characteristics into a Mouse Model of Cyclical Intermittent Hypoxia. J. Appl. Physiol. 2015, 118, 544–557. [Google Scholar] [CrossRef]
- Chopra, S.; Polotsky, V.Y.; Jun, J.C. Sleep Apnea Research in Animals. Past, Present, and Future. Am. J. Respir. Cell Mol. Biol. 2016, 54, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Dewan, N.A.; Nieto, F.J.; Somers, V.K. Intermittent Hypoxemia and OSA: Implications for Comorbidities. Chest 2015, 147, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochmans-Lemoine, A.; Villalpando, G.; Gonzales, M.; Valverde, I.; Soria, R.; Joseph, V. Divergent Physiological Responses in Laboratory Rats and Mice Raised at High Altitude. J. Exp. Biol. 2015, 218, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Jochmans-Lemoine, A.; Shahare, M.; Soliz, J.; Joseph, V. HIF1α and Physiological Responses to Hypoxia Are Correlated in Mice but Not in Rats. J. Exp. Biol. 2016, 219, 3952–3961. [Google Scholar] [CrossRef] [Green Version]
- Arias-Reyes, C.; Soliz, J.; Joseph, V. Mice and Rats Display Different Ventilatory, Hematological, and Metabolic Features of Acclimatization to Hypoxia. Front. Physiol. 2021, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Kimoff, R.J. Sleep Fragmentation in Obstructive Sleep Apnea. Sleep 1996, 19 (Suppl. 9), S61–S66. [Google Scholar] [CrossRef] [Green Version]
- Montgomery-Downs, H.E.; Crabtree, V.M.; Gozal, D. Cognition, Sleep and Respiration in at-Risk Children Treated for Obstructive Sleep Apnoea. Eur. Respir. J. 2005, 25, 336–342. [Google Scholar] [CrossRef]
- Ramesh, V.; Nair, D.; Zhang, S.X.L.; Hakim, F.; Kaushal, N.; Kayali, F.; Wang, Y.; Li, R.C.; Carreras, A.; Gozal, D. Disrupted Sleep without Sleep Curtailment Induces Sleepiness and Cognitive Dysfunction via the Tumor Necrosis Factor-α Pathway. J. Neuroinflamm. 2012, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- McKenna, J.T.; Tartar, J.L.; Ward, C.P.; Thakkar, M.M.; Cordeira, J.W.; McCarley, R.W.; Strecker, R.E. Sleep Fragmentation Elevates Behavioral, Electrographic and Neurochemical Measures of Sleepiness. Neuroscience 2007, 146, 1462–1473. [Google Scholar] [CrossRef] [Green Version]
- Sinton, C.M.; Kovakkattu, D.; Friese, R.S. Validation of a Novel Method to Interrupt Sleep in the Mouse. J. Neurosci. Methods 2009, 184, 71–78. [Google Scholar] [CrossRef]
- Tartar, J.L.; Ward, C.P.; McKenna, J.T.; Thakkar, M.; Arrigoni, E.; McCarley, R.W.; Brown, R.E.; Strecker, R.E. Hippocampal Synaptic Plasticity and Spatial Learning Are Impaired in a Rat Model of Sleep Fragmentation. Eur. J. Neurosci. 2006, 23, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.P.; McCoy, J.G.; McKenna, J.T.; Connolly, N.P.; McCarley, R.W.; Strecker, R.E. Spatial Learning and Memory Deficits Following Exposure to 24 h of Sleep Fragmentation or Intermittent Hypoxia in a Rat Model of Obstructive Sleep Apnea. Brain Res. 2009, 1294, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, V.; Kaushal, N.; Gozal, D. Sleep Fragmentation Differentially Modifies EEG Delta Power during Slow Wave Sleep in Socially Isolated and Paired Mice. Sleep Sci. 2009, 2, 64–75. [Google Scholar]
- McAlpine, C.S.; Swirski, F.K. Circadian Influence on Metabolism and Inflammation in Atherosclerosis. Circ. Res. 2016, 119, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Maki, K.A.; Burke, L.A.; Calik, M.W.; Watanabe-Chailland, M.; Sweeney, D.; Romick-Rosendale, L.E.; Green, S.J.; Fink, A.M. Sleep Fragmentation Increases Blood Pressure and Is Associated with Alterations in the Gut Microbiome and Fecal Metabolome in Rats. Physiol. Genom. 2020, 52, 280–292. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Almendros, I.; Díaz-García, E.; Toledano, V.; Casitas, R.; Galera, R.; López-Collazo, E.; Farre, R.; Gozal, D.; García-Rio, F. Differential Effect of Intermittent Hypoxia and Sleep Fragmentation on PD-1/PD-L1 Upregulation. Sleep 2020, 43, zsz285. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Kockanek, K.D.; Xu, J.; Arias, E. Mortality in the United States; NCHS Data Brief 2020; NCHS: Hyattsville, MD, USA, No. 355; 2020; pp. 1–8. [Google Scholar]
- Long, J.M.; Holtzman, D.M. Leading Edge Review Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Bobinski, M.; Wegiel, J.; Tarnawski, M.; Bobinski, M.; Reisberg, B.; de Leon, M.J.; Miller, D.C.; Wisniewski, H.M. Relationships between Regional Neuronal Loss and Neurofibrillary Changes in the Hippocampal Formation and Duration and Severity of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 1997, 56, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Silbert, L.C.; Quinn, J.F.; Moore, M.M.; Corbridge, E.; Ball, M.J.; Murdoch, G.; Sexton, G.; Kaye, J.A. Changes in Premorbid Brain Volume Predict Alzheimer’s Disease Pathology. Neurology 2003, 61, 487–492. [Google Scholar] [CrossRef]
- Alzheimer’s Disease Fact Sheet|National Institute on Aging. Available online: https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet (accessed on 29 September 2021).
