
Mechanisms, Diagnosis and Treatment of Bone Metastases


{kind=link}
{kind=link}
Abstract
:1. Introduction
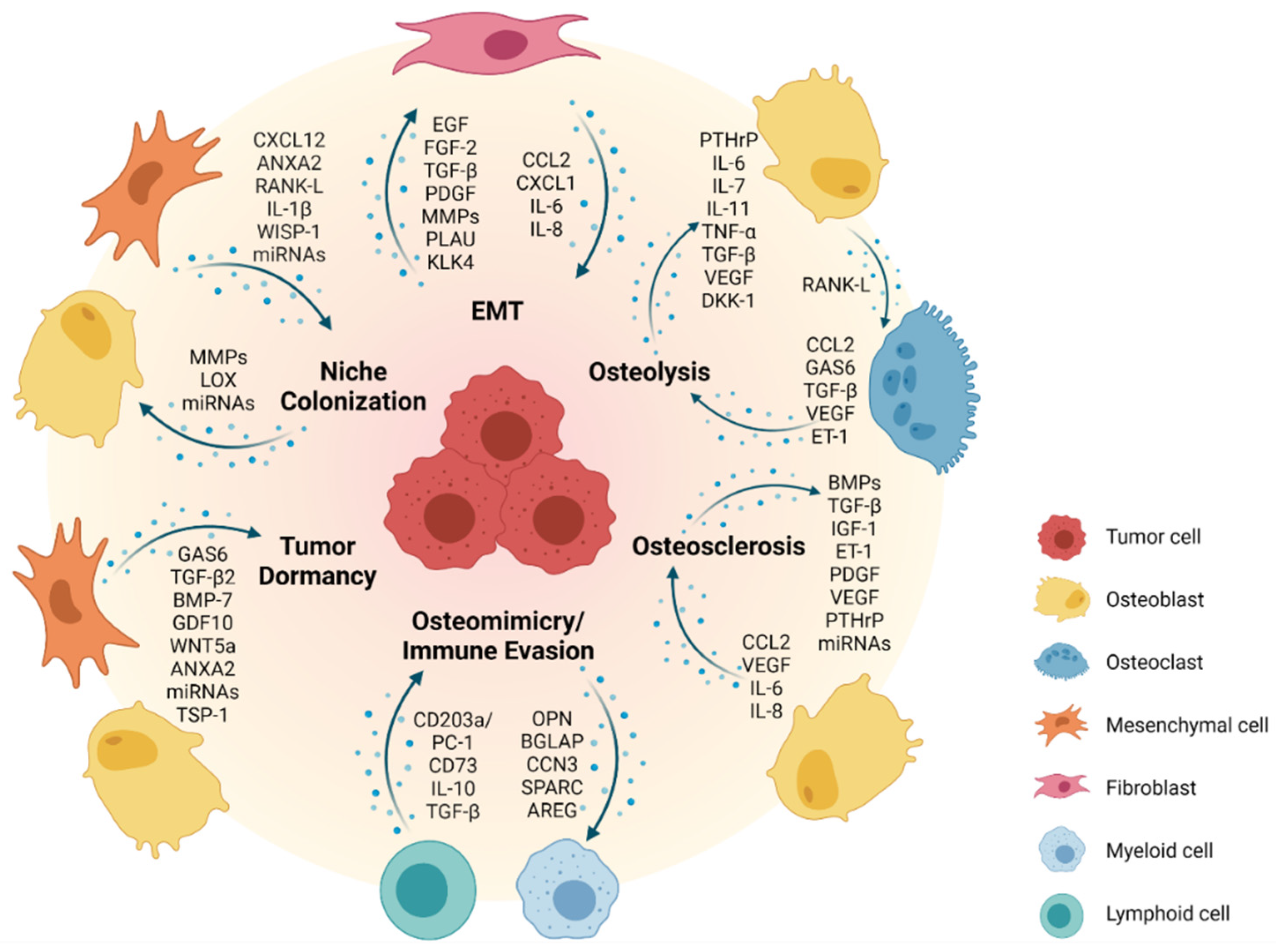
2. Mechanisms of Metastasis
2.1. EMT—The First Step towards Formation of Metastasis
2.2. Formation of a Pre-Metastatic Niche and Bone Colonization
2.3. Metastatic Dormancy and Reactivation in the Bone Niche
2.4. Reconstruction: Osteoblastic versus Osteolytic Bone Metastases
2.5. Bone Microenvironment and Bone Sarcomas
2.6. Bone Metastases in Extraosseous Pediatric Solid Tumors
3. Diagnosis and Therapy of Bone Metastases
3.1. Approaches to Biologically Targeted Therapy
3.2. Potentials for Immunotherapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dillekas, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [Green Version]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Stamatopoulos, A.; Stamatopoulos, T.; Gamie, Z.; Kenanidis, E.; Ribeiro, R.D.C.; Rankin, K.S.; Gerrand, C.; Dalgarno, K.; Tsiridis, E. Mesenchymal stromal cells for bone sarcoma treatment: Roadmap to clinical practice. J. Bone Oncol. 2019, 16, 100231. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.F.; Shen, J.; Li, X.; Rengan, R.; Silvestris, N.; Wang, M.; Derosa, L.; Zheng, X.; Belli, A.; Zhang, X.L.; et al. Incidence of patients with bone metastases at diagnosis of solid tumors in adults: A large population-based study. Ann. Transl. Med. 2020, 8, 482. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.M.; Lange, P.H.; Porter, M.P.; Lin, D.W.; Ellis, W.J.; Gallaher, I.S.; Vessella, R.L. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin. Cancer Res. 2009, 15, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Tjensvoll, K.; Oltedal, S.; Heikkilä, R.; Kvaløy, J.T.; Gilje, B.; Reuben, J.M.; Smaaland, R.; Nordgård, O. Persistent tumor cells in bone marrow of non-metastatic breast cancer patients after primary surgery are associated with inferior outcome. BMC Cancer 2012, 12, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochheuser, C.; Windt, L.J.; Kunze, N.Y.; de Vos, D.L.; Tytgat, G.A.M.; Voermans, C.; Timmerman, I. Mesenchymal Stromal Cells in Neuroblastoma: Exploring Crosstalk and Therapeutic Implications. Stem Cells Dev. 2021, 30, 59–78. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Zhang, G.; Li, S.; Duan, J.; Cheng, J.; Ding, G.; Zhou, C.; Zhang, J.; Luo, P.; et al. Osteosarcoma metastasis: Prospective role of ezrin. Tumour Biol. 2014, 35, 5055–5059. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Coleman, R.E.; Michalski, J.M.; Nilsson, S.; O’Sullivan, J.M.; Parker, C.; Widmark, A.; Thuresson, M.; Xu, L.; Germino, J.; et al. Hematologic Safety of Radium-223 Dichloride: Baseline Prognostic Factors Associated With Myelosuppression in the ALSYMPCA Trial. Clin. Genitourin Cancer 2017, 15, 42–52.e8. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.E.; Croucher, P.I.; Padhani, A.R.; Clézardin, P.; Chow, E.; Fallon, M.; Guise, T.; Colangeli, S.; Capanna, R.; Costa, L. Bone metastases. Nat. Rev. Dis. Primers 2020, 6, 83. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Massagué, J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer 2015, 1, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X. Interactions between cancer cells and bone microenvironment promote bone metastasis in prostate cancer. Cancer Commun. 2019, 39, 76. [Google Scholar] [CrossRef] [Green Version]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef]
- Clezardin, P.; Coleman, R.; Puppo, M.; Ottewell, P.; Bonnelye, E.; Paycha, F.; Confavreux, C.B.; Holen, I. Bone metastasis: Mechanisms, therapies, and biomarkers. Physiol. Rev. 2021, 101, 797–855. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Wang, N.; Meek, J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocr.-Relat. Cancer 2014, 21, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Fidler, I.J. Metastasis: Quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2′-deoxyuridine. J. Natl. Cancer Inst. 1970, 45, 773–782. [Google Scholar] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zhang, C.; Guo, Q.; Xu, Y.; Feng, G.; Li, L.; Han, X.; Lu, F.; Ma, Y.; Wang, X.; et al. The homogeneous and heterogeneous risk factors for the morbidity and prognosis of bone metastasis in patients with prostate cancer. Cancer Manag. Res. 2018, 10, 1639–1646. [Google Scholar] [CrossRef] [Green Version]
- Sathiakumar, N.; Delzell, E.; Morrisey, M.A.; Falkson, C.; Yong, M.; Chia, V.; Blackburn, J.; Arora, T.; Kilgore, M.L. Mortality following bone metastasis and skeletal-related events among men with prostate cancer: A population-based analysis of US Medicare beneficiaries, 1999–2006. Prostate Cancer Prostatic Dis. 2011, 14, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Bado, I.L.; Hu, J.; Wan, Y.W.; Wu, L.; Wang, H.; Gao, Y.; Jeong, H.H.; Xu, Z.; Hao, X.; et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 2021, 184, 2471–2486.e20. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Ottewell, P.D.; O’Donnell, L.; Holen, I. Molecular alterations that drive breast cancer metastasis to bone. Bonekey Rep. 2015, 4, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Thompson, E.W.; Paik, S.; Brünner, N.; Sommers, C.L.; Zugmaier, G.; Clarke, R.; Shima, T.B.; Torri, J.; Donahue, S.; Lippman, M.E.; et al. Association of increased basement membrane invasiveness with absence of estrogen receptor and expression of vimentin in human breast cancer cell lines. J. Cell. Physiol. 1992, 150, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.L.; Heckford, S.E.; Skerker, J.M.; Worland, P.; Torri, J.A.; Thompson, E.W.; Byers, S.W.; Gelmann, E.P. Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res. 1992, 52, 5190–5197. [Google Scholar] [PubMed]
- Gilles, C.; Polette, M.; Zahm, J.M.; Tournier, J.M.; Volders, L.; Foidart, J.M.; Birembaut, P. Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci 1999, 112 Pt. 24, 4615–4625. [Google Scholar] [CrossRef]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Hazan, R.B.; Phillips, G.R.; Qiao, R.F.; Norton, L.; Aaronson, S.A. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 2000, 148, 779–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, K.; van Bokhoven, A.; van Leenders, G.J.; Ruijter, E.T.; Jansen, C.F.; Bussemakers, M.J.; Schalken, J.A. Cadherin switching in human prostate cancer progression. Cancer Res. 2000, 60, 3650–3654. [Google Scholar]
- Jaggi, M.; Nazemi, T.; Abrahams, N.A.; Baker, J.J.; Galich, A.; Smith, L.M.; Balaji, K.C. N-cadherin switching occurs in high Gleason grade prostate cancer. Prostate 2006, 66, 193–199. [Google Scholar] [CrossRef] [PubMed]
- McAlhany, S.J.; Ayala, G.E.; Frolov, A.; Ressler, S.J.; Wheeler, T.M.; Watson, J.E.; Collins, C.; Rowley, D.R. Decreased stromal expression and increased epithelial expression of WFDC1/ps20 in prostate cancer is associated with reduced recurrence-free survival. Prostate 2004, 61, 182–191. [Google Scholar] [CrossRef]
- Derycke, L.; De Wever, O.; Stove, V.; Vanhoecke, B.; Delanghe, J.; Depypere, H.; Bracke, M. Soluble N-cadherin in human biological fluids. Int. J. Cancer 2006, 119, 2895–2900. [Google Scholar] [CrossRef]
- Chunthapong, J.; Seftor, E.A.; Khalkhali-Ellis, Z.; Seftor, R.E.; Amir, S.; Lubaroff, D.M.; Heidger, P.M., Jr.; Hendrix, M.J. Dual roles of E-cadherin in prostate cancer invasion. J. Cell. Biochem. 2004, 91, 649–661. [Google Scholar] [CrossRef]
- Lu, Z.; Ghosh, S.; Wang, Z.; Hunter, T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4, 499–515. [Google Scholar] [CrossRef] [Green Version]
- Wells, C.M.; Ahmed, T.; Masters, J.R.; Jones, G.E. Rho family GTPases are activated during HGF-stimulated prostate cancer-cell scattering. Cell Motil. Cytoskelet. 2005, 62, 180–194. [Google Scholar] [CrossRef]
