
The Unfolded Protein Response as a Guardian of the Secretory Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Conventional Role of the Unfolded Protein Response (UPR) Is Proteostasis
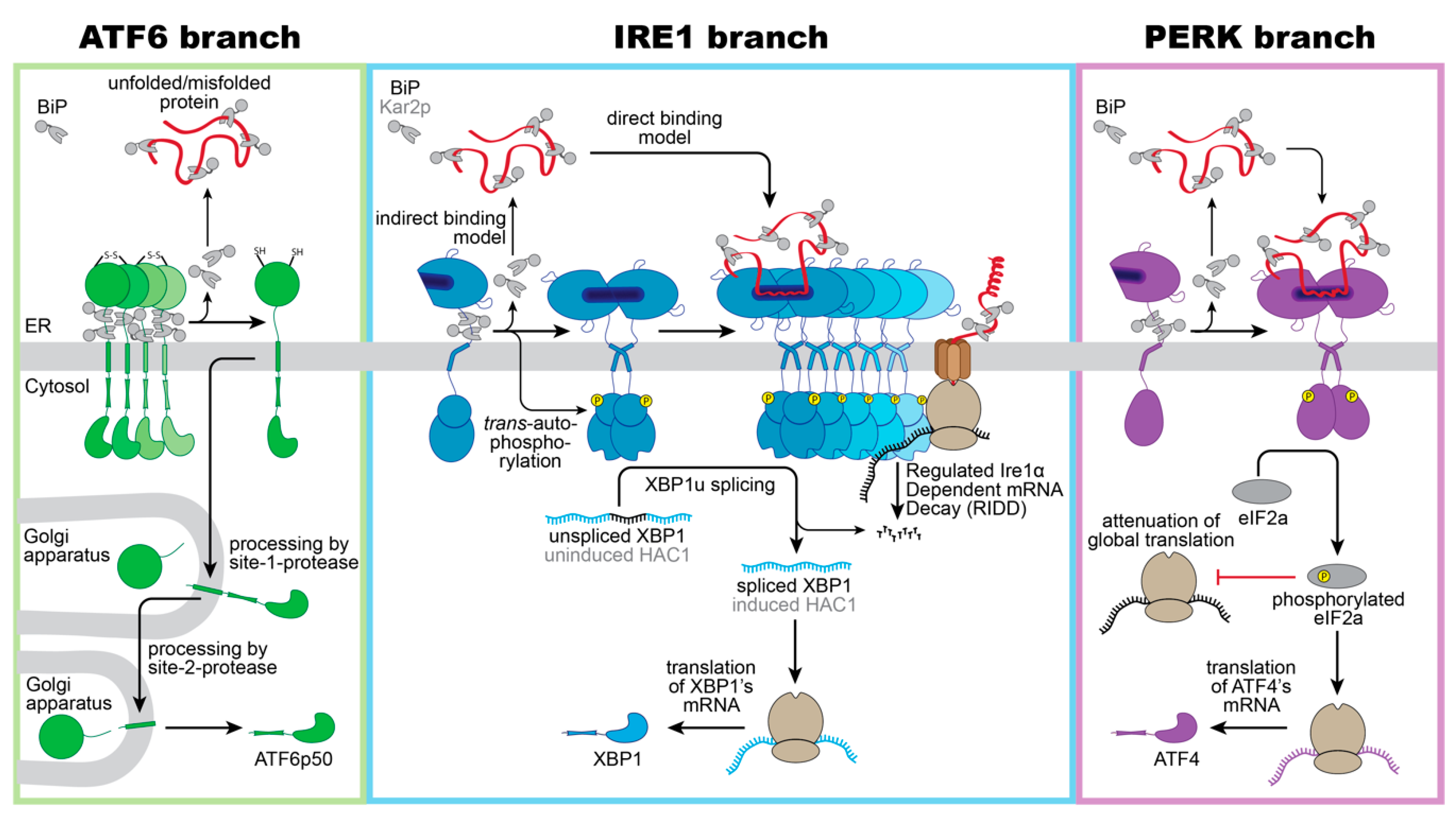
3. Three Musketeers—The Mammalian UPR Transducers
4. A Common Principle: The Oligomeric State Regulates Activity of UPR Transducers
5. Three Mechanisms of Sensing Unfolded Proteins by IRE1
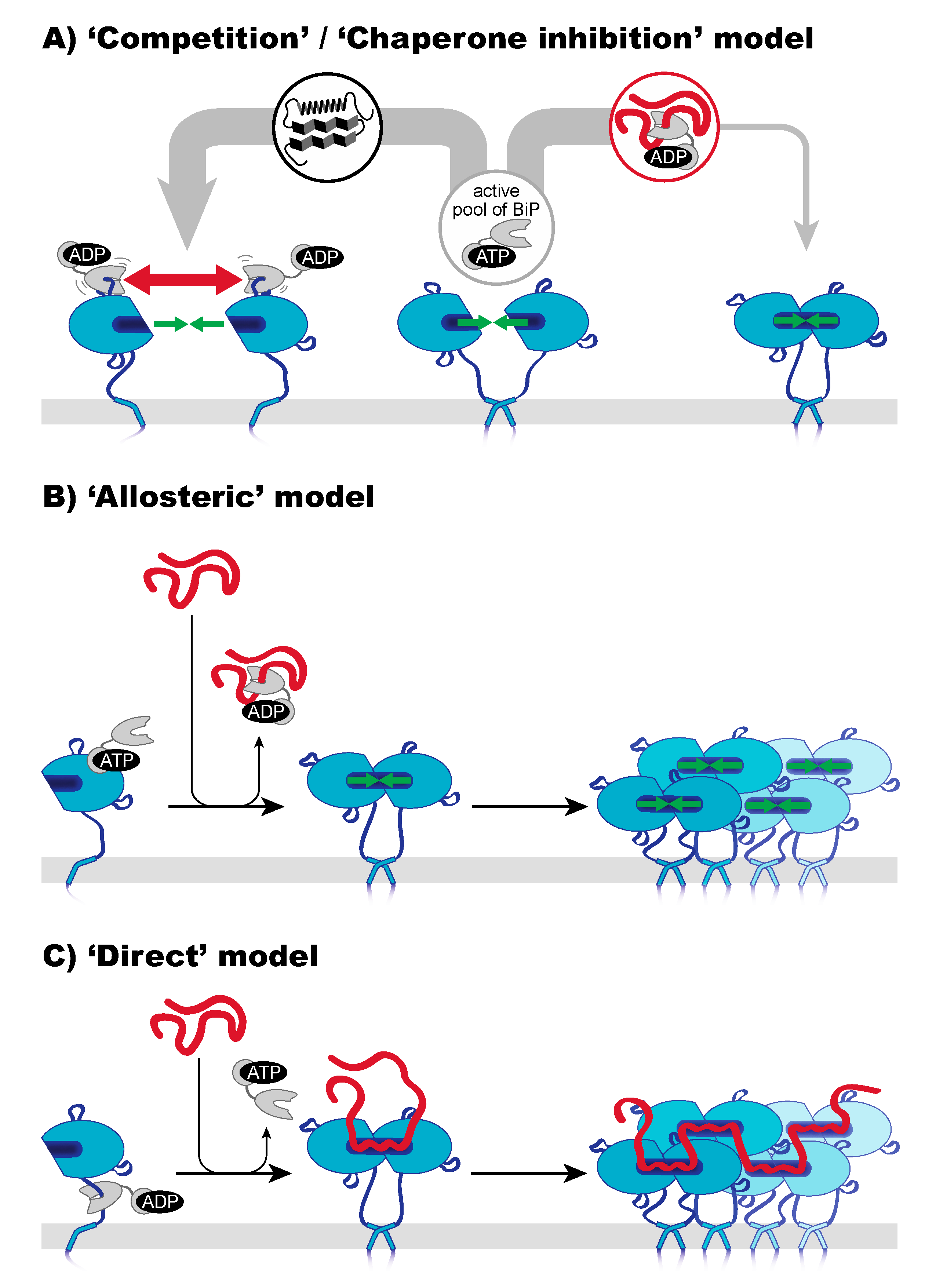
6. A Closer Look on Indirect Models of Sensing—Co-Opting BiP as a Sensor
7. A Closer Look on the Direct Model—Unfolded Proteins as Agonists
8. A Conserved Sensitivity of UPR Transducers for Lipid Bilayer Stress
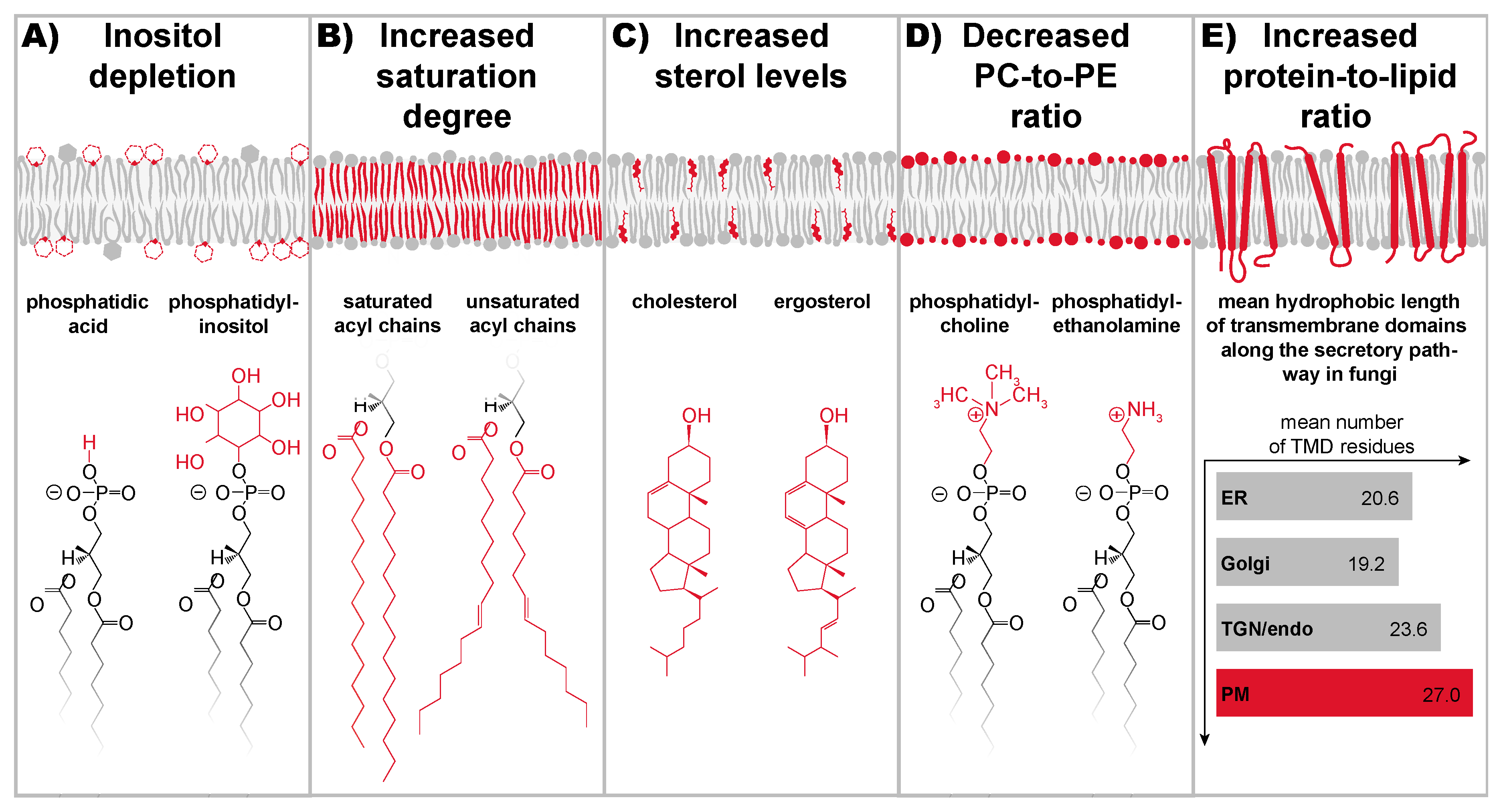
9. A Stunning Variety of Signals Cause Lipid Bilayer Stress
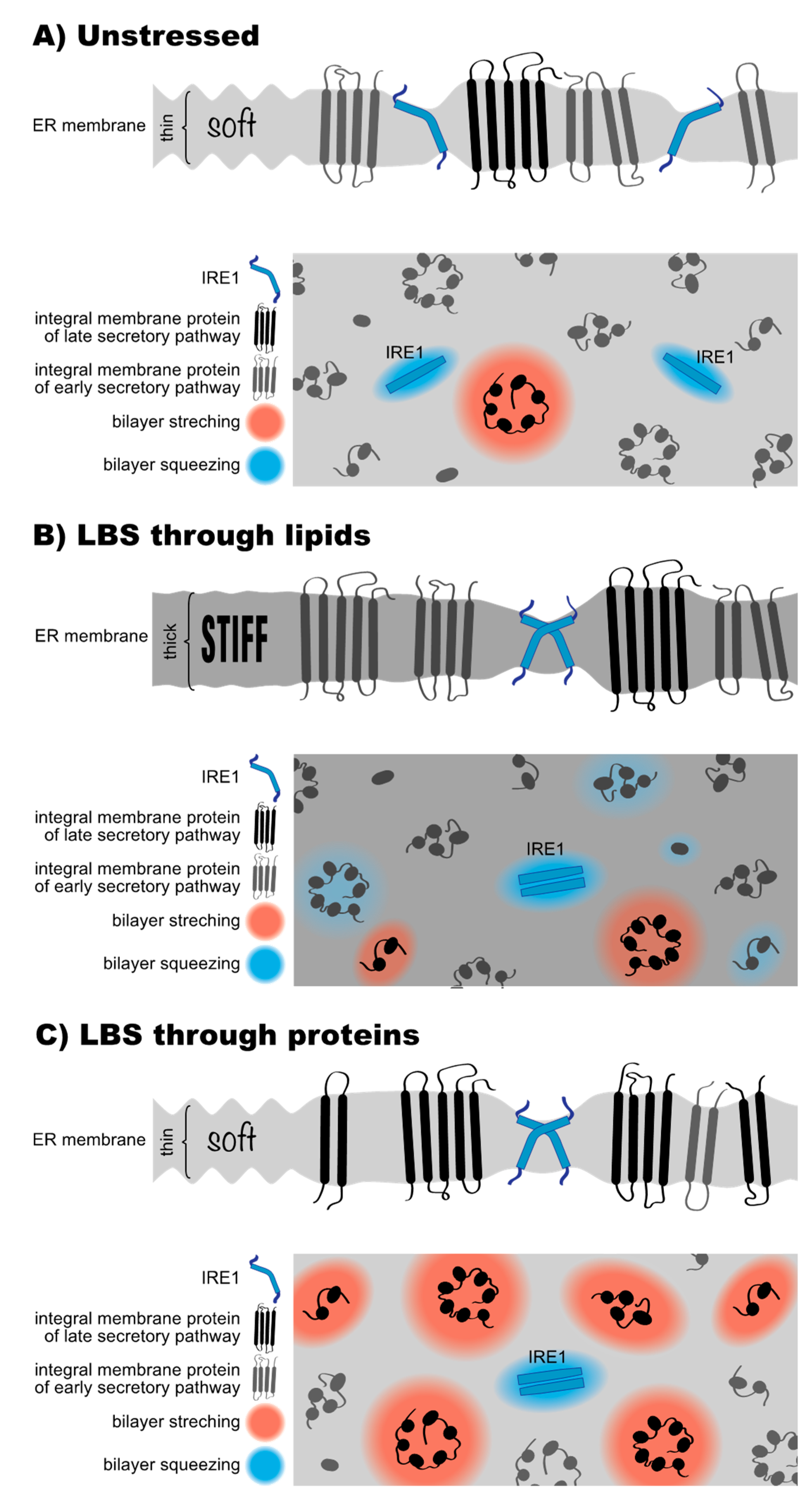
10. How UPR Transducers Sense Membrane Stiffening from Lipid Bilayer Stress
11. What Is Proteotoxic Stress and What Is Lipid Bilayer Stress after All?
12. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2018, 70, 24–31. [Google Scholar] [CrossRef]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef] [Green Version]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Hibi, T.; Dosch, H.-M. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur. J. Immunol. 1986, 16, 139–145. [Google Scholar] [CrossRef]
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Kasai, A.; Omori, H.; Takino, T.; Sugihara, M.; Umemoto, T.; Hamasaki, M.; Hatta, T.; Natsume, T.; Morimoto, R.I.; et al. ERdj8 governs the size of autophagosomes during the formation process. J. Cell Biol. 2020, 219, e201903127. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Zanetti, G.; Pahuja, K.B.; Studer, S.; Shim, S.; Schekman, R. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 2012, 14, 20–28. [Google Scholar] [CrossRef]
- Herzig, Y.; Sharpe, H.J.; Elbaz, Y.; Munro, S.; Schuldiner, M. A systematic approach to pair secretory cargo receptors with their cargo suggests a mechanism for cargo selection by Erv14. PLoS Biol. 2012, 10, e1001329. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Stevens, T.J.; Munro, S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010, 142, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroga, R.; Trenchi, A.; Montoro, A.G.; Taubas, J.V.; Maccioni, H.J.F. Short length transmembrane domains having voluminous exoplasmic halves determine retention of Type II membrane proteins in the Golgi complex. J. Cell Sci. 2013, 126, 5344–5349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, H.-J.; Orlowski, A.; Rog, T.; Nyholm, T.K.M.; Chai, W.; Feizi, T.; Lingwood, D.; Vattulainen, I.; Simons, K. Lateral sorting in model membranes by cholesterol-mediated hydrophobic matching. Proc. Natl. Acad. Sci. USA 2011, 108, 16628–16633. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008, 8, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinser, E.; Paltauf, F.; Daum, G. Sterol composition of yeast organelle membranes and subcellular distribution of enzymes involved in sterol metabolism. J. Bacteriol. 1993, 175, 2853–2858. [Google Scholar] [CrossRef] [Green Version]
- Covino, R.; Ballweg, S.; Stordeur, C.; Michaelis, J.B.; Puth, K.; Wernig, F.; Bahrami, A.; Ernst, A.M.; Hummer, G.; Ernst, R. A eukaryotic sensor for membrane lipid saturation. Mol. Cell 2016, 63, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Ernst, R.; Ejsing, C.S.; Antonny, B. Homeoviscous adaptation and the regulation of membrane lipids. J. Mol. Biol. 2016, 428, 4776–4791. [Google Scholar] [CrossRef] [Green Version]
- Holthuis, J.C.M.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef]