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Maccioni, R.B.; Farías, G.; Moraels, I.; Navarrete, L. The Revitalized Tau Hypothesis on Alzheimer Disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef]
- Ashraf, G.M.; Tarasov, V.V.; Makhmutova, A.; Chubarev, V.N.; Avila-Rodriguez, M.; Bachurin, S.O.; Aliev, G. The Possibility of an Infectious Etiology of Alzheimer Disease. Mol. Neurobiol. 2019, 56, 4479–4491. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Aβ and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-Driven Cerebral Amyloidosis in Transgenic Mice Reveals Early and Robust Pathology. EMBO Rep. 2006, 7, 940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 Function Impedes Tau Seeding in Neuritic Plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s Disease: Experimental Models and Reality. Acta Neuropathol. 2016, 133, 155–175. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP Mediates Rapid Microglial Response to Local Brain Injury in Vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial Interactions with Synapses Are Modulated by Visual Experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [Green Version]
- Schain, A.J.; Hill, R.A.; Grutzendler, J. Label-Free in Vivo Imaging of Myelinated Axons in Health and Disease with Spectral Confocal Reflectance Microscopy. Nat. Med. 2014, 20, 443–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M.-È. Microglia across the Lifespan: From Origin to Function in Brain Development, Plasticity and Cognition. J. Physiol. 2017, 595, 1929–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulland, T.K.; Colonna, M. TREM2—A Key Player in Microglial Biology and Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Emahazion, T.; Feuk, L.; Jobs, M.; Sawyer, S.L.; Fredman, D.; St Clair, D.; Prince, J.A.; Brookes, A.J. SNP Association Studies in Alzheimer’s Disease Highlight Problems for Complex Disease Analysis. Trends Genet. 2001, 17, 407–413. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-Derived ASC Specks Cross-Seed Amyloid-β in Alzheimer’s Disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Landel, V.; Baranger, K.; Virard, I.; Loriod, B.; Khrestchatisky, M.; Rivera, S.; Benech, P.; Féron, F. Temporal Gene Profiling of the 5XFAD Transgenic Mouse Model Highlights the Importance of Microglial Activation in Alzheimer’s Disease. Mol. Neurodegener. 2014, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia Constitute a Barrier That Prevents Neurotoxic Protofibrillar Aβ42 Hotspots around Plaques. Nat. Commun. 2015, 6, 6175. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-Mediated Early Microglial Response Limits Diffusion and Toxicity of Amyloid Plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate Inhibits Inflammasome Activation to Attenuate Alzheimer’s Disease Pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-Stimulating Factor 1 Receptor Signaling Is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating Microglia in Alzheimer’s Mice Prevents Neuronal Loss without Modulating Amyloid-b Pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [Green Version]
- Casali, B.T.; MacPherson, K.P.; Reed-Geaghan, E.G.; Landreth, G.E. Microglia Depletion Rapidly and Reversibly Alters Amyloid Pathology by Modification of Plaque Compaction and Morphologies. Neurobiol. Dis. 2020, 142, 104956. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Serrano, J.R.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 Deficiency Attenuates Neuroinflammation and Protects against Neurodegeneration in a Mouse Model of Tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley–Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Aβ Accumulation and Progressive Synaptic Loss, Gliosis, and Tangle Formation in AD Brain. Neurology 2004, 62, 925–931. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Bushong, E.A.; Price, D.L.; Smarr, B.L.; Phung, V.; Terada, M.; Ellisman, M.H.; Pekny, M. Redefining the Concept of Reactive Astrocytes as Cells That Remain within Their Unique Domains upon Reaction to Injury. Proc. Natl. Acad. Sci. USA 2006, 103, 17513–17518. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular Dissection of Reactive Astrogliosis and Glial Scar Formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2009, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.A.; Ao, Y.; Sofroniew, M.v. Heterogeneity of Reactive Astrocytes. Neurosci. Lett. 2014, 565, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Jakobs, T.C. Structural Remodeling of Astrocytes in the Injured CNS. Neuroscientist 2011, 18, 567–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive Glia Not Only Associates with Plaques but Also Parallels Tangles in Alzheimer’s Disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes Accumulate Aβ42 and Give Rise to Astrocytic Amyloid Plaques in Alzheimer Disease Brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Wegiel, J. Spatial Relationships between Astrocytes and Classical Plaque Components. Neurobiol. Aging 1991, 12, 593–600. [Google Scholar] [CrossRef]