- Alexander, N.R.; Tran, N.L.; Rekapally, H.; Summers, C.E.; Glackin, C.; Heimark, R.L. N-cadherin gene expression in prostate carcinoma is modulated by integrin-dependent nuclear translocation of Twist1. Cancer Res. 2006, 66, 3365–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, W.K.; Ling, M.T.; Lee, T.W.; Lau, T.C.; Zhou, C.; Zhang, X.; Chua, C.W.; Chan, K.W.; Chan, F.L.; Glackin, C.; et al. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005, 65, 5153–5162. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Zerbini, L.F.; Otu, H.H.; Bhasin, M.; Yang, Q.; Joseph, M.G.; Grall, F.; Onatunde, T.; Correa, R.G.; Libermann, T.A. Reduced PDEF expression increases invasion and expression of mesenchymal genes in prostate cancer cells. Cancer Res. 2007, 67, 4219–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veveris-Lowe, T.L.; Lawrence, M.G.; Collard, R.L.; Bui, L.; Herington, A.C.; Nicol, D.L.; Clements, J.A. Kallikrein 4 (hK4) and prostate-specific antigen (PSA) are associated with the loss of E-cadherin and an epithelial-mesenchymal transition (EMT)-like effect in prostate cancer cells. Endocr.-Relat. Cancer 2005, 12, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitbread, A.K.; Veveris-Lowe, T.L.; Lawrence, M.G.; Nicol, D.L.; Clements, J.A. The role of kallikrein-related peptidases in prostate cancer: Potential involvement in an epithelial to mesenchymal transition. Biol. Chem. 2006, 387, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhong, C.; Frenkel, B.; Reddi, A.H.; Roy-Burman, P. Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res. 2005, 65, 5769–5777. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, R.; Xie, Z.H.; Odero-Marah, V.; Pathak, S.; Multani, A.; Chung, L.W.; Zhau, H.E. Prostate cancer metastasis: Role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate 2006, 66, 1664–1673. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Mazo, I.B.; von Andrian, U.H. Adhesion and homing of blood-borne cells in bone marrow microvessels. J. Leukoc. Biol. 1999, 66, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef]
- Ren, G.; Esposito, M.; Kang, Y. Bone metastasis and the metastatic niche. J. Mol. Med. 2015, 93, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massague, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 154, 1060–1073. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar]
- Wang, J.; Loberg, R.; Taichman, R.S. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006, 25, 573–587. [Google Scholar] [CrossRef]
- Hung, C.S.; Su, H.Y.; Liang, H.H.; Lai, C.W.; Chang, Y.C.; Ho, Y.S.; Wu, C.H.; Ho, J.D.; Wei, P.L.; Chang, Y.J. High-level expression of CXCR4 in breast cancer is associated with early distant and bone metastases. Tumour Biol. 2014, 35, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Wang, J.; Lee, E.; McGee, S.; Berry, J.E.; Yumoto, K.; Dai, J.; Keller, E.T.; Shiozawa, Y.; Taichman, R.S. Annexin 2-CXCL12 interactions regulate metastatic cell targeting and growth in the bone marrow. Mol. Cancer Res. 2015, 13, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, G.C.; Zhau, H.E.; Wang, R.; Rogatko, A.; Feng, X.; Zayzafoon, M.; Liu, Y.; Farach-Carson, M.C.; You, S.; Kim, J.; et al. RANK- and c-Met-mediated signal network promotes prostate cancer metastatic colonization. Endocr.-Relat. Cancer 2014, 21, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Woodward, J.K.; Holen, I.; Coleman, R.E.; Buttle, D.J. The roles of proteolytic enzymes in the development of tumour-induced bone disease in breast and prostate cancer. Bone 2007, 41, 912–927. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Casimiro, S.; Mohammad, K.S.; Pires, R.; Tato-Costa, J.; Alho, I.; Teixeira, R.; Carvalho, A.; Ribeiro, S.; Lipton, A.; Guise, T.A.; et al. RANKL/RANK/MMP-1 molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells in vitro. PLoS ONE 2013, 8, e63153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R.; Rumney, R.M.H.; Schoof, E.M.; Perryman, L.; Hoye, A.M.; Agrawal, A.; Bird, D.; Latif, N.A.; Forrest, H.; Evans, H.R.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Zoni, E.; van der Pluijm, G. The role of microRNAs in bone metastasis. J. Bone Oncol. 2016, 5, 104–108. [Google Scholar] [CrossRef]
- Ell, B.; Mercatali, L.; Ibrahim, T.; Campbell, N.; Schwarzenbach, H.; Pantel, K.; Amadori, D.; Kang, Y. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell 2013, 24, 542–556. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.P.; De, S.; Vasanji, A.; Brainard, J.; Byzova, T.V. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene 2007, 26, 6238–6243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8, R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.C.; Chen, P.C.; Lin, Y.F.; Su, C.M.; Liu, J.F.; Lin, T.H.; Chuang, S.M.; Tang, C.H. Osteoblast-secreted WISP-1 promotes adherence of prostate cancer cells to bone via the VCAM-1/integrin alpha4beta1 system. Cancer Lett 2018, 426, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Eyre, R.; Alferez, D.G.; Santiago-Gomez, A.; Spence, K.; McConnell, J.C.; Hart, C.; Simoes, B.M.; Lefley, D.; Tulotta, C.; Storer, J.; et al. Microenvironmental IL1beta promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat. Commun. 2019, 10, 5016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra373. [Google Scholar] [CrossRef]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.T.; Smit, D.J.; Taipaleenmäki, H. The Endosteal Niche in Breast Cancer Bone Metastasis. Front. Oncol. 2020, 10, 335. [Google Scholar] [CrossRef] [Green Version]
- Mayhew, V.; Omokehinde, T.; Johnson, R.W. Tumor dormancy in bone. Cancer Rep. 2020, 3, e1156. [Google Scholar] [CrossRef]
- Templeton, Z.S.; Lie, W.R.; Wang, W.; Rosenberg-Hasson, Y.; Alluri, R.V.; Tamaresis, J.S.; Bachmann, M.H.; Lee, K.; Maloney, W.J.; Contag, C.H.; et al. Breast Cancer Cell Colonization of the Human Bone Marrow Adipose Tissue Niche. Neoplasia 2015, 17, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and disseminated tumour cells—Mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Rucci, N.; Teti, A. Osteomimicry: How the Seed Grows in the Soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef]
- Reinstein, Z.Z.; Pamarthy, S.; Sagar, V.; Costa, R.; Abdulkadir, S.A.; Giles, F.J.; Carneiro, B.A. Overcoming immunosuppression in bone metastases. Crit. Rev. Oncol. Hematol. 2017, 117, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pan, H.; Shen, L. Pan-Cancer Analyses Reveal Prognostic Value of Osteomimicry Across 20 Solid Cancer Types. Front. Mol. Biosci. 2020, 7, 576269. [Google Scholar] [CrossRef]
- Byrne, N.M.; Summers, M.A.; McDonald, M.M. Tumor Cell Dormancy and Reactivation in Bone: Skeletal Biology and Therapeutic Opportunities. JBMR Plus 2019, 3, e10125. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Gomatou, G.; Syrigos, N.; Vathiotis, I.A.; Kotteas, E.A. Tumor Dormancy: Implications for Invasion and Metastasis. Int. J. Mol. Sci. 2021, 22, 4862. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [Green Version]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Kan, C.; Vargas, G.; Pape, F.L.; Clézardin, P. Cancer Cell Colonisation in the Bone Microenvironment. Int. J. Mol. Sci. 2016, 17, 1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Prunier, C.; Baker, D.; Ten Dijke, P.; Ritsma, L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Dai, Y.; Yang, Q.; Zhang, X.; Guo, W.; Ye, L.; Huang, S.; Chen, X.; Lai, Y.; Du, H.; et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J. Exp. Med. 2019, 216, 428–449. [Google Scholar] [CrossRef]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Nobre, A.R.; Risson, E.; Singh, D.K.; Di Martino, J.S.; Cheung, J.F.; Wang, J.; Johnson, J.; Russnes, H.G.; Bravo-Cordero, J.J.; Birbrair, A.; et al. Bone marrow NG2+/Nestin+ mesenchymal stem cells drive DTC dormancy via TGF-β2. Nat. Cancer 2021, 2, 327–339. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Shupp, A.B.; Kolb, A.D.; Mukhopadhyay, D.; Bussard, K.M. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers 2018, 10, 182. [Google Scholar] [CrossRef] [Green Version]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Reeves, K.J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin. Cancer Res. 2014, 20, 2922–2932. [Google Scholar] [CrossRef] [Green Version]