- Ernst, R.; Ballweg, S.; Levental, I. Cellular mechanisms of physicochemical membrane homeostasis. Curr. Opin. Cell Biol. 2018, 53, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Bigay, J.; Antonny, B. Curvature, lipid packing, and electrostatics of membrane organelles: Defining cellular territories in determining specificity. Dev. Cell 2012, 23, 886–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Biol. Nat. Rev. Mol. Cell 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.-J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Maiuolo, J.; Bulotta, S.; Verderio, C.; Benfante, R.; Borgese, N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. USA 2011, 108, 7832–7837. [Google Scholar] [CrossRef] [Green Version]
- Brewer, J.W.; Jackowski, S. UPR-Mediated membrane biogenesis in B Cells. Biochem. Res. Int. 2012, 2012, 738471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, Q.G.; Kimata, Y. Multiple ways for stress sensing and regulation of the endoplasmic reticulum-stress sensors. Cell Struct. Funct. 2021, 46, 37–49. [Google Scholar] [CrossRef]
- Covino, R.; Hummer, G.; Ernst, R. Integrated functions of membrane property sensors and a hidden side of the unfolded protein response. Mol. Cell 2018, 71, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Feng, D.; Li, Y.; Iida, K.; McGrath, B.; Cavener, D.R. PERK EIF2AK3 control of pancreatic β cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006, 4, 491–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Assaad, W.; Buteau, J.; Peyot, M.-L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Koba, H.; Jin, S.; Imada, N.; Ishikawa, T.; Ninagawa, S.; Okada, T.; Sakuma, T.; Yamamoto, T.; Mori, K. Reinvestigation of disulfide-bonded oligomeric forms of the unfolded protein response transducer ATF6. Cell Struct. Funct. 2020, 45, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [Green Version]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.-Y.; Kaufman, R.J. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [Green Version]
- Oka, O.B.; Lith, M.; Rudolf, J.; Tungkum, W.; Pringle, M.A.; Bulleid, N.J. ER p18 regulates activation of ATF 6α during unfolded protein response. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Ellis, R.E.; Lee, K.; Liu, C.-Y.; Yang, K.; Solomon, A.; Yoshida, H.; Morimoto, R.; Kurnit, D.M.; Mori, K.; et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001, 107, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Jurkin, J.; Henkel, T.; Nielsen, A.F.; Minnich, M.; Popow, J.; Kaufmann, T.; Heindl, K.; Hoffmann, T.; Busslinger, M.; Martinez, J. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014, 33, 2922–2936. [Google Scholar] [CrossRef] [PubMed]
- Peschek, J.; Acosta-Alvear, D.; Mendez, A.S.; Walter, P. A conformational RNA zipper promotes intron ejection during non-conventional XBP 1 mRNA splicing. EMBO Rep. 2015, 16, 1688–1698. [Google Scholar] [CrossRef]
- Sidrauski, C.; Cox, J.S.; Walter, P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 1996, 87, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Karagöz, G.E.; Fröhlich, F.; Li, H.; Walther, T.C.; Walter, P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 2018, 7, e43036. [Google Scholar] [CrossRef] [PubMed]
- Bae, D.; Moore, K.A.; Mella, J.M.; Hayashi, S.Y.; Hollien, J. Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J. Cell Biol. 2019, 218, 1118–1127. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-Mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Welihinda, A.A.; Kaufman, R.J. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem. 1996, 271, 18181–18187. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, A.; Appathurai, S.; Plumb, R.; Mariappan, M. Dynamic changes in complexes of IRE1α, PERK, and ATF6α during endoplasmic reticulum stress. Mol. Biol. Cell 2018, 29, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Belyy, V.; Tran, N.