- Overmyer, M.; Helisalmi, S.; Soininen, H.; Laakso, M.; Riekkinen Sr., P.; Alafuzoff, I. Reactive Microglia in Aging and Dementia: An Immunohistochemical Study of Postmortem Human Brain Tissue. Acta Neuropathol. 1999, 97, 383–392. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A. Astroglia in Dementia and Alzheimer’s Disease. Cell Death Differ. 2008, 16, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Simpson, J.E.; Ince, P.G.; Lace, G.; Forster, G.; Shaw, P.J.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S.B. Astrocyte Phenotype in Relation to Alzheimer-Type Pathology in the Ageing Brain. Neurobiol. Aging 2010, 31, 578–590. [Google Scholar] [CrossRef]
- Senitz, D.; Reichenbach, A.; Smith Jr, T. Surface Complexity of Human Neocortical Astrocytic Cells: Changes with Development, Aging, and Dementia. J. Hirnforsch 1995, 36, 531–537. [Google Scholar]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant Astroglial Atrophy and Astrogliosis in a Triple Transgenic Animal Model of Alzheimer’s Disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; van Leuven, F. Focal Glial Activation Coincides with Increased BACE1 Activation and Precedes Amyloid Plaque Deposition in APP [V717I] Transgenic Mice. J. Neuroinflamm. 2005, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.-Y.; Vadhwana, B.; Verkhratsky, A.; Rodríguez, J.J. Early Astrocytic Atrophy in the Entorhinal Cortex of a Triple Transgenic Animal Model of Alzheimer’s Disease. ASN Neuro 2011, 3, 271–279. [Google Scholar] [CrossRef]
- Carter, S.F.; Schöll, M.; Almkvist, O.; Wall, A.; Engler, H.; Långström, B.; Nordberg, A. Evidence for Astrocytosis in Prodromal Alzheimer Disease Provided by 11C-Deuterium-L-Deprenyl: A Multitracer PET Paradigm Combining 11C-Pittsburgh Compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Vieitez, E.; Ni, R.; Gulyás, B.; Tóth, M.; Häggkvist, J.; Halldin, C.; Voytenko, L.; Marutle, A.; Nordberg, A. Astrocytosis Precedes Amyloid Plaque Deposition in Alzheimer APPswe Transgenic Mouse Brain: A Correlative Positron Emission Tomography and in Vitro Imaging Study. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1119–1132. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, F.; Abbasi, H.; Sadeghi, M.; Imani, M.M. The Effect of Obstructive Sleep Apnea Syndrome on Serum S100B and NSE Levels: A Systematic Review and Meta-Analysis of Observational Studies. BMC Pulm. Med. 2020, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Macey, P.M.; Sarma, M.K.; Prasad, J.P.; Ogren, J.A.; Aysola, R.; Harper, R.M.; Thomas, M.A. Obstructive Sleep Apnea Is Associated with Altered Midbrain Chemical Concentrations. Neuroscience 2017, 363, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Macheda, T.; Roberts, K.; Lyons, D.N.; Higgins, E.; Ritter, K.J.; Lin, A.-L.; Alilain, W.J.; Bachstetter, A.D. Chronic Intermittent Hypoxia Induces Robust Astrogliosis in an Alzheimer’s Disease-Relevant Mouse Model HHS Public Access. Neuroscience 2019, 398, 55–63. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 Inflammasome Activation Drives Tau Pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; Mcmanus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Michael, I.; Heneka, T.; et al. Soluble Aβ Oligomers and Protofibrils Induce NLRP3 Inflammasome Activation in Microglia. J. Neurochem. 2020, 155, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, S.F.; King, A.D.; O’Donnell, C.; Roche, H.M.; Ryan, S. Mechanisms of Intermittent Hypoxia-Mediated Macrophage Activation—Potential Therapeutic Targets for Obstructive Sleep Apnoea. J. Sleep Res. 2021, 30, e13202. [Google Scholar] [CrossRef]
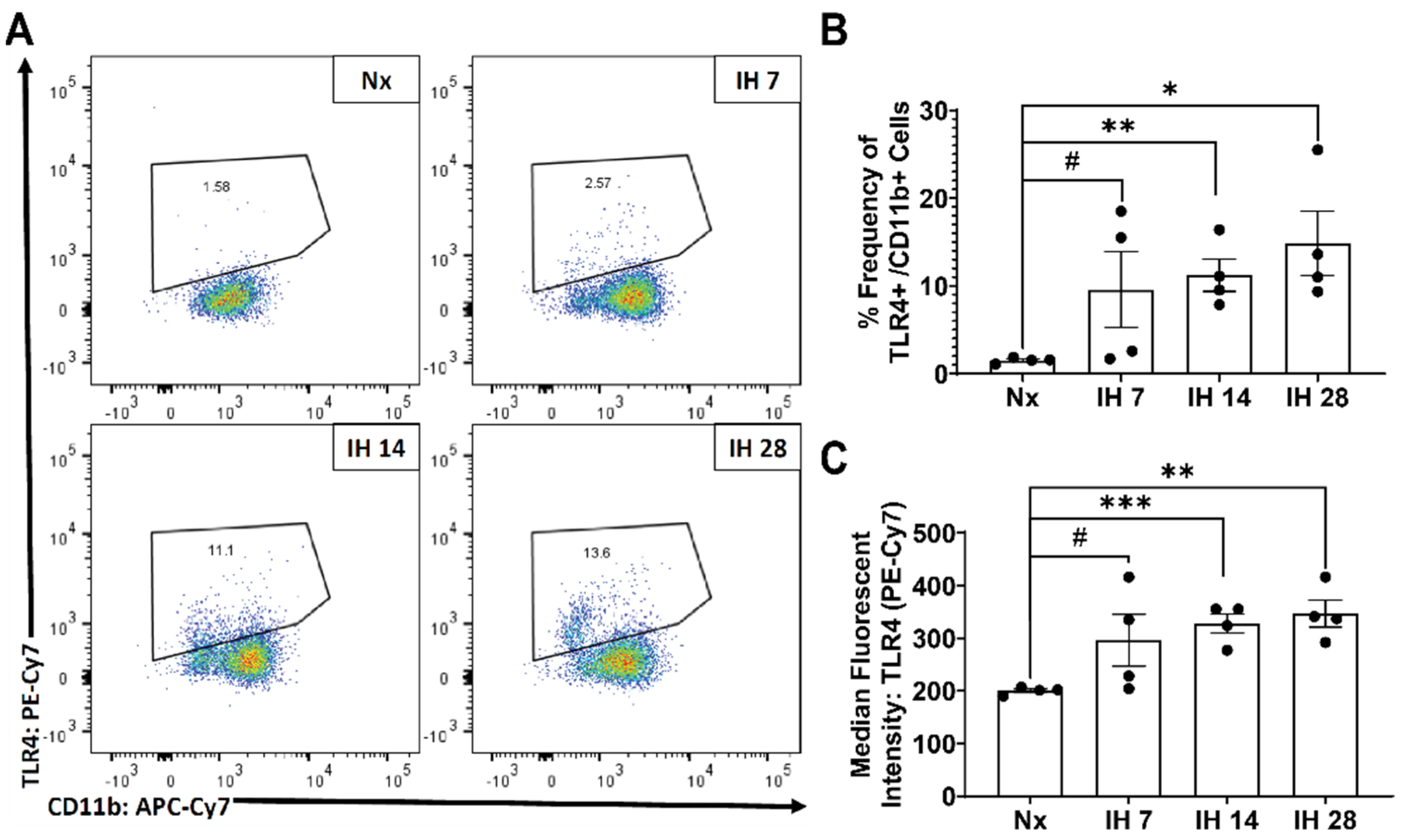