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int. J. Cancer 2015, 137, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; He, Y.; Yu, X. Bone Marrow Adipocyte: An Intimate Partner With Tumor Cells in Bone Metastasis. Front. Endocrinol. 2018, 9, 339. [Google Scholar] [CrossRef]
- Phadke, P.A.; Mercer, R.R.; Harms, J.F.; Jia, Y.; Frost, A.R.; Jewell, J.L.; Bussard, K.M.; Nelson, S.; Moore, C.; Kappes, J.C.; et al. Kinetics of metastatic breast cancer cell trafficking in bone. Clin. Cancer Res. 2006, 12, 1431–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, H.A.; Cream, L. Biology of bone metastases: Causes and consequences. Clin. Breast Cancer 2007, 7 (Suppl. 1), S7–S13. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Joeckel, E.; Haber, T.; Prawitt, D.; Junker, K.; Hampel, C.; Thuroff, J.W.; Roos, F.C.; Brenner, W. High calcium concentration in bones promotes bone metastasis in renal cell carcinomas expressing calcium-sensing receptor. Mol. Cancer 2014, 13, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Takyar, F.M.; Swan, K.; Jeong, J.; VanHouten, J.; Sullivan, C.; Dann, P.; Yu, H.; Fiaschi-Taesch, N.; Chang, W.; et al. Calcium-Sensing Receptor Promotes Breast Cancer by Stimulating Intracrine Actions of Parathyroid Hormone-Related Protein. Cancer Res. 2016, 76, 5348–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, J.; Inada, K.; Yamashita, S.; Nakashima, Y.; Matsuo, S.; Ogawa, M. Tissue-type plasminogen activator is involved in skeletal metastasis from human breast cancer. Int. J. Clin. Lab. Res. 1992, 21, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Lev, D.C.; Kim, S.J.; Onn, A.; Stone, V.; Nam, D.H.; Yazici, S.; Fidler, I.J.; Price, J.E. Inhibition of platelet-derived growth factor receptor signaling restricts the growth of human breast cancer in the bone of nude mice. Clin. Cancer Res. 2005, 11, 306–314. [Google Scholar]
- Zhang, X.H.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massagué, J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Vander Ark, A.; Lee, P.; Hostetter, G.; Bhowmick, N.A.; Matrisian, L.M.; Williams, B.O.; Miranti, C.K.; Li, X. Myeloid-specific TGF-β signaling in bone promotes basic-FGF and breast cancer bone metastasis. Oncogene 2016, 35, 2370–2378. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Coenegrachts, L.; Maes, C.; Torrekens, S.; Van Looveren, R.; Mazzone, M.; Guise, T.A.; Bouillon, R.; Stassen, J.M.; Carmeliet, P.; Carmeliet, G. Anti-placental growth factor reduces bone metastasis by blocking tumor cell engraftment and osteoclast differentiation. Cancer Res. 2010, 70, 6537–6547. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhou, H.; Fong-Yee, C.; Modzelewski, J.R.; Seibel, M.J.; Dunstan, C.R. Bone resorption increases tumour growth in a mouse model of osteosclerotic breast cancer metastasis. Clin. Exp. Metastasis 2008, 25, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhou, H.; Ooi, L.L.; Snir, A.D.; Dunstan, C.R.; Seibel, M.J. Vitamin D deficiency promotes prostate cancer growth in bone. Prostate 2011, 71, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Ooi, L.L.; Zhou, H.; Kalak, R.; Zheng, Y.; Conigrave, A.D.; Seibel, M.J.; Dunstan, C.R. Vitamin D deficiency promotes human breast cancer growth in a murine model of bone metastasis. Cancer Res. 2010, 70, 1835–1844. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, J.A.; Vessella, R.L.; Quinn, J.E.; Odman, A.M.; Zhang, J.; Keller, E.T.; Kostenuik, P.J.; Dunstan, C.R.; Corey, E. The effect of osteoprotegerin administration on the intra-tibial growth of the osteoblastic LuCaP 23.1 prostate cancer xenograft. Clin. Exp. Metastasis 2004, 21, 381–387. [Google Scholar] [CrossRef]
- Corey, E.; Brown, L.G.; Quinn, J.E.; Poot, M.; Roudier, M.P.; Higano, C.S.; Vessella, R.L. Zoledronic acid exhibits inhibitory effects on osteoblastic and osteolytic metastases of prostate cancer. Clin. Cancer Res. 2003, 9, 295–306. [Google Scholar]
- Corey, E.; Brown, L.G.; Kiefer, J.A.; Quinn, J.E.; Pitts, T.E.; Blair, J.M.; Vessella, R.L. Osteoprotegerin in prostate cancer bone metastasis. Cancer Res. 2005, 65, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, T.; Lundholm, M.; Widmark, A.; Persson, E. Tumor Cell-Derived Exosomes from the Prostate Cancer Cell Line TRAMP-C1 Impair Osteoclast Formation and Differentiation. PLoS ONE 2016, 11, e0166284. [Google Scholar] [CrossRef]
- Nierste, B.A.; Glackin, C.A.; Kirshner, J. Dkk-1 and IL-7 in plasma of patients with multiple myeloma prevent differentiation of mesenchymal stem cells into osteoblasts. Am. J. Blood Res. 2014, 4, 73–85. [Google Scholar] [PubMed]
- Abe, M.; Hiura, K.; Ozaki, S.; Kido, S.; Matsumoto, T. Vicious cycle between myeloma cell binding to bone marrow stromal cells via VLA-4-VCAM-1 adhesion and macrophage inflammatory protein-1alpha and MIP-1beta production. J. Bone Miner. Metab. 2009, 27, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Body, J.J.; Casimiro, S.; Costa, L. Targeting bone metastases in prostate cancer: Improving clinical outcome. Nat. Rev. Urol. 2015, 12, 340–356. [Google Scholar] [CrossRef]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: Evidence from in vivo models. J. Bone Miner. Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logothetis, C.J.; Lin, S.H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer 2005, 5, 21–28. [Google Scholar] [CrossRef]
- Yang, J.; Fizazi, K.; Peleg, S.; Sikes, C.R.; Raymond, A.K.; Jamal, N.; Hu, M.; Olive, M.; Martinez, L.A.; Wood, C.G.; et al. Prostate cancer cells induce osteoblast differentiation through a Cbfa1-dependent pathway. Cancer Res. 2001, 61, 5652–5659. [Google Scholar] [PubMed]
- Fizazi, K.; Yang, J.; Peleg, S.; Sikes, C.R.; Kreimann, E.L.; Daliani, D.; Olive, M.; Raymond, K.A.; Janus, T.J.; Logothetis, C.J.; et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clin. Cancer Res. 2003, 9, 2587–2597. [Google Scholar]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [Green Version]
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Clines, G.A.; Mohammad, K.S.; Bao, Y.; Stephens, O.W.; Suva, L.J.; Shaughnessy, J.D., Jr.; Fox, J.W.; Chirgwin, J.M.; Guise, T.A. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol. Endocrinol. 2007, 21, 486–498. [Google Scholar] [CrossRef] [Green Version]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Itoh, T.; Ito, Y.; Ohtsuki, Y.; Ando, M.; Tsukamasa, Y.; Yamada, N.; Naoe, T.; Akao, Y. Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. J. Mol. Histol. 2012, 43, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Ochi, H.; Sunamura, S.; Kosaka, N.; Mabuchi, Y.; Fukuda, T.; Yao, K.; Kanda, H.; Ae, K.; Okawa, A.; et al. Cancer-secreted hsa-miR-940 induces an osteoblastic phenotype in the bone metastatic microenvironment via targeting ARHGAP1 and FAM134A. Proc. Natl. Acad. Sci. USA 2018, 115, 2204–2209. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Li, S.L.; Ma, Y.Y.; Diao, Y.J.; Yang, L.; Su, M.Q.; Li, Z.; Ji, Y.; Wang, J.; Lei, L.; et al. Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Oncotarget 2017, 8, 94834–94849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenta, R.; Sotiriou, E.; Pitulis, N.; Thyphronitis, G.; Koutsilieris, M. Prostate cancer cell survival pathways activated by bone metastasis microenvironment. J. Musculoskelet. Neuronal Interact. 2005, 5, 135–144. [Google Scholar]