-H.; Walter, P. Quantitative microscopy reveals dynamics and fate of clustered IRE1α. Proc. Natl. Acad. Sci. USA 2020, 117, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, D.; Kimata, Y.; Takeuchi, M.; Kohno, K. An essential dimer-forming subregion of the endoplasmic reticulum stress sensor Ire1. Biochem. J. 2005, 391, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.K.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M.U. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4, e03522. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Schroder, M.; Kaufman, R.J. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef] [Green Version]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar] [CrossRef] [Green Version]
- Kimata, Y.; Ishiwata-Kimata, Y.; Ito, T.; Hirata, A.; Suzuki, T.; Oikawa, D.; Takeuchi, M.; Kohno, K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol. 2007, 179, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragón, T.; van Anken, E.; Pincus, D.; Serafimova, I.M.; Korennykh, A.V.; Rubio, C.A.; Walter, P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 2009, 457, 736–740. [Google Scholar] [CrossRef] [Green Version]
- Karagöz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 2017, 6, e30700. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Ali, M.M.U. Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 2015, 34, 1589–1600. [Google Scholar] [CrossRef]
- Sato, Y.; Nadanaka, S.; Okada, T.; Okawa, K.; Mori, K. Luminal Domain of ATF6 Alone Is Sufficient for sensing endoplasmic reticulum stress and subsequent transport to the golgi apparatus. Cell Struct. Funct. 2011, 36, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef]
- Amin-Wetzel, N.; Saunders, R.A.; Kamphuis, M.J.; Rato, C.; Preissler, S.; Harding, H.P.; Ron, D. A J-Protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 2017, 171, 1625–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin-Wetzel, N.; Neidhardt, L.; Yan, Y.; Mayer, M.P.; Ron, D. Unstructured regions in IRE1α specify BiP-mediated destabilisation of the luminal domain dimer and repression of the UPR. Elife 2019, 8, e50793. [Google Scholar] [CrossRef]
- Liu, C.Y.; Wong, H.N.; Schauerte, J.A.; Kaufman, R.J. The protein kinase/endoribonuclease IRE1α that signals the unfolded protein response has a luminal N-terminal ligand-independent dimerization domain. J. Biol. Chem. 2002, 277, 18346–18356. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.C.; Nowak, P.R.; Larburu, N.; Adams, C.J.; Ali, M.M.U. In vitro FRET analysis of IRE1 and BiP association and dissociation upon endoplasmic reticulum stress. Elife 2018, 7, e30257. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [Green Version]
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The unfolded protein response: Detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, a033886. [Google Scholar] [CrossRef] [Green Version]
- Preissler, S.; Ron, D. Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.A.; Dorner, A.J.; Edwards, C.A.; Hendershott, L.M.; Kaufman, R.J. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J. Biol. Chem. 1997, 272, 4327–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preissler, S.; Rato, C.; Yan, Y.; Perera, L.A.; Czako, A.; Ron, D. Calcium depletion challenges endoplasmic reticulum proteostasis by destabilising BiP-substrate complexes. Elife 2020, 9, e62601. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, S.; Appathurai, S.; Sundaram, A.; Plumb, R.; Mariappan, M. A Molecular Mechanism for turning off IRE1α signaling during endoplasmic reticulum stress. Cell Rep. 2020, 33, 108563. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.; Chevalier, M.W.; Aragón, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef] [Green Version]