- Smith, S.M.C.; Friedle, S.A.; Watters, J.J. Chronic Intermittent Hypoxia Exerts CNS Region-Specific Effects on Rat Microglial Inflammatory and TLR4 Gene Expression. PLoS ONE 2013, 8, e81584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, D.; Dayyat, E.A.; Zhang, S.X.; Wang, Y.; Gozal, D. Intermittent Hypoxia-Induced Cognitive Deficits Are Mediated by NADPH Oxidase Activity in a Murine Model of Sleep Apnea. PLoS ONE 2011, 6, e19847. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.; Baril, A.A.; Gagnon, J.F.; Fortin, M.; Décary, A.; Lafond, C.; Desautels, A.; Montplaisir, J.; Gosselin, N. Cognitive Impairment in Obstructive Sleep Apnea. Pathol. Biol. 2014, 62, 233–240. [Google Scholar] [CrossRef]
- Osorio, R.S.; Gumb, T.; Pirraglia, E.; Varga, A.W.; Lu, S.; Lim, J.; Wohlleber, M.E.; Ducca, E.L.; Koushyk, V.; Glodzik, L.; et al. Sleep-Disordered Breathing Advances Cognitive Decline in the Elderly. Neurology 2015, 84, 1964–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, N.C.; Tien, K.J.; Yang, C.M.; Wang, J.J.; Weng, S.F. Increased Risk of Parkinson’s Disease in Patients with Obstructive Sleep Apnea: A Population-Based, Propensity Score-Matched, Longitudinal Follow-up Study. Medicine 2016, 95, e2293. [Google Scholar] [CrossRef]
- Chou, P.-S.; Lai, C.-L.; Chou, Y.-H.; Chang, W.-P. Sleep Apnea and the Subsequent Risk of Parkinson’s Disease: A 3-Year Nationwide Population-Based Study. Neuropsychiatr. Dis. Treat. 2017, 13, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Ancoli-Israel, S.; Klauber, M.R.; Butters, N.; Parker, L.; Kripke, D.F. Dementia in Institutionalized Elderly: Relation to Sleep Apnea. J. Am. Geriatr. Soc. 1991, 39, 258–263. [Google Scholar] [CrossRef]
- Ju, Y.-E.S.; Finn, M.B.; Sutphen, C.L.; Herries, E.M.; Jerome, G.M.; Ladenson, J.H.; Crimmins, D.L.; Fagan, A.M.; Holtzman, D.M. Obstructive Sleep Apnea Decreases Central Nervous System–Derived Proteins in the Cerebrospinal Fluid. Ann. Neurol. 2016, 80, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Liguori, C.; Mercuri, N.B.; Izzi, F.; Romigi, A.; Cordella, A.; Sancesario, G.; Placidi, F. Obstructive Sleep Apnea Is Associated With Early but Possibly Modifiable Alzheimer’s Disease Biomarkers Changes. Sleep 2017, 40, zsx011. [Google Scholar] [CrossRef] [Green Version]
- Liguori, C.; Mercuri, N.B.; Nuccetelli, M.; Izzi, F.; Cordella, A.; Bernardini, S.; Placidi, F. Obstructive Sleep Apnea May Induce Orexinergic System and Cerebral β-Amyloid Metabolism Dysregulation: Is It a Further Proof for Alzheimer’s Disease Risk? Sleep Med. 2019, 56, 171–176. [Google Scholar] [CrossRef]
- Mullins, A.E.; Kam, K.; Parekh, A.; Bubu, O.M.; Osorio, R.S.; Varga, A.W. Obstructive Sleep Apnea and Its Treatment in Aging: Effects on Alzheimer’s Disease Biomarkers, Cognition, Brain Structure and Neurophysiology. Neurobiol. Dis. 2020, 145, 105054. [Google Scholar] [CrossRef]
- Bu, X.-L.; Liu, Y.-H.; Wang, Q.-H.; Jiao, S.-S.; Zeng, F.; Yao, X.-Q.; Gao, D.; Chen, J.-C.; Wang, Y.-J. Serum Amyloid-Beta Levels Are Increased in Patients with Obstructive Sleep Apnea Syndrome. Sci. Rep. 2015, 5, 13917. [Google Scholar] [CrossRef] [Green Version]
- Motamedi, V.; Kanefsky, R.; Matsangas, P.; Mithani, S.; Jeromin, A.; Brock, M.S.; Mysliwiec, V.; Gill, J. Elevated Tau and Interleukin-6 Concentrations in Adults with Obstructive Sleep Apnea. Sleep Med. 2018, 43, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.P.; Yager, C.; Brandt, J.; Smith, G.S.; Zhou, Y.; Mathur, A.; Kumar, A.; Brašić, J.R.; Wong, D.F.; Wu, M.N. Objectively Measured Sleep and β-Amyloid Burden in Older Adults: A Pilot Study. SAGE Open Med. 2014, 2, 211454652. [Google Scholar] [CrossRef]
- Yun, C.-H.; Lee, H.-Y.; Lee, S.K.; Kim, H.; Seo, H.S.; Bang, S.A.; Kim, S.E.; Greve, D.N.; Au, R.; Shin, C.; et al. Amyloid Burden in Obstructive Sleep Apnea. J. Alzheimer’s Dis. 2017, 59, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Elias, A.; Cummins, T.; Tyrrell, R.; Lamb, F.; Dore, V.; Williams, R.; Rosenfeld, J.V.; Hopwood, M.; Villemagne, V.L.; Rowe, C.C. Risk of Alzheimer’s Disease in Obstructive Sleep Apnea Syndrome: Amyloid-β and Tau Imaging. J. Alzheimer’s Dis. 2018, 66, 733–741. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Rehel, S.; Kuhn, E.; Landeau, B.; Moulinet, I.; Touron, E.; Ourry, V.; le Du, G.; Mézenge, F.; Tomadesso, C.; et al. Association of Sleep-Disordered Breathing with Alzheimer Disease Biomarkers in Community-Dwelling Older Adults: A Secondary Analysis of a Randomized Clinical Trial. JAMA Neurol. 2020, 77, 716–724. [Google Scholar] [CrossRef]