- Yi, B.; Williams, P.J.; Niewolna, M.; Wang, Y.; Yoneda, T. Tumor-derived platelet-derived growth factor-BB plays a critical role in osteosclerotic bone metastasis in an animal model of human breast cancer. Cancer Res. 2002, 62, 917–923. [Google Scholar] [PubMed]
- Stern, P.H.; Tatrai, A.; Semler, D.E.; Lee, S.K.; Lakatos, P.; Strieleman, P.J.; Tarjan, G.; Sanders, J.L. Endothelin receptors, second messengers, and actions in bone. J. Nutr. 1995, 125, 2028s–2032s. [Google Scholar] [CrossRef]
- Saidak, Z.; Le Henaff, C.; Azzi, S.; Marty, C.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. Wnt/β-catenin signaling mediates osteoblast differentiation triggered by peptide-induced α5β1 integrin priming in mesenchymal skeletal cells. J. Biol. Chem. 2015, 290, 6903–6912. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Shi, H.Y.; Stock, S.R.; Stern, P.H.; Zhang, M. Regulation of breast cancer-induced bone lesions by β-catenin protein signaling. J. Biol. Chem. 2011, 286, 42575–42584. [Google Scholar] [CrossRef] [Green Version]
- Bu, G.; Lu, W.; Liu, C.C.; Selander, K.; Yoneda, T.; Hall, C.; Keller, E.T.; Li, Y. Breast cancer-derived Dickkopf1 inhibits osteoblast differentiation and osteoprotegerin expression: Implication for breast cancer osteolytic bone metastases. Int. J. Cancer 2008, 123, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- Velletri, T.; Xie, N.; Wang, Y.; Huang, Y.; Yang, Q.; Chen, X.; Chen, Q.; Shou, P.; Gan, Y.; Cao, G.; et al. P53 functional abnormality in mesenchymal stem cells promotes osteosarcoma development. Cell Death Dis. 2016, 7, e2015. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Li, Y.; Zhang, J.; Li, J.; Gao, J. Combined analysis of DNA methylation and gene expression profiles of osteosarcoma identified several prognosis signatures. Gene 2018, 650, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Mohseny, A.B.; Szuhai, K.; Romeo, S.; Buddingh, E.P.; Briaire-de Bruijn, I.; de Jong, D.; van Pel, M.; Cleton-Jansen, A.M.; Hogendoorn, P.C. Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. J. Pathol. 2009, 219, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Pietrovito, L.; Leo, A.; Gori, V.; Lulli, M.; Parri, M.; Becherucci, V.; Piccini, L.; Bambi, F.; Taddei, M.L.; Chiarugi, P. Bone marrow-derived mesenchymal stem cells promote invasiveness and transendothelial migration of osteosarcoma cells via a mesenchymal to amoeboid transition. Mol. Oncol. 2018, 12, 659–676. [Google Scholar] [CrossRef]
- Sun, T.; Zhong, X.; Song, H.; Liu, J.; Li, J.; Leung, F.; Lu, W.W.; Liu, Z.L. Anoikis resistant mediated by FASN promoted growth and metastasis of osteosarcoma. Cell Death Dis. 2019, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, K.C.; Penfornis, P.; Dhule, S.; Guillonneau, F.; Adams, K.V.; Mo, Y.Y.; Xu, R.; Liu, Y.; Watabe, K.; Vemuri, M.C.; et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget 2015, 6, 4953–4967. [Google Scholar] [CrossRef] [Green Version]
- Avnet, S.; Di Pompo, G.; Chano, T.; Errani, C.; Ibrahim-Hashim, A.; Gillies, R.J.; Donati, D.M.; Baldini, N. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-κB activation. Int. J. Cancer 2017, 140, 1331–1345. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Zheng, J.; Yu, P.; Xu, L.; Jiang, P.; Gao, J.; Wang, H.; Zhang, Y. Transforming growth factor β1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma. Stem Cells 2013, 31, 433–446. [Google Scholar] [CrossRef]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wang, D.; Li, N. MicroRNA-20b Downregulates HIF-1α and Inhibits the Proliferation and Invasion of Osteosarcoma Cells. Oncol. Res. 2016, 23, 257–266. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, C.; Wang, K.; Liu, X.; Liu, Q. MicroRNA-33b Inhibits the Proliferation and Migration of Osteosarcoma Cells via Targeting Hypoxia-Inducible Factor-1α. Oncol. Res. 2017, 25, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, S.; Chen, J.; Ren, P.; Hu, Y.; Cao, Z.; Sun, H.; Ding, Y. Oxymatrine induces mitochondria dependent apoptosis in human osteosarcoma MNNG/HOS cells through inhibition of PI3K/Akt pathway. Tumour Biol. 2014, 35, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Jin, A. ZIC2 promotes viability and invasion of human osteosarcoma cells by suppressing SHIP2 expression and activating PI3K/AKT pathways. J. Cell. Biochem. 2018, 119, 2248–2257. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.H.; Wang, S.Q.; Zhang, M.H.; Yu, F.B.; Zhang, L.; Yu, Z.X.; Kuang, Y. Knockdown of galectin-1 suppresses the growth and invasion of osteosarcoma cells through inhibition of the MAPK/ERK pathway. Oncol. Rep. 2014, 32, 1497–1504. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Park, B.S.; Kang, H.K.; Park, H.R.; Yu, S.B.; Kim, I.R. Delphinidin induces apoptosis and inhibits epithelial-to-mesenchymal transition via the ERK/p38 MAPK-signaling pathway in human osteosarcoma cell lines. Environ. Toxicol. 2018, 33, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.M.; Fuchs, B. Hedgehog signaling inhibitors as anti-cancer agents in osteosarcoma. Cancers 2015, 7, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.H.; Wang, W.; Yeung, W.; Deng, Y.; Yuan, P.; Mak, K.K. Hedgehog signaling induces osteosarcoma development through Yap1 and H19 overexpression. Oncogene 2014, 33, 4857–4866. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Jia, Q.; Wu, M.S.; Xie, X.; Wang, Y.; Song, G.; Zou, C.Y.; Tang, Q.; Lu, J.; Huang, G.; et al. Degalactotigonin, a Natural Compound from Solanum nigrum L., Inhibits Growth and Metastasis of Osteosarcoma through GSK3β Inactivation-Mediated Repression of the Hedgehog/Gli1 Pathway. Clin. Cancer Res. 2018, 24, 130–144. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, K.; Bakiri, L.; Wolff, L.I.; Linder, M.; Mikels-Vigdal, A.; Patiño-García, A.; Lecanda, F.; Hartmann, C.; Sibilia, M.; Wagner, E.F. Wnt signaling and Loxl2 promote aggressive osteosarcoma. Cell Res. 2020, 30, 885–901. [Google Scholar] [CrossRef]
- Gustafson, C.T.; Mamo, T.; Shogren, K.L.; Maran, A.; Yaszemski, M.J. FH535 Suppresses Osteosarcoma Growth In Vitro and Inhibits Wnt Signaling through Tankyrases. Front. Pharmacol. 2017, 8, 285. [Google Scholar] [CrossRef] [Green Version]
- Vega, O.A.; Lucero, C.M.J.; Araya, H.F.; Jerez, S.; Tapia, J.C.; Antonelli, M.; Salazar-Onfray, F.; Las Heras, F.; Thaler, R.; Riester, S.M.; et al. Wnt/β-Catenin Signaling Activates Expression of the Bone-Related Transcription Factor RUNX2 in Select Human Osteosarcoma Cell Types. J. Cell. Biochem. 2017, 118, 3662–3674. [Google Scholar] [CrossRef]
- Tian, H.; Zhou, T.; Chen, H.; Li, C.; Jiang, Z.; Lao, L.; Kahn, S.A.; Duarte, M.E.L.; Zhao, J.; Daubs, M.D.; et al. Bone morphogenetic protein-2 promotes osteosarcoma growth by promoting epithelial-mesenchymal transition (EMT) through the Wnt/β-catenin signaling pathway. J. Orthop. Res. 2019, 37, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Erkizan, H.V.; Uversky, V.N.; Toretsky, J.A. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing’s sarcoma. Clin. Cancer Res. 2010, 16, 4077–4083. [Google Scholar] [CrossRef] [Green Version]
- Katschnig, A.M.; Kauer, M.O.; Schwentner, R.; Tomazou, E.M.; Mutz, C.N.; Linder, M.; Sibilia, M.; Alonso, J.; Aryee, D.N.T.; Kovar, H. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 2017, 36, 5995–6005. [Google Scholar] [CrossRef] [Green Version]
- Chansky, H.A.; Barahmand-Pour, F.; Mei, Q.; Kahn-Farooqi, W.; Zielinska-Kwiatkowska, A.; Blackburn, M.; Chansky, K.; Conrad, E.U., 3rd; Bruckner, J.D.; Greenlee, T.K.; et al. Targeting of EWS/FLI-1 by RNA interference attenuates the tumor phenotype of Ewing’s sarcoma cells in vitro. J. Orthop. Res. 2004, 22, 910–917. [Google Scholar] [CrossRef]