- Kimata, Y.; Oikawa, D.; Shimizu, Y.; Ishiwata-Kimata, Y.; Kohno, K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J. Cell Biol. 2004, 167, 445–456. [Google Scholar] [CrossRef]
- Matlack, K.E.S.; Misselwitz, B.; Plath, K.; Rapoport, T.A. BiP acts as a molecular ratchet during posttranslational transport of prepro-α factor across the ER membrane. Cell 1999, 97, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Larburu, N.; Adams, C.J.; Chen, C.-S.; Nowak, P.R.; Ali, M.M.U. Mechanism of Hsp70 specialized interactions in protein translocation and the unfolded protein response. Open Biol. 2020, 10, 200089. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Gaudet, R.; Tortorella, D.; Wang, E.W.; Ploegh, H.L.; Wiley, D.C. Antigen presentation subverted: Structure of the human cytomegalovirus protein US2 bound to the class I molecule HLA-A2. Proc. Natl. Acad. Sci. USA 2001, 98, 6794–6799. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 2009, 315, 2496–2504. [Google Scholar] [CrossRef]
- Mathuranyanon, R.; Tsukamoto, T.; Takeuchi, A.; Ishiwata-Kimata, Y.; Tuchiya, Y.; Kohno, K.; Kimata, Y. Tight regulation of the unfolded protein sensor Ire1 by its intramolecularly antagonizing subdomain. J. Cell Sci. 2015, 128, 1762–1772. [Google Scholar] [CrossRef] [Green Version]
- Kimata, Y.; Kohno, K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 2011, 23, 135–142. [Google Scholar] [CrossRef]
- Marcinowski, M.; Höller, M.; Feige, M.J.; Baerend, D.; Lamb, D.C.; Buchner, J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 2011, 18, 150–158. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Duarte, A.M.S.; Akoury, E.; Ippel, H.; Biernat, J.; Morán Luengo, T.; Radli, M.; Didenko, T.; Nordhues, B.A.; Veprintsev, D.B.; et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 2014, 156, 963–974. [Google Scholar] [CrossRef] [Green Version]
- Woehlbier, U.; Hetz, C. Modulating stress responses by the UPRosome: A matter of life and death. Trends Biochem. Sci. 2011, 36, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nagata, K. Roles of the endoplasmic reticulum–resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J. Biol. Chem. 2019, 294, 2133–2141. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, D.; Rojas-Rivera, D.; Rodríguez, D.A.; Groenendyk, J.; Köhler, A.; Lebeaupin, C.; Ito, S.; Urra, H.; Carreras-Sureda, A.; Hazari, Y.; et al. Interactome screening identifies the ER Luminal Chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1α. Mol. Cell 2018, 69, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eletto, D.; Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, I.-H.; Yoon, J.S.; Yang, Q.; Liu, Y.; Skach, W.; Thomas, P. A role for the ribosome-associated complex in activation of the IRE1 branch of UPR. Cell Rep. 2021, 35, 109217. [Google Scholar] [CrossRef]
- Sundaram, A.; Plumb, R.; Appathurai, S.; Mariappan, M. The Sec61 translocon limits IRE1α signaling during the unfolded protein response. Elife 2017, 6, e27187. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.-H.; Carter, S.D.; De Mazière, A.; Ashkenazi, A.; Klumperman, J.; Walter, P.; Jensen, G.J. The stress-sensing domain of activated IRE1α forms helical filaments in narrow ER membrane tubes. Science 2021, 374, 52–57. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Piccolis, M.; Bond, L.M.; Kampmann, M.; Pulimeno, P.; Chitraju, C.; Jayson, C.B.K.; Vaites, L.P.; Boland, S.; Lai, Z.W.; Gabriel, K.R.; et al. Probing the global cellular responses to lipotoxicity caused by saturated fatty acids. Mol. Cell 2019, 74, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [Green Version]
- Wigger, L.; Barovic, M.; Brunner, A.-D.; Marzetta, F.; Schöniger, E.; Mehl, F.; Kipke, N.; Friedland, D.; Burdet, F.; Kessler, C.; et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nat. Metab. 2021, 3, 1017–1031. [Google Scholar] [CrossRef]