- Boeve, B.F. Update on the Diagnosis and Management of Sleep Disturbances in Dementia. Sleep Med. Clin. 2008, 3, 347–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratti, L.; Viticchi, G.; Falsetti, L.; Cagnetti, C.; Luzzi, S.; Bartolini, M.; Provinciali, L.; Silvestrini, M. Vascular Impairment in Alzheimer’s Disease: The Role of Obstructive Sleep Apnea. J. Alzheimer’s Dis. 2014, 38, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Stark, C.D.; Stark, R.J. Sleep Apnoea and the Neurologist. Pract. Neurol. 2017, 17, 21–27. [Google Scholar] [CrossRef]
- Johnson, S.M.; Randhawa, K.S.; Epstein, J.J.; Gustafson, E.; Hocker, A.D.; Huxtable, A.G.; Baker, T.L.; Watters, J.J. Gestational Intermittent Hypoxia Increases Susceptibility to Neuroinflammation and Alters Respiratory Motor Control in Neonatal Rats. Respir. Physiol. Neurobiol. 2018, 256, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.E.S.; Videnovic, A.; Vaughn, B.V. Comorbid Sleep Disturbances in Neurologic Disorders. CONTINUUM Lifelong Learn. Neurol. 2017, 23, 1117–1131. [Google Scholar] [CrossRef]
- Snyder, B.; Shell, B.; Cunningham, J.T.; Cunningham, R.L. Chronic Intermittent Hypoxia Induces Oxidative Stress and Inflammation in Brain Regions Associated with Early-Stage Neurodegeneration. Physiol. Rep. 2017, 5, e13258. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Giles, W.H.; Croft, J.B.; Bliwise, D.L. Habitual Sleep Patterns and Risk for Stroke and Coronary Heart Disease: A 10-Year Follow-up from NHANES I. Neurology 1997, 48, 904–911. [Google Scholar] [CrossRef]
- Ayas, N.T.; White, D.P.; Al-Delaimy, W.K.; Manson, J.E.; Stampfer, M.J.; Speizer, F.E.; Patel, S.; Hu, F.B. A Prospective Study of Self-Reported Sleep Duration and Incident Diabetes in Women. Diabetes Care 2003, 26, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, K.; Tasali, E.; Leproult, R.; van Cauter, E. Effects of Poor and Short Sleep on Glucose Metabolism and Obesity Risk. Nat. Rev. Endocrinol. 2009, 5, 253–261. [Google Scholar] [CrossRef]
- Chen, J.-C.; Brunner, R.L.; Ren, H.; Wassertheil-Smoller, S.; Larson, J.C.; Levine, D.W.; Allison, M.; Naughton, M.J.; Stefanick, M.L. Sleep Duration and Risk of Ischemic Stroke in Postmenopausal Women. Stroke 2008, 39, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Shokri-Kojori, E.; Wang, G.; Wiers, C.; Demiral, S.; Guo, M.; Kim, S.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Holtzman, D. Bidirectional relationship between sleep and Alzheimer’s disease: Role of amyloid, tau, and other factors. Neuropsychopharmacology 2019, 45, 104–120. [Google Scholar] [CrossRef]
- Holth, J.; Fritschi, S.; Wang, C.; Pedersen, N.; Cirrito, J.; Mahan, T.; Finn, M.; Manis, M.; Geerling, J.; Fuller, P.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- Macey, P.M.; Kumar, R.; Woo, M.A.; Valladares, E.M.; Yan-Go, F.L.; Harper, R.M. Brain Structural Changes in Obstructive Sleep Apnea. Sleep 2008, 31, 967. [Google Scholar] [PubMed] [Green Version]
- Canessa, N.; Castronovo, V.; Cappa, S.F.; Aloia, M.S.; Marelli, S.; Falini, A.; Alemanno, F.; Ferini-Strambi, L. Obstructive Sleep Apnea: Brain Structural Changes and Neurocognitive Function before and after Treatment. Am. J. Respir. Crit. Care Med. 2012, 183, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.E.; BenediktsdÓttir, B.; Gislason, T.; Robinson, S.R. Neuropathological Investigation of Cell Layer Thickness and Myelination in the Hippocampus of People with Obstructive Sleep Apnea. Sleep 2019, 42, zsy199. [Google Scholar] [CrossRef] [Green Version]
- Giri, S.; Ranjan, A.; Kumar, A.; Amar, M.; Mallick, B.N. Rapid Eye Movement Sleep Deprivation Impairs Neuronal Plasticity and Reduces Hippocampal Neuronal Arborization in Male Albino Rats: Noradrenaline Is Involved in the Process. J. Neurosci. Res. 2021, 99, 1815–1834. [Google Scholar] [CrossRef]
- Biswas, S.; Mishra, P.; Mallick, B.N. Increased Apoptosis in Rat Brain after Rapid Eye Movement Sleep Loss. Neuroscience 2006, 142, 315–331. [Google Scholar] [CrossRef]
- Kamali, A.-M.; Noorafshan, A.; Karimi, F.; Karbalay-Doust, S.; Nami, M. The Impact of Chronic Sleep Restriction on Neuronal Number and Volumetric Correlates of the Dorsal Respiratory Nuclei in a Rat Model. Sleep 2017, 40, zsx072. [Google Scholar] [CrossRef] [Green Version]
- Noorafshan, A.; Karimi, F.; Karbalay-Doust, S.; Kamali, A.M. Using Curcumin to Prevent Structural and Behavioral Changes of Medial Prefrontal Cortex Induced by Sleep Deprivation in Rats. EXCLI J. 2017, 16, 520. [Google Scholar] [CrossRef]
