
A Cell-Based Platform for the Investigation of Immunoproteasome Subunit β5i Expression and Biology of β5i-Containing Proteasomes

 , , , ,
, , , ,
Abstract
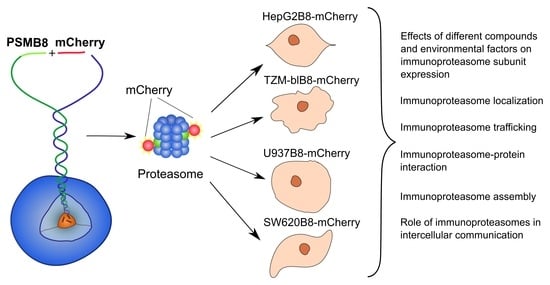
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Molecular Cloning
2.3. Transfection and Cell Sorting
2.4. Isolation of Genomic DNA, RNA, PCR and Real-Time PCR
2.5. RNAseq
2.6. Preparation of Cellular Lysates and Western Blotting
2.7. Immunoprecipitation of Proteasomes
2.8. Detection of Catalytically Active Proteasome Subunits
2.9. Determination of Proteasome Activity
2.10. Fluorescent and Confocal Microscopy
2.11. Flow Cytometry
2.12. Treatment of Cells with Anti-Cancer Drugs
2.13. Statistical Analyses
3. Results
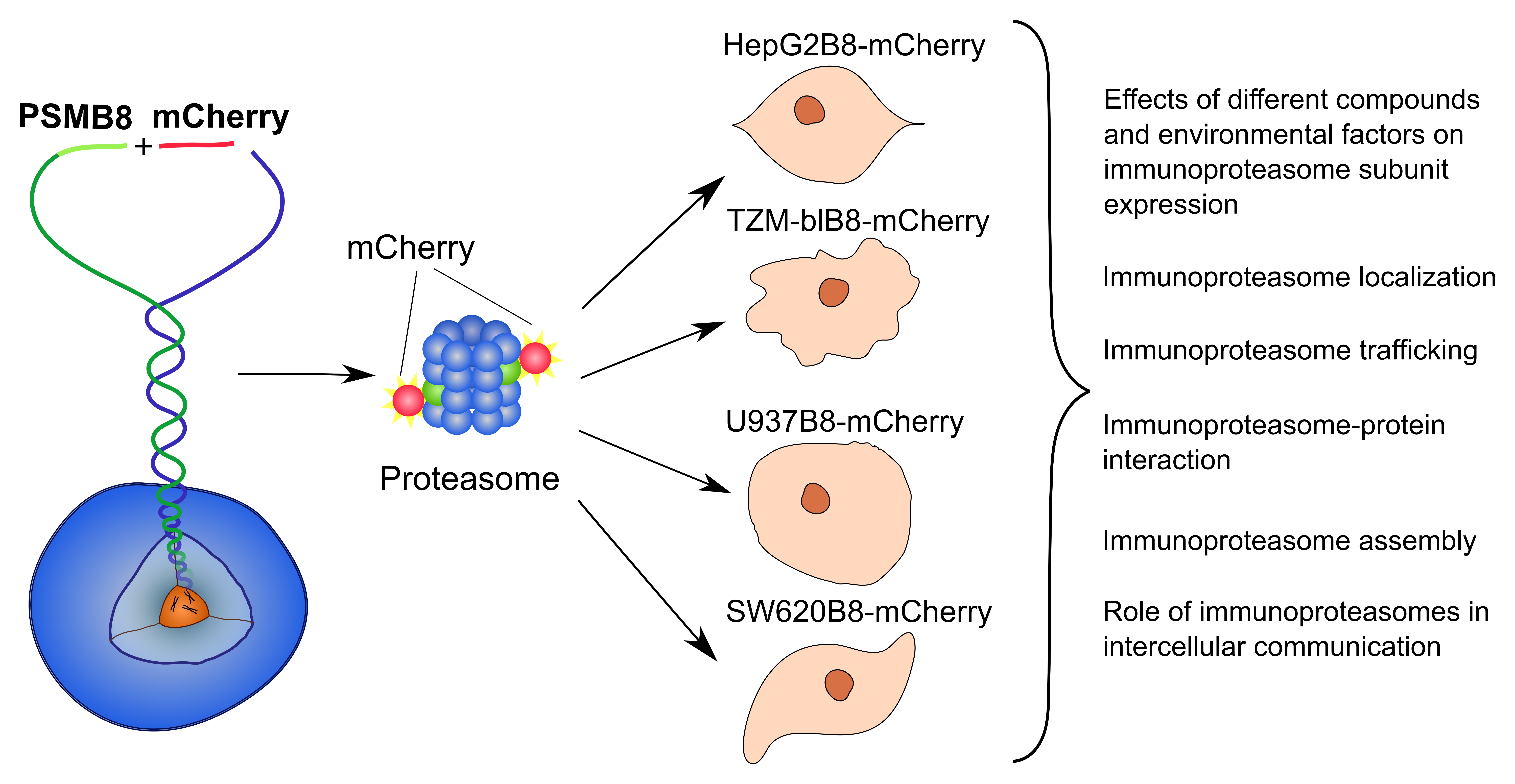