- Han, X.; Yang, J.; Yang, K.; Zhao, Z.; Abendschein, D.R.; Gross, R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: A shotgun lipidomics study. Biochemistry 2007, 46, 6417–6428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kartsoli, S.; Kostara, C.E.; Tsimihodimos, V.; Bairaktari, E.T.; Christodoulou, D.K. Lipidomics in non-alcoholic fatty liver disease. World J. Hepatol. 2020, 12, 436–450. [Google Scholar] [CrossRef]
- Han, X. Lipidomics for studying metabolism. Nat. Rev. Endocrinol. 2016, 12, 668–679. [Google Scholar] [CrossRef]
- Pu, Y.; Ruberti, C.; Angelos, E.R.; Brandizzi, F. AtIRE1C, an unconventional isoform of the UPR master regulator AtIRE1, is functionally associated with AtIRE1B in Arabidopsis gametogenesis. Plant Direct 2019, 3, e00187. [Google Scholar] [CrossRef]
- Pineau, L.; Colas, J.; Dupont, S.; Beney, L.; Fleurat-Lessard, P.; Berjeaud, J.M.; Bergès, T.; Ferreira, T. Lipid-induced ER stress: Synergistic effects of sterols and saturated fatty acids. Traffic 2009, 10, 673–690. [Google Scholar] [CrossRef]
- Hou, N.S.; Gutschmidt, A.; Choi, D.Y.; Pather, K.; Shi, X.; Watts, J.L.; Hoppe, T.; Taubert, S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E2271–E2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitai, Y.; Ariyama, H.; Kono, N.; Oikawa, D.; Iwawaki, T.; Arai, H. Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells 2013, 18, 798–809. [Google Scholar] [CrossRef]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.; Yap, W.S.; Xu, J.; Wu, H.; Koh, J.H.; Goh, W.W.B.; George, B.; Chong, S.C.; Taubert, S.; Thibault, G. Stress sensor Ire1 deploys a divergent transcriptional program in response to lipid bilayer stress. J. Cell Biol. 2020, 219, e201909165. [Google Scholar] [CrossRef] [PubMed]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hänelt, I.; Hummer, G.; Ernst, R. Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 2017, 67, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Väth, K.; Mattes, C.; Reinhard, J.; Covino, R.; Stumpf, H.; Hummer, G.; Ernst, R. Cysteine cross-linking in native membranes establishes the transmembrane architecture of Ire1. J. Cell Biol. 2021, 220, e202011078. [Google Scholar] [CrossRef]
- Ishiwata-Kimata, Y.; Promlek, T.; Kohno, K.; Kimata, Y. BiP-bound and nonclustered mode of Ire1 evokes a weak but sustained unfolded protein response. Genes Cells 2013, 18, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Lhoták, Š.; Sharma, A.M.; Austin, R.C. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J. Lipid Res. 2009, 50, 2486–2501. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Goldberg, E.; Goldberg, J. ER retention is imposed by COPII protein sorting and attenuated by 4-phenylbutyrate. eLife 2017, 6, e26624. [Google Scholar] [CrossRef]
- Jonikas, M.C.; Collins, S.R.; Denic, V.; Oh, E.; Quan, E.M.; Schmid, V.; Weibezahn, J.; Schwappach, B.; Walter, P.; Weissman, J.S.; et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 2009, 323, 1693–1697. [Google Scholar] [CrossRef] [Green Version]
- Surma, M.A.; Klose, C.; Peng, D.; Shales, M.; Mrejen, C.; Stefanko, A.; Braberg, H.; Gordon, D.E.; Vorkel, D.; Ejsing, C.S.; et al. A lipid E-MAP identifies Ubx2 as a critical regulator of lipid saturation and lipid bilayer stress. Mol. Cell 2013, 51, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.S.; Chapman, R.E.; Walter, P. The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol. Biol. Cell 1997, 8, 1805–1814. [Google Scholar] [CrossRef]