- Novati, A.; Hulshof, H.J.; Koolhaas, J.M.; Lucassen, P.J.; Meerlo, P. Chronic Sleep Restriction Causes a Decrease in Hippocampal Volume in Adolescent Rats, Which Is Not Explained by Changes in Glucocorticoid Levels or Neurogenesis. Neuroscience 2011, 190, 145–155. [Google Scholar] [CrossRef]
- Noorafshan, A.; Karimi, F.; Kamali, A.M.; Karbalay-Doust, S.; Nami, M. Restorative Effects of Curcumin on Sleep-Deprivation Induced Memory Impairments and Structural Changes of the Hippocampus in a Rat Model. Life Sci. 2017, 189, 63–70. [Google Scholar] [CrossRef]
- Sethi, M.; Joshi, S.S.; Webb, R.L.; Beckett, T.L.; Donohue, K.D.; Murphy, M.P.; O’Hara, B.F.; Duncan, M.J. Increased Fragmentation of Sleep–Wake Cycles in the 5XFAD Mouse Model of Alzheimer’s Disease. Neuroscience 2015, 290, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Punjabi, N.M. The Epidemiology of Adult Obstructive Sleep Apnea. Proc. Am. Thorac. Soc. 2008, 5, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Pien, G.W.; Weaver, T.E. Gender Differences in the Clinical Manifestation of Obstructive Sleep Apnea. Sleep Med. 2009, 10, 1075–1084. [Google Scholar] [CrossRef]
- Semelka, M.; Wilson, J.; Floyd, R. Diagnosis and Treatment of Obstructive Sleep Apnea in Adults. Am. Fam. Physician 2016, 94, 355–361. [Google Scholar] [PubMed]
- Khalyfa, A.; Kheirandish-Gozal, L.; Gozal, D. Exosome and Macrophage Crosstalk in Sleep-Disordered Breathing-Induced Metabolic Dysfunction. Int. J. Mol. Sci. 2018, 19, 3383. [Google Scholar] [CrossRef] [Green Version]
- Melamed, K.H.; Goldhaber, S.Z. Obstructive Sleep Apnea. Circulation 2015, 132, e114–e116. [Google Scholar] [CrossRef] [Green Version]
- Basoglu, O.K.; Tasbakan, M.S. Gender Differences in Clinical and Polysomnographic Features of Obstructive Sleep Apnea: A Clinical Study of 2827 Patients. Sleep Breath. 2017, 22, 241–249. [Google Scholar] [CrossRef]
- Helvaci, N.; Karabulut, E.; Demir, A.U.; Yildiz, B.O. Polycystic Ovary Syndrome and the Risk of Obstructive Sleep Apnea: A Meta-Analysis and Review of the Literature. Endocr. Connect. 2017, 6, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Randeva, H.S.; Tan, B.K.; Weickert, M.O.; Lois, K.; Nestler, J.E.; Sattar, N.; Lehnert, H. Cardiometabolic Aspects of the Polycystic Ovary Syndrome. Endocr. Rev. 2012, 33, 812–841. [Google Scholar] [CrossRef]
- Sathish, V.; Martin, Y.N.; Prakash, Y.S. Sex Steroid Signaling: Implications for Lung Diseases. Pharmacol. Ther. 2015, 150, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Young, T.; Finn, L.; Austin, D.; Peterson, A. Menopausal Status and Sleep-Disordered Breathing in the Wisconsin Sleep Cohort Study. Am. J. Respir. Crit. Care Med. 2003, 167, 1181–1185. [Google Scholar] [CrossRef]
- Young, T.; Rabago, D.; Zgierska, A.; Austin, D.; Finn, L. Objective and Subjective Sleep Quality in Premenopausal, Perimenopausal, and Postmenopausal Women in the Wisconsin Sleep Cohort Study. Sleep 2003, 26, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harbor Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [Green Version]
- Yaffe, K.; Laffan, A.M.; Harrison, S.L.; Redline, S.; Spira, A.P.; Ensrud, K.E.; Ancoli-Israel, S.; Stone, K.L. Sleep-Disordered Breathing, Hypoxia, and Risk of Mild Cognitive Impairment and Dementia in Older Women. JAMA 2011, 306, 613–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasgon, N.L.; Kenna, H.A. Insulin Resistance in Depressive Disorders and Alzheimer’s Disease: Revisiting the Missing Link Hypothesis. Neurobiol. Aging 2005, 26, 103–107. [Google Scholar] [CrossRef]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical Epidemiology of Alzheimer’s Disease: Assessing Sex and Gender Differences. Clin. Epidemiol. 2014, 2014, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L.; Prabhakar, N.R. Neural Regulation of Hypoxia-Inducible Factors and Redox State Drives the Pathogenesis of Hypertension in a Rodent Model of Sleep Apnea. J. Appl. Physiol. 2015, 119, 1152–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, A.M.; Mehra, R. Obstructive Sleep Apnea: Role of Intermittent Hypoxia and Inflammation. Semin. Respir. Crit. Care Med. 2014, 35, 531–544. [Google Scholar] [CrossRef]
- Hedner, J.; Bengtsson-Boström, K.; Peker, Y.; Grote, L.; Råstam, L.; Lindblad, U. Hypertension Prevalence in Obstructive Sleep Apnoea and Sex: A Population-Based Case–Control Study. Eur. Respir. J. 2006, 27, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Yin, G.; Zhang, P.; Song, Z.; Chen, Y.; Zhang, D.; Hu, W. Distinct Associations between Hypertension and Obstructive Sleep Apnea in Male and Female Patients. PLoS ONE 2014, 9, e113076. [Google Scholar] [CrossRef] [Green Version]