- Franzetti, G.A.; Laud-Duval, K.; van der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; de Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef] [Green Version]
- Adane, B.; Alexe, G.; Seong, B.K.A.; Lu, D.; Hwang, E.E.; Hnisz, D.; Lareau, C.A.; Ross, L.; Lin, S.; Dela Cruz, F.S.; et al. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cancer Cell 2021, 39, 827–844.e10. [Google Scholar] [CrossRef]
- Surdez, D.; Zaidi, S.; Grossetete, S.; Laud-Duval, K.; Ferre, A.S.; Mous, L.; Vourc’h, T.; Tirode, F.; Pierron, G.; Raynal, V.; et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell 2021, 39, 810–826.e9. [Google Scholar] [CrossRef] [PubMed]
- Bierbaumer, L.; Katschnig, A.M.; Radic-Sarikas, B.; Kauer, M.O.; Petro, J.A.; Högler, S.; Gurnhofer, E.; Pedot, G.; Schäfer, B.W.; Schwentner, R.; et al. YAP/TAZ inhibition reduces metastatic potential of Ewing sarcoma cells. Oncogenesis 2021, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Ban, J.; Bennani-Baiti, I.M.; Kauer, M.; Schaefer, K.L.; Poremba, C.; Jug, G.; Schwentner, R.; Smrzka, O.; Muehlbacher, K.; Aryee, D.N.; et al. EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Res. 2008, 68, 7100–7109. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.F.; Hughes, C.S.; Li, W.; He, J.Z.; Surdez, D.; El-Naggar, A.M.; Cheng, H.; Prudova, A.; Delaidelli, A.; Negri, G.L.; et al. Proteomic screens for suppressors of anoikis identify IL1RAP as a promising surface target in Ewing sarcoma. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Lissat, A.; Joerschke, M.; Shinde, D.A.; Braunschweig, T.; Meier, A.; Makowska, A.; Bortnick, R.; Henneke, P.; Herget, G.; Gorr, T.A.; et al. IL6 secreted by Ewing sarcoma tumor microenvironment confers anti-apoptotic and cell-disseminating paracrine responses in Ewing sarcoma cells. BMC Cancer 2015, 15, 552. [Google Scholar] [CrossRef] [PubMed]
- Rübe, C.E.; van Valen, F.; Wilfert, F.; Palm, J.; Schuck, A.; Willich, N.; Winkelmann, W.; Jürgens, H.; Rübe, C. Ewing’s sarcoma and peripheral primitive neuroectodermal tumor cells produce large quantities of bioactive tumor necrosis factor-alpha (TNF-alpha) after radiation exposure. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 1414–1425. [Google Scholar] [CrossRef]
- Miller, I.V.; Raposo, G.; Welsch, U.; Prazeres da Costa, O.; Thiel, U.; Lebar, M.; Maurer, M.; Bender, H.U.; von Luettichau, I.; Richter, G.H.; et al. First identification of Ewing’s sarcoma-derived extracellular vesicles and exploration of their biological and potential diagnostic implications. Biol. Cell 2013, 105, 289–303. [Google Scholar] [CrossRef]
- Villasante, A.; Marturano-Kruik, A.; Ambati, S.R.; Liu, Z.; Godier-Furnemont, A.; Parsa, H.; Lee, B.W.; Moore, M.A.; Vunjak-Novakovic, G. Recapitulating the Size and Cargo of Tumor Exosomes in a Tissue-Engineered Model. Theranostics 2016, 6, 1119–1130. [Google Scholar] [CrossRef]
- Li, X.; McGee-Lawrence, M.E.; Decker, M.; Westendorf, J.J. The Ewing’s sarcoma fusion protein, EWS-FLI, binds Runx2 and blocks osteoblast differentiation. J. Cell. Biochem. 2010, 111, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Chirgwin, J.M.; Guise, T.A. Molecular mechanisms of tumor-bone interactions in osteolytic metastases. Crit. Rev. Eukaryot. Gene Expr. 2000, 10, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Redini, F.; Heymann, D. Bone Tumor Environment as a Potential Therapeutic Target in Ewing Sarcoma. Front. Oncol. 2015, 5, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Park, C.J.; Jang, S.; Cho, Y.U.; Seo, J.J.; Im, H.J.; Koh, K.N.; Cho, K.J.; Song, J.S.; Seo, E.J. Clinical and Cytogenetic Profiles of Rhabdomyosarcoma with Bone Marrow Involvement in Korean Children: A 15-Year Single-Institution Experience. Ann. Lab. Med. 2018, 38, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Ting, S.C.; Kiefer, T.; Ehlert, K.; Goericke, S.L.; Hinze, R.; Ketteler, P.; Bechrakis, N.E.; Schildhaus, H.U. Bone metastasis of retinoblastoma five years after primary treatment. Am. J. Ophthalmol. Case Rep. 2020, 19, 100834. [Google Scholar] [CrossRef]
- Bernards, R. N-myc disrupts protein kinase C-mediated signal transduction in neuroblastoma. EMBO J. 1991, 10, 1119–1125. [Google Scholar] [CrossRef]
- Akeson, R.; Bernards, R. N-myc down regulates neural cell adhesion molecule expression in rat neuroblastoma. Mol. Cell. Biol. 1990, 10, 2012–2016. [Google Scholar] [CrossRef] [Green Version]
- Teitz, T.; Inoue, M.; Valentine, M.B.; Zhu, K.; Rehg, J.E.; Zhao, W.; Finkelstein, D.; Wang, Y.D.; Johnson, M.D.; Calabrese, C.; et al. Th-MYCN mice with caspase-8 deficiency develop advanced neuroblastoma with bone marrow metastasis. Cancer Res. 2013, 73, 4086–4097. [Google Scholar] [CrossRef] [Green Version]
- Hecht, M.; Schulte, J.H.; Eggert, A.; Wilting, J.; Schweigerer, L. The neurotrophin receptor TrkB cooperates with c-Met in enhancing neuroblastoma invasiveness. Carcinogenesis 2005, 26, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Muhlethaler-Mottet, A.; Liberman, J.; Ascencao, K.; Flahaut, M.; Balmas Bourloud, K.; Yan, P.; Jauquier, N.; Gross, N.; Joseph, J.M. The CXCR4/CXCR7/CXCL12 Axis Is Involved in a Secondary but Complex Control of Neuroblastoma Metastatic Cell Homing. PLoS ONE 2015, 10, e0125616. [Google Scholar] [CrossRef]
- Ulrich, H.; Ratajczak, M.Z.; Schneider, G.; Adinolfi, E.; Orioli, E.; Ferrazoli, E.G.; Glaser, T.; Correa-Velloso, J.; Martins, P.C.M.; Coutinho, F.; et al. Kinin and Purine Signaling Contributes to Neuroblastoma Metastasis. Front. Pharmacol. 2018, 9, 500. [Google Scholar] [CrossRef] [Green Version]
- Airoldi, I.; Cocco, C.; Morandi, F.; Prigione, I.; Pistoia, V. CXCR5 may be involved in the attraction of human metastatic neuroblastoma cells to the bone marrow. Cancer Immunol. Immunother. 2008, 57, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Nevo, I.; Sagi-Assif, O.; Meshel, T.; Ben-Baruch, A.; Johrer, K.; Greil, R.; Trejo, L.E.; Kharenko, O.; Feinmesser, M.; Yron, I.; et al. The involvement of the fractalkine receptor in the transmigration of neuroblastoma cells through bone-marrow endothelial cells. Cancer Lett. 2009, 273, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Silverman, A.M.; Nakata, R.; Shimada, H.; Sposto, R.; DeClerck, Y.A. A galectin-3-dependent pathway upregulates interleukin-6 in the microenvironment of human neuroblastoma. Cancer Res. 2012, 72, 2228–2238. [Google Scholar] [CrossRef] [Green Version]
- Shankar, V.; Hori, H.; Kihira, K.; Lei, Q.; Toyoda, H.; Iwamoto, S.; Komada, Y. Mesenchymal stromal cell secretome up-regulates 47 kDa CXCR4 expression, and induce invasiveness in neuroblastoma cell lines. PLoS ONE 2015, 10, e0120069. [Google Scholar] [CrossRef] [PubMed]
- Colletti, M.; Tomao, L.; Galardi, A.; Paolini, A.; Di Paolo, V.; De Stefanis, C.; Mascio, P.; Nazio, F.; Petrini, S.; Castellano, A.; et al. Neuroblastoma-secreted exosomes carrying miR-375 promote osteogenic differentiation of bone-marrow mesenchymal stromal cells. J. Extracell. Vesicles 2020, 9, 1774144. [Google Scholar] [CrossRef]
- Hochheuser, C.; van Zogchel, L.M.J.; Kleijer, M.; Kuijk, C.; Tol, S.; van der Schoot, C.E.; Voermans, C.; Tytgat, G.A.M.; Timmerman, I. The Metastatic Bone Marrow Niche in Neuroblastoma: Altered Phenotype and Function of Mesenchymal Stromal Cells. Cancers 2020, 12, 3231. [Google Scholar] [CrossRef]
- Morandi, F.; Marimpietri, D.; Horenstein, A.L.; Corrias, M.V.; Malavasi, F. Microvesicles expressing adenosinergic ectoenzymes and their potential role in modulating bone marrow infiltration by neuroblastoma cells. Oncoimmunology 2019, 8, e1574198. [Google Scholar] [CrossRef] [Green Version]