- Nikawa, J.; Yamashita, S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol. Microbiol. 1992, 6, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Henry, S.A.; Kohlwein, S.D.; Carman, G.M. Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 2012, 190, 317–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajoie, P.; Moir, R.D.; Willis, I.M.; Snapp, E.L. Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Mol. Biol. Cell 2012, 23, 955–964. [Google Scholar] [CrossRef]
- Promlek, T.; Ishiwata-Kimata, Y.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 2011, 22, 3520–3532. [Google Scholar] [CrossRef]
- Michell, R.H. Do inositol supplements enhance phosphatidylinositol supply and thus support endoplasmic reticulum function? Br. J. Nutr. 2018, 120, 301–316. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.M.; Takagi, H.; Kimata, Y. Categorization of endoplasmic reticulum stress as accumulation of unfolded proteins or membrane lipid aberrancy using yeast Ire1 mutants. Biosci. Biotechnol. Biochem. 2019, 83, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.-P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef] [Green Version]
- Ariyama, H.; Kono, N.; Matsuda, S.; Inoue, T.; Arai, H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J. Biol. Chem. 2010, 285, 22027–22035. [Google Scholar] [CrossRef] [Green Version]
- Pineau, L.; Bonifait, L.; Berjeaud, J.M.; Alimardani-Theuil, P.; Berges, T.; Ferreira, T. A lipid-mediated quality control process in the Golgi apparatus in yeast. Mol Biol Cell 2008, 19, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Zhao, Z.; Zhang, L.; Shi, L.; Shahriar, S.; Chan, R.B.; Di Paolo, G.; Min, W. Metabolic activity induces membrane phase separation in endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2017, 114, 13394–13399. [Google Scholar] [CrossRef] [Green Version]
- Kono, N.; Amin-Wetzel, N.; Ron, D. Generic membrane-spanning features endow IRE1α with responsiveness to membrane aberrancy. Mol. Biol. Cell 2017, 28, 2318–2332. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.; Li, L.; Liu, Y.; Pu, J.; Zhang, S.; Yu, J.; Zhao, J.; Liu, P. Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology 2011, 152, 2206–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguil, J.; Pineau, L.; Rowland Snyder, E.C.; Dupont, S.; Beney, L.; Gil, A.; Frapper, G.; Ferreira, T. Modulation of lipid-induced er stress by fatty acid shape. Traffic 2011, 12, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on cholesterol homeostasis: The central role of scap. Annu. Rev. Biochem. 2018, 87, 783–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 is an ER membrane sensor that is central to cholesterol homeostasis. Cell 2017, 171, 1094–1109.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, A.; Kwartler, C.S.; Kaw, K.; Li, Y.; Kaw, A.; Chen, J.; LeMaire, S.A.; Shen, Y.H.; Milewicz, D.M. Cholesterol-induced phenotypic modulation of smooth muscle cells to macrophage/fibroblast–like cells is driven by an unfolded protein response. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 302–316. [Google Scholar] [CrossRef]
- Feng, B.; Yaol, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef]
- Tufanli, O.; Akillilar, P.T.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Çimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, J.; Mattes, C.; Väth, K.; Radanović, T.; Surma, M.A.; Klose, C.; Ernst, R. A Quantitative analysis of cellular lipid compositions during acute proteotoxic er stress reveals specificity in the production of asymmetric lipids. Front. Cell Dev. Biol. 2020, 8, 756. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D. Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes. Biophys. J. 2007, 93, 3884–3899. [Google Scholar] [CrossRef] [Green Version]