- Knight, W.D.; Little, J.T.; Carreno, F.R.; Toney, G.M.; Mifflin, S.W.; Cunningham, J.T. Chronic Intermittent Hypoxia Increases Blood Pressure and Expression of FosB/ΔFosB in Central Autonomic Regions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R131–R139. [Google Scholar] [CrossRef] [Green Version]
- Hinojosa-Laborde, C.; Mifflin, S.W. Sex Differences in Blood Pressure Response to Intermittent Hypoxia in Rats. Hypertension 2005, 46, 1016–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, E.; Bakker, J.P.; Clarke, D.N.; Csizmadia, E.; Kocher, O.; Veves, A.; Tecilazich, F.; O’Donnell, C.P.; Ferran, C.; Malhotra, A. Molecular Biomarkers of Vascular Dysfunction in Obstructive Sleep Apnea. PLoS ONE 2013, 8, e70559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamberger, M.E.; Landreth, G.E. Inflammation, Apoptosis, and Alzheimer’s Disease. Neuroscientist 2002, 8, 276–283. [Google Scholar] [CrossRef]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, Immunity, and Hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, M.; Snyder, H.M.; Carrillo, M.C.; Fazio, S.; Kim, H.; Johns, H. Summary of the Evidence on Modifiable Risk Factors for Cognitive Decline and Dementia: A Population-Based Perspective. Alzheimer’s Dement. 2015, 11, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Kheirandish-Gozal, L.; Sans Capdevila, O.; Kheirandish, E.; Gozal, D. Elevated Serum Aminotransferase Levels in Children at Risk for Obstructive Sleep Apnea. Chest 2008, 133, 92–99. [Google Scholar] [CrossRef]
- Kheirandish-Gozal, L.; Philby, M.F.; Alonso-Álvarez, M.L.; Terán-Santos, J.; Gozal, D. Biomarkers of Alzheimer Disease in Children with Obstructive Sleep Apnea: Effect of Adenotonsillectomy. Sleep 2016, 39, 1225–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpagnano, G.E.; Kharitonov, S.A.; Resta, O.; Foschino-Barbaro, M.P.; Gramiccioni, E.; Barnes, P.J. Increased 8-Isoprostane and Interleukin-6 in Breath Condensate of Obstructive Sleep Apnea Patients. Chest 2002, 122, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Bouloukaki, I.; Mermigkis, C.; Tzanakis, N.; Kallergis, E.; Moniaki, V.; Mauroudi, E.; Schiza, S.E. Evaluation of Inflammatory Markers in a Large Sample of Obstructive Sleep Apnea Patients without Comorbidities. Mediat. Inflamm. 2017, 2017, 4573756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Peña Bravo, M.; Serpero, L.D.; Barceló, A.; Barbé, F.; Agustí, A.; Gozal, D. Inflammatory Proteins in Patients with Obstructive Sleep Apnea with and without Daytime Sleepiness. Sleep Breath. 2007, 11, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Minoguchi, K.; Tazaki, T.; Yokoe, T.; Minoguchi, H.; Watanabe, Y.; Yamamoto, M.; Adachi, M. Elevated Production of Tumor Necrosis Factor-α by Monocytes in Patients With Obstructive Sleep Apnea Syndrome. Chest 2004, 126, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, T.U.; Kokturk, O.; Bukan, N.; Bilgihan, A. The Relationship between Serum Cytokine Levels with Obesity and Obstructive Sleep Apnea Syndrome. Cytokine 2004, 28, 87–91. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Papanicolaou, D.A.; Bixler, E.O.; Kales, A.; Tyson, K.; Chrousos, G.P. Elevation of Plasma Cytokines in Disorders of Excessive Daytime Sleepiness: Role of Sleep Disturbance and Obesity. J. Clin. Endocrinol. Metab. 1997, 82, 1313–1316. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Papanicolaou, D.A.; Bixler, E.O.; Hopper, K.; Lotsikas, A.; Lin, H.-M.; Kales, A.; Chrousos, G.P. Sleep Apnea and Daytime Sleepiness and Fatigue: Relation to Visceral Obesity, Insulin Resistance, and Hypercytokinemia. J. Clin. Endocrinol. Metab. 2000, 85, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Briançon-Marjollet, A.; Monneret, D.; Henri, M.; Hazane-Puch, F.; Pepin, J.-L.; Faure, P.; Godin-Ribuot, D. Endothelin Regulates Intermittent Hypoxia-Induced Lipolytic Remodelling of Adipose Tissue and Phosphorylation of Hormone-Sensitive Lipase. J. Physiol. 2016, 594, 1727–1740. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, I.; Williams, S.C.R.; Morrella, M.J. The Impact of Sleep and Hypoxia on the Brain: Potential Mechanisms for the Effects of Obstructive Sleep Apnea. Curr. Opin. Pulm. Med. 2014, 20, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Sapin, E.; Peyron, C.; Roche, F.; Gay, N.; Carcenac, C.; Savasta, M.; Levy, P.; Dematteis, M. Chronic Intermittent Hypoxia Induces Chronic Low-Grade Neuroinflammation in the Dorsal Hippocampus of Mice. Sleep 2015, 38, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