- Morandi, F.; Scaruffi, P.; Gallo, F.; Stigliani, S.; Moretti, S.; Bonassi, S.; Gambini, C.; Mazzocco, K.; Fardin, P.; Haupt, R.; et al. Bone marrow-infiltrating human neuroblastoma cells express high levels of calprotectin and HLA-G proteins. PLoS ONE 2012, 7, e29922. [Google Scholar] [CrossRef] [Green Version]
- Anract, P.; Biau, D.; Boudou-Rouquette, P. Metastatic fractures of long limb bones. Orthop. Traumatol. Surg. Res. 2017, 103, S41–s51. [Google Scholar] [CrossRef] [PubMed]
- Montilla-Soler, J.L.; Makanji, R. Skeletal Scintigraphy. Cancer Control 2017, 24, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Grootjans, W.; Serém, S.J.; Gomes, M.I.; Heijmen, L.; Bulten, B.F.; Mijnheere, E.P.; Hermsen, R.; van den Broek, W.J. Half-time bone scintigraphy in prostate and breast cancer patients. Q. J. Nucl. Med. Mol. Imaging 2018, 62, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Edenbrandt, L.; Mizokami, A. Bone scan index: A new biomarker of bone metastasis in patients with prostate cancer. Int. J. Urol. 2017, 24, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, G.P.; Schoenberg, S.O.; Reiser, M.F.; Baur-Melnyk, A. Whole-body MR imaging of bone marrow. Eur. J. Radiol. 2005, 55, 33–40. [Google Scholar] [CrossRef]
- Arita, Y.; Takahara, T.; Yoshida, S.; Kwee, T.C.; Yajima, S.; Ishii, C.; Ishii, R.; Okuda, S.; Jinzaki, M.; Fujii, Y. Quantitative Assessment of Bone Metastasis in Prostate Cancer Using Synthetic Magnetic Resonance Imaging. Investig. Radiol. 2019, 54, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, M. (18)F-Sodium Fluoride PET/CT and PET/MR Imaging of Bone and Joint Disorders. PET Clin. 2018, 13, 477–490. [Google Scholar] [CrossRef]
- Chen, B.; Wei, P.; Macapinlac, H.A.; Lu, Y. Comparison of 18F-Fluciclovine PET/CT and 99mTc-MDP bone scan in detection of bone metastasis in prostate cancer. Nucl. Med. Commun. 2019, 40, 940–946. [Google Scholar] [CrossRef]
- Shimozuma, K.; Sonoo, H.; Fukunaga, M.; Ichihara, K.; Aoyama, T.; Tanaka, K. Biochemical markers of bone turnover in breast cancer patients with bone metastases: A preliminary report. Jpn. J. Clin. Oncol. 1999, 29, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Chew, C.K.; Clarke, B.L. Biochemical Testing Relevant to Bone. Endocrinol. Metab. Clin. N. Am. 2017, 46, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Katagiri, H.; Murata, H.; Wasa, J.; Miyagi, M.; Honda, Y.; Takahashi, M. Surgery for femoral metastases. Bone Jt. J. 2020, 102-B, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Saravana-Bawan, S.; David, E.; Sahgal, A.; Chow, E. Palliation of bone metastases-exploring options beyond radiotherapy. Ann. Palliat. Med. 2019, 8, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Wenger, M. Vertebroplasty for metastasis. Med. Oncol. 2003, 20, 203–209. [Google Scholar] [CrossRef]
- Zakaly, H.M.H.; Mostafa, M.Y.A.; Deryabina, D.; Zhukovsky, M. Comparative studies on the potential use of (177)Lu-based radiopharmaceuticals for the palliative therapy of bone metastases. Int. J. Radiat. Biol. 2020, 96, 779–789. [Google Scholar] [CrossRef]
- Shore, N.D. Radium-223 dichloride for metastatic castration-resistant prostate cancer: The urologist’s perspective. Urology 2015, 85, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Kyriakopoulos, C.E.; Chen, Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Gdowski, A.S.; Ranjan, A.; Sarker, M.R.; Vishwanatha, J.K. Bone-targeted cabazitaxel nanoparticles for metastatic prostate cancer skeletal lesions and pain. Nanomedicine 2017, 12, 2083–2095. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Sakamoto, S.; Kawahara, T.; Uemura, K.; Yokomizo, Y.; Uemura, H. Correlation between Automated Bone Scan Index Change after Cabazitaxel and Survival among Men with Castration-Resistant Prostate Cancer. Urol. Int. 2019, 103, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Achterrath, W.; Hegewisch-Becker, S.; Mross, K.; Zander, A.R. Current options in treatment of anthracycline-resistant breast cancer. Cancer Treat. Rev. 1999, 25, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, R.; Wang, Z.; Yang, Y.; Wang, M.; Zou, W. Efficacy and Safety of Vinorelbine Plus Cisplatin vs. Gemcitabine Plus Cisplatin for Treatment of Metastatic Triple-Negative Breast Cancer After Failure with Anthracyclines and Taxanes. Med. Sci. Monit. 2017, 23, 4657–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amari, M.; Ishida, T.; Takeda, M.; Ohuchi, N. Capecitabine monotherapy is efficient and safe in all line settings in patients with metastatic and advanced breast cancer. Jpn. J. Clin. Oncol. 2010, 40, 188–193. [Google Scholar] [CrossRef]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Lucas, K. Current therapeutic approaches in metastatic and recurrent ewing sarcoma. Sarcoma 2011, 2011, 863210. [Google Scholar] [CrossRef] [Green Version]
- Khanna, N.; Pandey, A.; Bajpai, J. Metastatic Ewing’s Sarcoma: Revisiting the “Evidence on the Fence”. Indian J. Med. Paediatr. Oncol. 2017, 38, 173–181. [Google Scholar] [CrossRef]
- Mercadante, S.; Fulfaro, F. Management of painful bone metastases. Curr. Opin. Oncol. 2007, 19, 308–314. [Google Scholar] [CrossRef]
- Nanni, P.; Landuzzi, L.; Manara, M.C.; Righi, A.; Nicoletti, G.; Cristalli, C.; Pasello, M.; Parra, A.; Carrabotta, M.; Ferracin, M.; et al. Bone sarcoma patient-derived xenografts are faithful and stable preclinical models for molecular and therapeutic investigations. Sci. Rep. 2019, 9, 12174. [Google Scholar] [CrossRef]
- Braekeveldt, N.; Wigerup, C.; Gisselsson, D.; Mohlin, S.; Merselius, M.; Beckman, S.; Jonson, T.; Borjesson, A.; Backman, T.; Tadeo, I.; et al. Neuroblastoma patient-derived orthotopic xenografts retain metastatic patterns and geno- and phenotypes of patient tumours. Int. J. Cancer 2015, 136, E252–E261. [Google Scholar] [CrossRef] [Green Version]
- Rea, D.; Del Vecchio, V.; Palma, G.; Barbieri, A.; Falco, M.; Luciano, A.; De Biase, D.; Perdona, S.; Facchini, G.; Arra, C. Mouse Models in Prostate Cancer Translational Research: From Xenograft to PDX. BioMed Res. Int. 2016, 2016, 9750795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, J.A.; Harb, J.F.; Barroso, U., Jr.; He, Z.; Grignon, D.J.; Cher, M.L. Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res. 1999, 59, 1987–1993. [Google Scholar] [PubMed]
- Schuster, J.; Zhang, J.; Longo, M. A novel human osteoblast-derived severe combined immunodeficiency mouse model of bone metastasis. J. Neurosurg. Spine 2006, 4, 388–391. [Google Scholar] [CrossRef]
- Li, F.X.; Liu, J.J.; Xu, F.; Lin, X.; Zhong, J.Y.; Wu, F.; Yuan, L.Q. Role of tumor-derived exosomes in bone metastasis. Oncol. Lett. 2019, 18, 3935–3945. [Google Scholar] [CrossRef]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holen, I.; Lefley, D.V.; Francis, S.E.; Rennicks, S.; Bradbury, S.; Coleman, R.E.; Ottewell, P. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 2016, 7, 75571–75584. [Google Scholar] [CrossRef] [Green Version]
- Sawada, Y.; Kikugawa, T.; Iio, H.; Sakakibara, I.; Yoshida, S.; Ikedo, A.; Yanagihara, Y.; Saeki, N.; Gyorffy, B.; Kishida, T.; et al. GPRC5A facilitates cell proliferation through cell cycle regulation and correlates with bone metastasis in prostate cancer. Int. J. Cancer 2020, 146, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.S.; Hostetter, G.; Tang, L.; Frank, S.B.; Saboda, K.; Mehra, R.; Wang, L.; Li, X.; Keller, E.T.; Miranti, C.K. Notch3 promotes prostate cancer-induced bone lesion development via MMP-3. Oncogene 2020, 39, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Berish, R.B.; Ali, A.N.; Telmer, P.G.; Ronald, J.A.; Leong, H.S. Translational models of prostate cancer bone metastasis. Nat. Rev. Urol. 2018, 15, 403–421. [Google Scholar] [CrossRef] [PubMed]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J. Bone Oncol. 2019, 15, 100205. [Google Scholar] [CrossRef]