- Van Den Brink-Van Der Laan, E.; Antoinette Killian, J.; De Kruijff, B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim. Biophys. Acta-Biomembr. 2004, 1666, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Radanović, T.; Reinhard, J.; Ballweg, S.; Pesek, K.; Ernst, R. An emerging group of membrane property sensors controls the physical state of organellar membranes to maintain their identity. BioEssays 2018, 40, e1700250. [Google Scholar] [CrossRef]
- Phillips, R.; Ursell, T.; Wiggins, P.; Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, G.; Shui, G.; Kim, W.; McAlister, G.C.; Ismail, N.; Gygi, S.P.; Wenk, M.R.; Ng, D.T.W. The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol. Cell 2012, 48, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; van der Veen, J.N.; Vance, J.E.; Thiesen, A.; Vance, D.E.; Jacobs, R.L. Lack of phosphatidylethanolamine N-methyltransferase alters hepatic phospholipid composition and induces endoplasmic reticulum stress. Biochim. Biophys. Acta 2015, 1852, 2689–2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumann, H.A. Depletion of phosphatidylcholine in yeast induces shortening and increased saturation of the lipid acyl chains: Evidence for regulation of intrinsic membrane curvature in a eukaryote. Mol. Biol. Cell 2005, 17, 1006–1017. [Google Scholar] [CrossRef]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Noyon, C.; Ruysschaert, J.-M.; Van Antwerpen, P.; Govaerts, C. Phosphatidylethanolamine is a key regulator of membrane fluidity in eukaryotic cells. J. Biol. Chem. 2016, 291, 3658–3667. [Google Scholar] [CrossRef] [Green Version]
- Shyu, P.; Ng, B.S.H.; Ho, N.; Chaw, R.; Seah, Y.L.; Marvalim, C.; Thibault, G. Membrane phospholipid alteration causes chronic ER stress through early degradation of homeostatic ER-resident proteins. Sci. Rep. 2019, 9, 8637. [Google Scholar] [CrossRef] [Green Version]
- Waite, K.A.; Cabilio, N.R.; Vance, D.E. Choline deficiency–induced liver damage is reversible in Pemt−/−Mice. J. Nutr. 2002, 132, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Breslow, D.K.; Collins, S.R.; Bodenmiller, B.; Aebersold, R.; Simons, K.; Shevchenko, A.; Ejsing, C.S.; Weissman, J.S. Orm family proteins mediate sphingolipid homeostasis. Nature 2010, 463, 1048–1053. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Huang, C.; Polu, S.R.; Schneiter, R.; Chang, A. Regulation of sphingolipid synthesis through Orm1 and Orm2 in yeast. J. Cell Sci. 2012, 125, 2428–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Lone, M.A.; Schneiter, R.; Chang, A. Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proc. Natl. Acad. Sci. USA 2010, 107, 5851–5856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micoogullari, Y.; Basu, S.S.; Ang, J.; Weisshaar, N.; Schmitt, N.D.; Abdelmoula, W.M.; Lopez, B.; Agar, J.N.; Agar, N.; Hanna, J. Dysregulation of very-long-chain fatty acid metabolism causes membrane saturation and induction of the unfolded protein response. Mol. Biol. Cell 2020, 31, 7–17. [Google Scholar] [CrossRef]
- Erdbrügger, P.; Fröhlich, F. The role of very long chain fatty acids in yeast physiology and human diseases. Biol. Chem. 2020, 402, 25–38. [Google Scholar] [CrossRef]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, I.G.; Nomura, D.K.; Forbes, D.J.; Niwa, M. The UPR Activator ATF6 Responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev. Cell 2018, 46, 327–343. [Google Scholar] [CrossRef] [Green Version]
- Contreras, F.-X.; Ernst, A.M.; Haberkant, P.; Björkholm, P.; Lindahl, E.; Gönen, B.; Tischer, C.; Elofsson, A.; von Heijne, G.; Thiele, C.; et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 2012, 481, 525–529. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, I.; Ohvo-Rekilä, H.; Slotte, J.P.; Johnson, A.E.; von Heijne, G. Inhibition of protein translocation across the endoplasmic reticulum membrane by sterols. J. Biol. Chem. 2001, 276, 41748–41754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Rapoport, T.A. Translocation of proteins through a distorted lipid bilayer. Trends Cell Biol. 2021, 31, 473–484. [Google Scholar] [CrossRef]