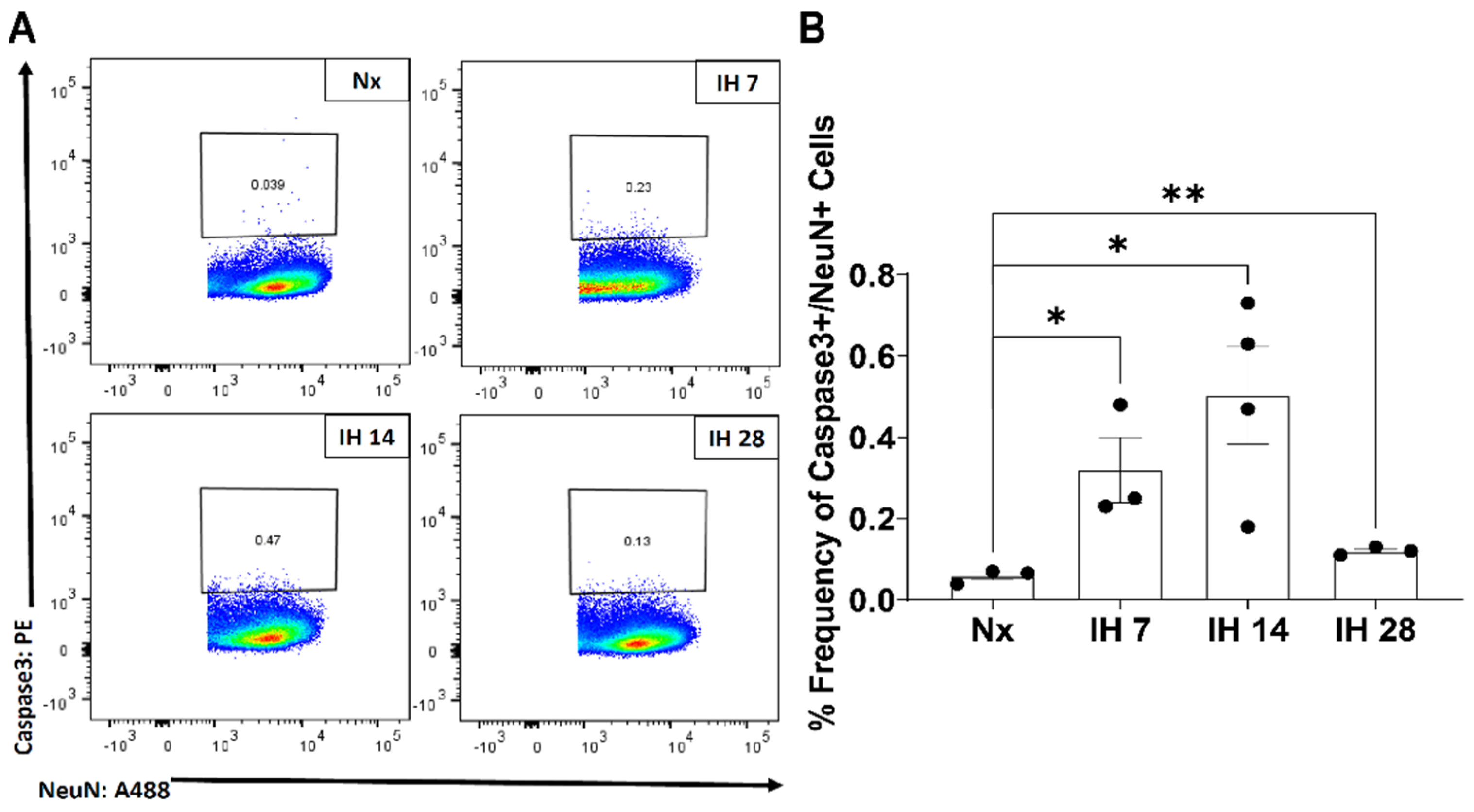
- Gozal, D.; Row, B.W.; Kheirandish, L.; Liu, R.; Guo, S.Z.; Qiang, F.; Brittian, K.R. Increased Susceptibility to Intermittent Hypoxia in Aging Rats: Changes in Proteasomal Activity, Neuronal Apoptosis and Spatial Function. J. Neurochem. 2003, 86, 1545–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitz, C.; Mayeux, R. Alzheimer Disease: Epidemiology, Diagnostic Criteria, Risk Factors and Biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; del Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, J.T.; Knight, W.D.; Mifflin, S.W.; Nestler, E.J. An Essential Role for ΔFosB in the Median Preoptic Nucleus in the Sustained Hypertensive Effects of Chronic Intermittent Hypoxia. Hypertension 2012, 60, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-C.; Wang, Y.-J.; Tseng, G.-F. Ascorbic Acid and α-Tocopherol Supplement Starting Prenatally Enhances the Resistance of Nucleus Tractus Solitarius Neurons to Hypobaric Hypoxic Challenge. Brain Struct. Funct. 2011, 216, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Kheirandish, L.; Row, B.W.; Li, R.C.; Brittian, K.R.; Gozal, D. Apolipoprotein E-Deficient Mice Exhibit Increased Vulnerability to Intermittent Hypoxia-Induced Spatial Learning Deficits. Sleep 2005, 28, 1412–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagaito, Y.; Polotsky, V.Y.; Campen, M.J.; Wilson, J.A.; Balbir, A.; Smith, P.L.; Schwartz, A.R.; O’Donnell, C.P. A Model of Sleep-Disordered Breathing in the C57BL/6J Mouse. J. Appl. Physiol. 2001, 91, 2758–2766. [Google Scholar] [CrossRef]
- Smith, S.M.C.; Kimyon, R.S.; Watters, J.J. Cell-Type-Specific Jumonji Histone Demethylase Gene Expression in the Healthy Rat CNS: Detection by a Novel Flow Cytometry Method. ASN Neuro 2014, 6, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lykidis, D.; van Noorden, S.; Armstrong, A.; Spencer-Dene, B.; Li, J.; Zhuang, Z.; Stamp, G.W.H. Novel Zinc-Based Fixative for High Quality DNA, RNA and Protein Analysis. Nucleic Acids Res. 2007, 35, e85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, U.B.; Owens, D.M.; Pedersen, S.; Christensen, R. Zinc Fixation Preserves Flow Cytometry Scatter and Fluorescence Parameters and Allows Simultaneous Analysis of DNA Content and Synthesis, and Intracellular and Surface Epitopes. Cytom. Part A 2010, 77, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, E.A.; Wang, T.; Vanderplow, A.M.; Cherukuri, S.; Cahill, M.E.; Watters, J.J. Neonatal Intermittent Hypoxia Induces Lasting Sex-Specific Augmentation of Rat Microglial Cytokine Expression. Front. Immunol. 2019, 10, 1479. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alzheimer’s Disease | Sleep Disordered Breathing | |
|---|---|---|
| Neuroinflammation | X | X |
| Inflammasome Activation | X | ? |
| Hypoxia | X | |
| Tau Pathology | X | |
| Amyloid β Plaques | X | ? |
| Hypertension | X | X |
| Sex Differences | X | X |
| Neuronal Apoptosis | X | X |
| Microglial Activation | X | X |
| Astrocyte Activation | X | X |
| Sleep Disturbances | X | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulland, T.K.; Ewald, A.C.; Knutson, A.O.; Marino, K.M.; Smith, S.M.C.; Watters, J.J. Alzheimer’s Disease, Sleep Disordered Breathing, and Microglia: Puzzling out a Common Link. Cells 2021, 10, 2907. https://doi.org/10.3390/cells10112907
Ulland TK, Ewald AC, Knutson AO, Marino KM, Smith SMC, Watters JJ. Alzheimer’s Disease, Sleep Disordered Breathing, and Microglia: Puzzling out a Common Link. Cells. 2021; 10(11):2907. https://doi.org/10.3390/cells10112907
Chicago/Turabian StyleUlland, Tyler K., Andrea C. Ewald, Andrew O. Knutson, Kaitlyn M. Marino, Stephanie M. C. Smith, and Jyoti J. Watters. 2021. "Alzheimer’s Disease, Sleep Disordered Breathing, and Microglia: Puzzling out a Common Link" Cells 10, no. 11: 2907. https://doi.org/10.3390/cells10112907
APA StyleUlland, T. K., Ewald, A. C., Knutson, A. O., Marino, K. M., Smith, S. M. C., & Watters, J. J. (2021). Alzheimer’s Disease, Sleep Disordered Breathing, and Microglia: Puzzling out a Common Link. Cells, 10(11), 2907. https://doi.org/10.3390/cells10112907