- Anastasilakis, A.D.; Polyzos, S.A.; Yavropoulou, M.P.; Makras, P. Combination and sequential treatment in women with postmenopausal osteoporosis. Expert Opin. Pharmacother. 2020, 21, 477–490. [Google Scholar] [CrossRef]
- Marocco, C.; Zimatore, G.; Mocini, E.; Fornari, R.; Iolascon, G.; Gallotta, M.C.; Bimonte, V.M.; Baldari, C.; Lenzi, A.; Migliaccio, S. Efficacy of Denosumab Therapy Following Treatment with Bisphosphonates in Women with Osteoporosis: A Cohort Study. Int. J. Environ. Res. Public Health 2021, 18, 1728. [Google Scholar] [CrossRef]
- Li, S.; Chen, P.; Pei, Y.; Zheng, K.; Wang, W.; Qiu, E.; Zhang, X. Addition of Zoledronate to Chemotherapy in Patients with Osteosarcoma Treated with Limb-Sparing Surgery: A Phase III Clinical Trial. Med. Sci. Monit. 2019, 25, 1429–1438. [Google Scholar] [CrossRef]
- Dirksen, U.; Koch, R.; Bhadri, V.; Brichard, B.; Butterfass-Bahloul, T.; Cyprova, S.; Gelderblom, H.; Hauser, P.; Havemann, L.; Hjorth, L.; et al. Efficacy of maintenance therapy with zoledronic acid in patients with localized Ewing sarcoma: Report from the international Ewing 2008 trial. J. Clin. Oncol. 2020, 38, 11523. [Google Scholar] [CrossRef]
- Gallo, L.H.; Ko, J.; Donoghue, D.J. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle 2017, 16, 634–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Liu, Z.; Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 2020, 19, 146. [Google Scholar] [CrossRef]
- Toscani, D.; Craviotto, L.; Giuliani, N. The Role of Proteasome Inhibitors in Multiple Myeloma Bone Disease and Bone Metastasis: Effects on Osteoblasts and Osteocytes. Appl. Sci. 2021, 11, 4642. [Google Scholar] [CrossRef]
- Garrett, I.R.; Chen, D.; Gutierrez, G.; Zhao, M.; Escobedo, A.; Rossini, G.; Harris, S.E.; Gallwitz, W.; Kim, K.B.; Hu, S.; et al. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J. Clin. Investig. 2003, 111, 1771–1782. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Bonomini, S.; Crugnola, M.; Mancini, C.; Martella, E.; Ferrari, L.; Tabilio, A.; et al. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood 2007, 110, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Fan, R.; Lei, L.; Lei, L.; Wang, Y.; Lv, N.; Chen, P.; Williamson, R.A.; Wang, B.; Hu, J. Cell cycle exit during bortezomib-induced osteogenic differentiation of mesenchymal stem cells was mediated by Xbp1s-upregulated p21(Cip1) and p27(Kip1). J. Cell. Mol. Med. 2020, 24, 9428–9438. [Google Scholar] [CrossRef]
- Zhang, D.; De Veirman, K.; Fan, R.; Jian, Q.; Zhang, Y.; Lei, L.; Evans, H.; Wang, Y.; Lei, L.; Wang, B.; et al. ER stress arm XBP1s plays a pivotal role in proteasome inhibition-induced bone formation. Stem Cell Res. Ther. 2020, 11, 516. [Google Scholar] [CrossRef]
- Jones, M.D.; Liu, J.C.; Barthel, T.K.; Hussain, S.; Lovria, E.; Cheng, D.; Schoonmaker, J.A.; Mulay, S.; Ayers, D.C.; Bouxsein, M.L.; et al. A proteasome inhibitor, bortezomib, inhibits breast cancer growth and reduces osteolysis by downregulating metastatic genes. Clin. Cancer Res. 2010, 16, 4978–4989. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, J.; Li, X.; Xing, L.; Ding, Y.; Shi, P.; Zhang, Y.; Guo, S.; Shu, X.; Shan, B. Bortezomib prevents oncogenesis and bone metastasis of prostate cancer by inhibiting WWP1, Smurf1 and Smurf2. Int. J. Oncol. 2014, 45, 1469–1478. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Ebetino, F.H.; Boeckman, R.K., Jr.; Srinivasan, V.; Tao, J.; Sawyer, T.K.; Li, J.; Yao, Z.; Boyce, B.F. Targeting anti-cancer agents to bone using bisphosphonates. Bone 2020, 138, 115492. [Google Scholar] [CrossRef]
- Russow, G.; Jahn, D.; Appelt, J.; Mardian, S.; Tsitsilonis, S.; Keller, J. Anabolic Therapies in Osteoporosis and Bone Regeneration. Int. J. Mol. Sci. 2018, 20, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, R.; Hauser, S.; Neuber, C.; Laube, M.; Schulze, S.; Rammelt, S.; Pietzsch, J. Adjuvant Drug-Assisted Bone Healing: Advances and Challenges in Drug Delivery Approaches. Pharmaceutics 2020, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Romero-Moreno, R.; Curtis, K.J.; Coughlin, T.R.; Miranda-Vergara, M.C.; Dutta, S.; Natarajan, A.; Facchine, B.A.; Jackson, K.M.; Nystrom, L.; Li, J.; et al. The CXCL5/CXCR2 axis is sufficient to promote breast cancer colonization during bone metastasis. Nat. Commun. 2019, 10, 4404. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Burner, D.N.; Mendoza, T.R.; Muldong, M.T.; Arreola, C.; Wu, C.N.; Cacalano, N.A.; Kulidjian, A.A.; Kane, C.J.; Jamieson, C.A.M. Establishment and Analysis of Three-Dimensional (3D) Organoids Derived from Patient Prostate Cancer Bone Metastasis Specimens and their Xenografts. J. Vis. Exp. 2020, 156, e60367. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Ha, L.; Cheng, G.; Wan, Y.; Xia, Y.; Sosnoski, D.M.; Mastro, A.M.; Zheng, S.Y. A Spontaneous 3D Bone-On-a-Chip for Bone Metastasis Study of Breast Cancer Cells. Small 2018, 14, e1702787. [Google Scholar] [CrossRef]
- Cui, H.; Esworthy, T.; Zhou, X.; Hann, S.Y.; Glazer, R.I.; Li, R.; Zhang, L.G. Engineering a Novel 3D Printed Vascularized Tissue Model for Investigating Breast Cancer Metastasis to Bone. Adv. Healthc. Mater. 2020, 9, e1900924. [Google Scholar] [CrossRef]
- Zhu, W.; Holmes, B.; Glazer, R.I.; Zhang, L.G. 3D printed nanocomposite matrix for the study of breast cancer bone metastasis. Nanomedicine 2016, 12, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Aveic, S.; Janssen, S.; Nasehi, R.; Seidelmann, M.; Vogt, M.; Pantile, M.; Rutten, S.; Fischer, H. A 3D printed in vitro bone model for the assessment of molecular and cellular cues in metastatic neuroblastoma. Biomater. Sci. 2021, 9, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Nowsheen, S.; Aziz, K.; Georgakilas, A.G. Toxicity and adverse effects of Tamoxifen and other anti-estrogen drugs. Pharmacol. Ther. 2013, 139, 392–404. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.; Yardley, D.; Burris, H.; De Boer, R.; Amadori, D.; McIntyre, K.; Ejlertsen, B.; Gnant, M.; Jonat, W.; Pritchard, K.I.; et al. Comparative Efficacy and Safety of Adjuvant Letrozole Versus Anastrozole in Postmenopausal Patients With Hormone Receptor-Positive, Node-Positive Early Breast Cancer: Final Results of the Randomized Phase III Femara Versus Anastrozole Clinical Evaluation (FACE) Trial. J. Clin. Oncol. 2017, 35, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-β in bone metastasis: Novel therapeutic perspectives. Bonekey Rep. 2012, 1, 96. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, A.; Agyin, J.K.; Wang, L.; Tang, Y.; Lei, X.; Story, B.M.; Cornell, J.E.; Pollock, B.H.; Mundy, G.R.; Sun, L.Z. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006, 66, 6714–6721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Wang, Q.; Hu, G.; Van Poznak, C.; Fleisher, M.; Reiss, M.; Massagué, J.; Kang, Y. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 2009, 23, 1882–1894. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, J.; Chang, A.; Cheng, D.; Huang, S.; Wu, D.; Sirkisoon, S.; Yang, S.; Lin, H.K.; Lo, H.W.; et al. Her2 promotes early dissemination of breast cancer by suppressing the p38 pathway through Skp2-mediated proteasomal degradation of Tpl2. Oncogene 2020, 39, 7034–7050. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diepenbruck, M.; Tiede, S.; Saxena, M.; Ivanek, R.; Kalathur, R.K.R.; Lüönd, F.; Meyer-Schaller, N.; Christofori, G. miR-1199-5p and Zeb1 function in a double-negative feedback loop potentially coordinating EMT and tumour metastasis. Nat. Commun. 2017, 8, 1168. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; Abdelghafer, A.; Markovic, M.; Yavner, J.; Melam, A.; Salama, N.N.; Azab, A.K. Targeting E-selectin to Tackle Cancer Using Uproleselan. Cancers 2021, 13, 335. [Google Scholar] [CrossRef]