- Ronchi, P.; Colombo, S.; Francolini, M.; Borgese, N. Transmembrane domain–dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 2008, 181, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Stanzione, F.; Oak, A.; Kim, G.H.; Yerneni, S.; Qi, L.; Sum, A.K.; Chan, C. Intrinsic structural features of the human IRE1α transmembrane domain sense membrane lipid saturation. Cell Rep. 2019, 27, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Ballweg, S.; Sezgin, E.; Doktorova, M.; Covino, R.; Reinhard, J.; Wunnicke, D.; Hänelt, I.; Levental, I.; Hummer, G.; Ernst, R. Regulation of lipid saturation without sensing membrane fluidity. Nat. Commun. 2020, 11, 756. [Google Scholar] [CrossRef] [Green Version]
- Cornell, R.B. Membrane lipids assist catalysis by CTP: Phosphocholine cytidylyltransferase. J. Mol. Biol. 2020, 432, 5023–5042. [Google Scholar] [CrossRef] [PubMed]
- Cornell, R.B. Membrane lipid compositional sensing by the inducible amphipathic helix of CCT. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Carman, G.M.; Han, G.-S. Fat-regulating phosphatidic acid phosphatase: A review of its roles and regulation in lipid homeostasis. J. Lipid Res. 2019, 60, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Karanasios, E.; Han, G.-S.; Xu, Z.; Carman, G.M.; Siniossoglou, S. A phosphorylation-regulated amphipathic helix controls the membrane translocation and function of the yeast phosphatidate phosphatase. Proc. Natl. Acad. Sci. USA 2010, 107, 17539–17544. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, H.F.; Gecht, M.; Fischer, S.C.; Seybert, A.; Frangakis, A.S.; Stelzer, E.H.K.; Covino, R.; Hummer, G.; Ernst, R. The molecular recognition of phosphatidic acid by an amphipathic helix in Opi1. J. Cell Biol. 2018, 217, 3109–3126. [Google Scholar] [CrossRef] [Green Version]
- Chua, N.K.; Howe, V.; Jatana, N.; Thukral, L.; Brown, A.J. A conserved degron containing an amphipathic helix regulates the cholesterol-mediated turnover of human squalene monooxygenase, a rate-limiting enzyme in cholesterol synthesis. J. Biol. Chem. 2017, 292, 19959–19973. [Google Scholar] [CrossRef] [Green Version]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plumb, R.; Zhang, Z.-R.; Appathurai, S.; Mariappan, M. A functional link between the co-translational protein translocation pathway and the UPR. Elife 2015, 4, e07426. [Google Scholar] [CrossRef] [PubMed]
- Pagant, S.; Wu, A.; Edwards, S.; Diehl, F.; Miller, E.A. Sec24 is a coincidence detector that simultaneously binds two signals to drive ER export. Curr. Biol. 2015, 25, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Oda, K.; Yokota, S.; Takatsuki, A.; Ikehara, Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 1988, 263, 18545–18552. [Google Scholar] [CrossRef]
- Liu, E.S.; Ou, J.H.; Lee, A.S. Brefeldin A as a regulator of grp78 gene expression in mammalian cells. J. Biol. Chem. 1992, 267, 7128–7133. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radanović, T.; Ernst, R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells 2021, 10, 2965. https://doi.org/10.3390/cells10112965
Radanović T, Ernst R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells. 2021; 10(11):2965. https://doi.org/10.3390/cells10112965
Chicago/Turabian StyleRadanović, Toni, and Robert Ernst. 2021. "The Unfolded Protein Response as a Guardian of the Secretory Pathway" Cells 10, no. 11: 2965. https://doi.org/10.3390/cells10112965
APA StyleRadanović, T., & Ernst, R. (2021). The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells, 10(11), 2965. https://doi.org/10.3390/cells10112965