- Esposito, M.; Kang, Y. Targeting tumor-stromal interactions in bone metastasis. Pharmacol. Ther. 2014, 141, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, D.; Maier, P.; Laufs, S.; Wenz, F.; Zeller, W.J.; Fruehauf, S.; Allgayer, H. CXCR4 Expression and Treatment with SDF-1α or Plerixafor Modulate Proliferation and Chemosensitivity of Colon Cancer Cells. Transl. Oncol. 2013, 6, 124–132. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Orfi, L.; Szabadkai, I.; Daub, H.; Kéri, G.; Ullrich, A. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res. 2019, 21, 31. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Gilkes, D.M. The Contribution of the Immune System in Bone Metastasis Pathogenesis. Int. J. Mol. Sci. 2019, 20, 999. [Google Scholar] [CrossRef] [Green Version]
- Kahkonen, T.E.; Halleen, J.M.; Bernoulli, J. Osteoimmuno-Oncology: Therapeutic Opportunities for Targeting Immune Cells in Bone Metastasis. Cells 2021, 10, 1529. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Takegahara, N.; Kim, H.; Choi, Y. Updating osteoimmunology: Regulation of bone cells by innate and adaptive immunity. Nat. Rev. Rheumatol. 2018, 14, 146–156. [Google Scholar] [CrossRef]
- Mendoza-Reinoso, V.; McCauley, L.K.; Fournier, P.G.J. Contribution of Macrophages and T Cells in Skeletal Metastasis. Cancers 2020, 12, 1014. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Rackov, G.; Izquierdo, E.; Alonso, V.; Gortazar, A.R.; Escribese, M.M. Targeting Macrophages: Friends or Foes in Disease? Front. Pharmacol. 2019, 10, 1255. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Hensel, J.A.; Chanda, D.; Harris, B.A.; Siegal, G.P.; Maheshwari, A.; Ponnazhagan, S. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J. Immunol. 2012, 189, 4258–4265. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Y.; Li, C.J.; Yiang, G.T.; Cheng, Y.L.; Tsai, A.P.; Hou, Y.T.; Ho, Y.C.; Hou, M.F.; Chu, P.Y. Molecular Regulation of Bone Metastasis Pathogenesis. Cell. Physiol. Biochem. 2018, 46, 1423–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Wang, L.; Dai, J.; Kryczek, I.; Wei, S.; Vatan, L.; Altuwaijri, S.; Sparwasser, T.; Wang, G.; Keller, E.T.; et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology 2012, 1, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Dahlberg, C.I.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [Green Version]
- Romee, R.; Leong, J.W.; Fehniger, T.A. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica 2014, 2014, 205796. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Dong, R.; Gong, Y.; Meng, W.; Yuan, M.; Zhu, H.; Ying, M.; He, Q.; Cao, J.; Yang, B. The involvement of M2 macrophage polarization inhibition in fenretinide-mediated chemopreventive effects on colon cancer. Cancer Lett. 2017, 388, 43–53. [Google Scholar] [CrossRef]
- Edwards, J.P.; Emens, L.A. The multikinase inhibitor sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE(2) in murine macrophages. Int. Immunopharmacol. 2010, 10, 1220–1228. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.; Martin, K.; Theurich, S.; von Bergwelt-Baildon, M.; Zippelius, A. Cancer chemotherapy agents target intratumoral dendritic cells to potentiate antitumor immunity. Oncoimmunology 2014, 3, e954460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; He, Y.; Zhao, Q.; Yu, X. Immune Cells Act as Promising Targets for the Treatment of Bone Metastasis. Recent Pat. Anticancer Drug Discov. 2017, 12, 221–233. [Google Scholar] [CrossRef]
- Rennard, S.I.; Dale, D.C.; Donohue, J.F.; Kanniess, F.; Magnussen, H.; Sutherland, E.R.; Watz, H.; Lu, S.; Stryszak, P.; Rosenberg, E.; et al. CXCR2 Antagonist MK-7123. A Phase 2 Proof-of-Concept Trial for Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1001–1011. [Google Scholar] [CrossRef]
- Benevides, L.; da Fonseca, D.M.; Donate, P.B.; Tiezzi, D.G.; De Carvalho, D.D.; de Andrade, J.M.; Martins, G.A.; Silva, J.S. IL17 Promotes Mammary Tumor Progression by Changing the Behavior of Tumor Cells and Eliciting Tumorigenic Neutrophils Recruitment. Cancer Res. 2015, 75, 3788–3799. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Aleza, C.; Nguyen, B.; Yoldi, G.; Ciscar, M.; Barranco, A.; Hernandez-Jimenez, E.; Maetens, M.; Salgado, R.; Zafeiroglou, M.; Pellegrini, P.; et al. Inhibition of RANK signaling in breast cancer induces an anti-tumor immune response orchestrated by CD8+ T cells. Nat. Commun. 2020, 11, 6335. [Google Scholar] [CrossRef]
- Jiao, S.; Subudhi, S.K.; Aparicio, A.; Ge, Z.; Guan, B.; Miura, Y.; Sharma, P. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell 2019, 179, 1177–1190.e13. [Google Scholar] [CrossRef]
- Moseley, K.F.; Naidoo, J.; Bingham, C.O.; Carducci, M.A.; Forde, P.M.; Gibney, G.T.; Lipson, E.J.; Shah, A.A.; Sharfman, W.H.; Cappelli, L.C. Immune-related adverse events with immune checkpoint inhibitors affecting the skeleton: A seminal case series. J. Immunother. Cancer 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Hilal, T.; Bansal, P.; Kelemen, K.; Slack, J. Nivolumab-associated bone marrow necrosis. Ann. Oncol. 2018, 29, 513–514. [Google Scholar] [CrossRef]
- Pillai, S.G.; Li, S.; Siddappa, C.M.; Ellis, M.J.; Watson, M.A.; Aft, R. Identifying biomarkers of breast cancer micrometastatic disease in bone marrow using a patient-derived xenograft mouse model. Breast Cancer Res. 2018, 20, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Manna, F.; De Menna, M.; Patel, N.; Karkampouna, S.; De Filippo, M.R.; Klima, I.; Kloen, P.; Beimers, L.; Thalmann, G.N.; Pelger, R.C.M.; et al. Dual-mTOR Inhibitor Rapalink-1 Reduces Prostate Cancer Patient-Derived Xenograft Growth and Alters Tumor Heterogeneity. Front. Oncol. 2020, 10, 1012. [Google Scholar] [CrossRef] [PubMed]
- Karkampouna, S.; De Filippo, M.R.; Ng, C.K.Y.; Klima, I.; Zoni, E.; Spahn, M.; Stein, F.; Haberkant, P.; Thalmann, G.N.; Kruithof-de Julio, M. Stroma Transcriptomic and Proteomic Profile of Prostate Cancer Metastasis Xenograft Models Reveals Prognostic Value of Stroma Signatures. Cancers 2020, 12, 3786. [Google Scholar] [CrossRef]
- Lefley, D.; Howard, F.; Arshad, F.; Bradbury, S.; Brown, H.; Tulotta, C.; Eyre, R.; Alferez, D.; Wilkinson, J.M.; Holen, I.; et al. Development of clinically relevant in vivo metastasis models using human bone discs and breast cancer patient-derived xenografts. Breast Cancer Res. 2019, 21, 130. [Google Scholar] [CrossRef]
- Katti, K.S.; Jasuja, H.; Kar, S.; Katti, D.R. Nanostructured Biomaterials for In Vitro Models of Bone Metastasis Cancer. Curr. Opin. Biomed. Eng. 2021, 17, 100254. [Google Scholar] [CrossRef]
- Hughes, A.M.; Kolb, A.D.; Shupp, A.B.; Shine, K.M.; Bussard, K.M. Printing the Pathway Forward in Bone Metastatic Cancer Research: Applications of 3D Engineered Models and Bioprinted Scaffolds to Recapitulate the Bone-Tumor Niche. Cancers 2021, 13, 507. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ban, J.; Fock, V.; Aryee, D.N.T.; Kovar, H. Mechanisms, Diagnosis and Treatment of Bone Metastases. Cells 2021, 10, 2944. https://doi.org/10.3390/cells10112944
Ban J, Fock V, Aryee DNT, Kovar H. Mechanisms, Diagnosis and Treatment of Bone Metastases. Cells. 2021; 10(11):2944. https://doi.org/10.3390/cells10112944
Chicago/Turabian StyleBan, Jozef, Valerie Fock, Dave N. T. Aryee, and Heinrich Kovar. 2021. "Mechanisms, Diagnosis and Treatment of Bone Metastases" Cells 10, no. 11: 2944. https://doi.org/10.3390/cells10112944
APA StyleBan, J., Fock, V., Aryee, D. N. T., & Kovar, H. (2021). Mechanisms, Diagnosis and Treatment of Bone Metastases. Cells, 10(11), 2944. https://doi.org/10.3390/cells10112944