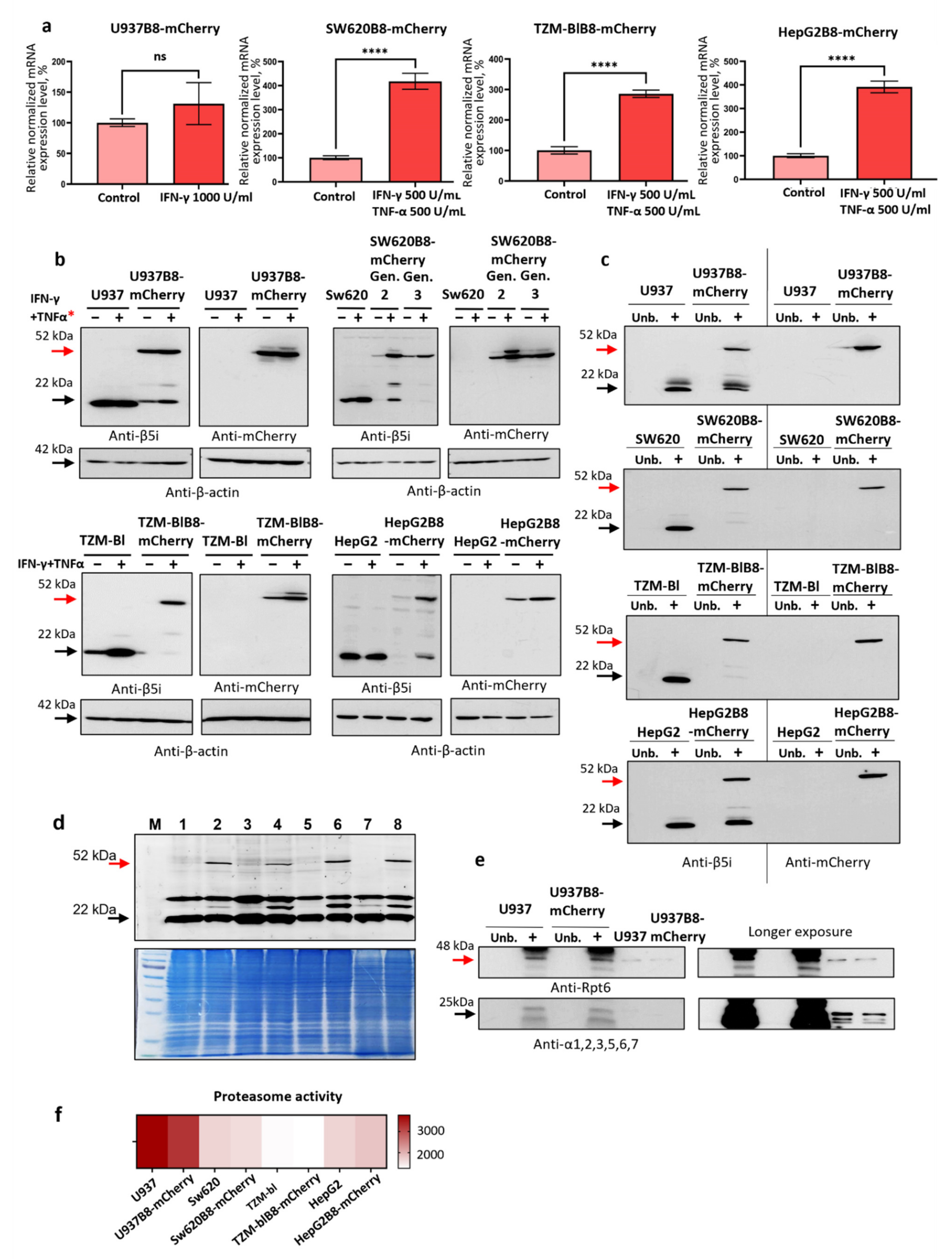
3.1. The mCherry Gene Is Integrated into Genome and Expressed in Modified Cells
3.2. The Control and Modified Cell Lines Demonstrate Marginal Differences in Gene Expression
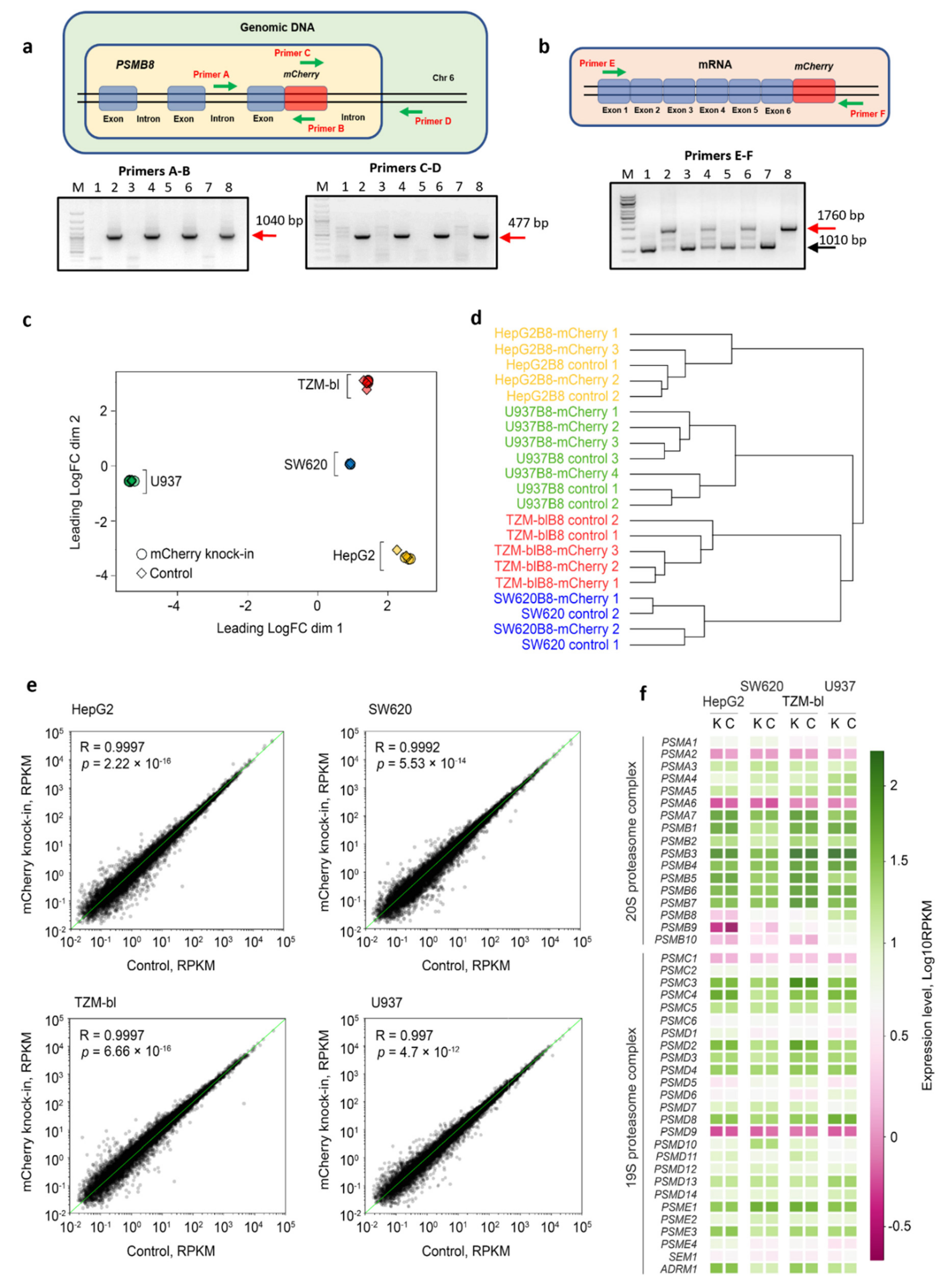
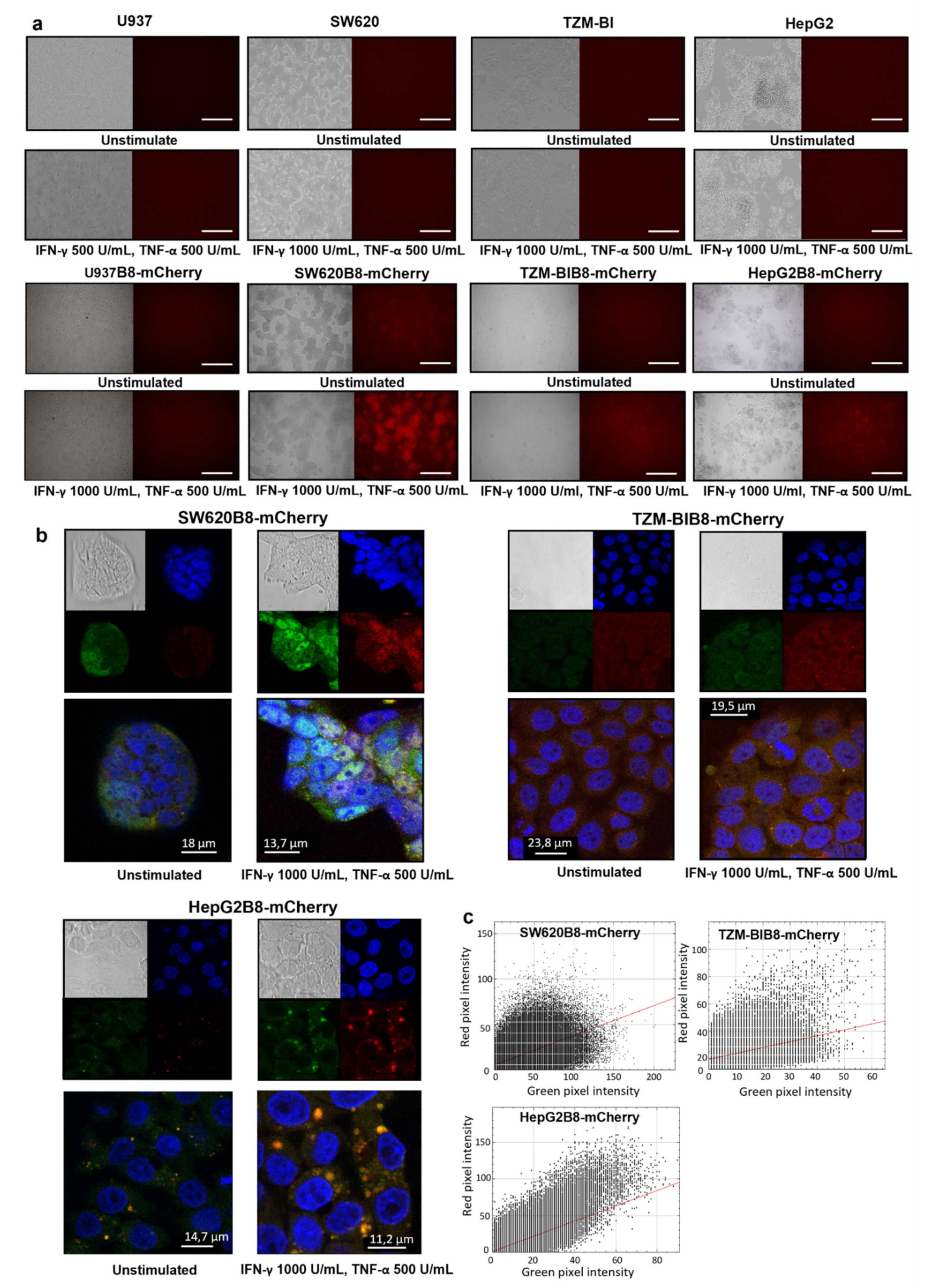
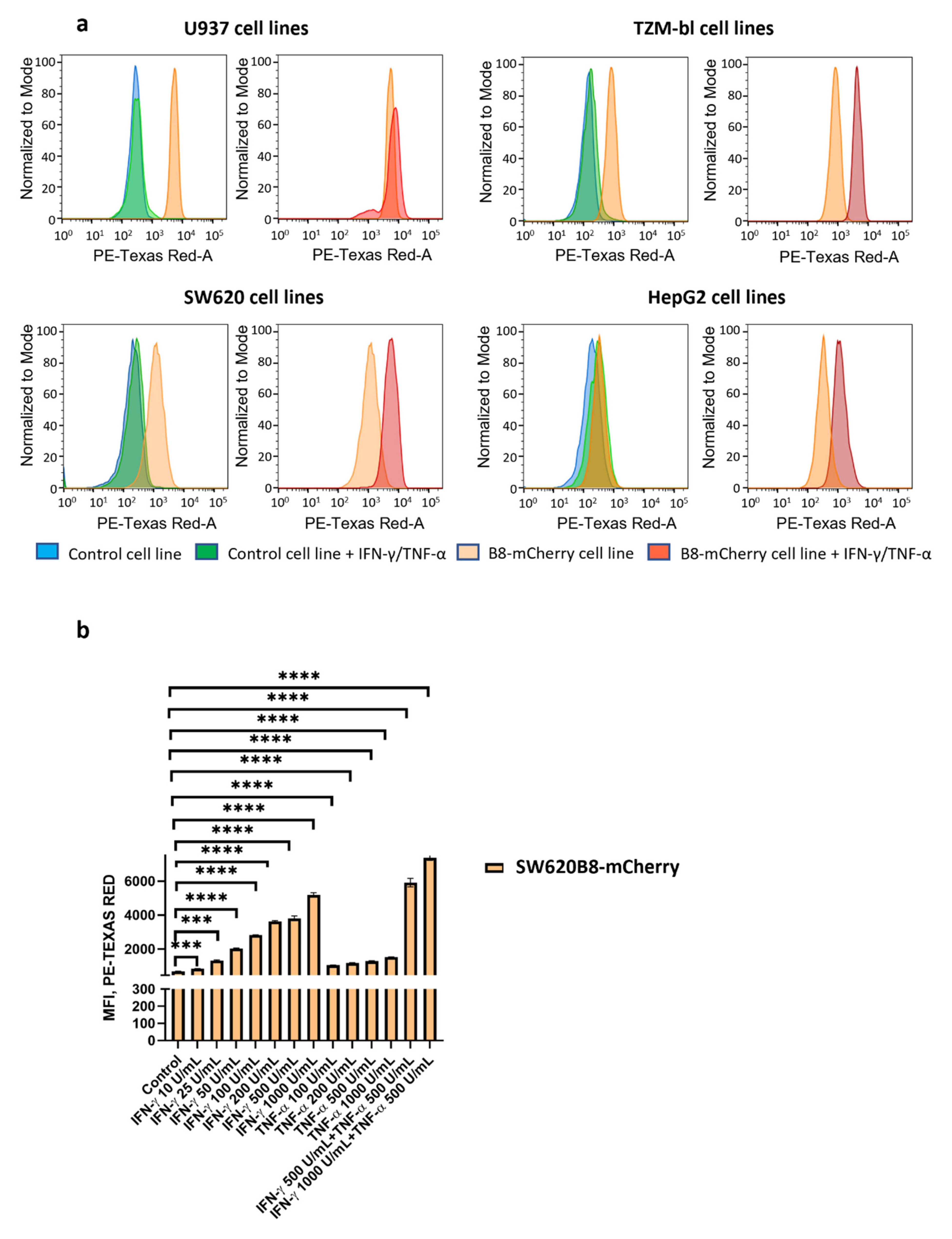
3.3. IFN-γ and TNF-α Stimulate Expression of β5i-mCherry Protein in Modified Cells
3.4. The β5i-mCherry Chimera Is Integrated into the Proteasome and Is an Active Proteasome Subunit in Modified Cells
- (i)
- the chimeric subunit is integrated and is catalytically active within the proteasomes in the modified cells;
- (ii)
- chimeric subunits are unlikely to hamper the association of 20S proteasomes with 19S regulators;
- (iii)
- the integration of β5i-mCherry into proteasomes has minimal influence on proteasome activity in the modified cells.
3.5. Proteasomes with β5i-mCherry Subunit Are Localized in the Nuclei and Cytoplasm of Modified Cells
3.6. Obtained Cell Lines Could Be Used for Quantitative Assessment of the Effects of Different Substances on β5i Subunit Expression
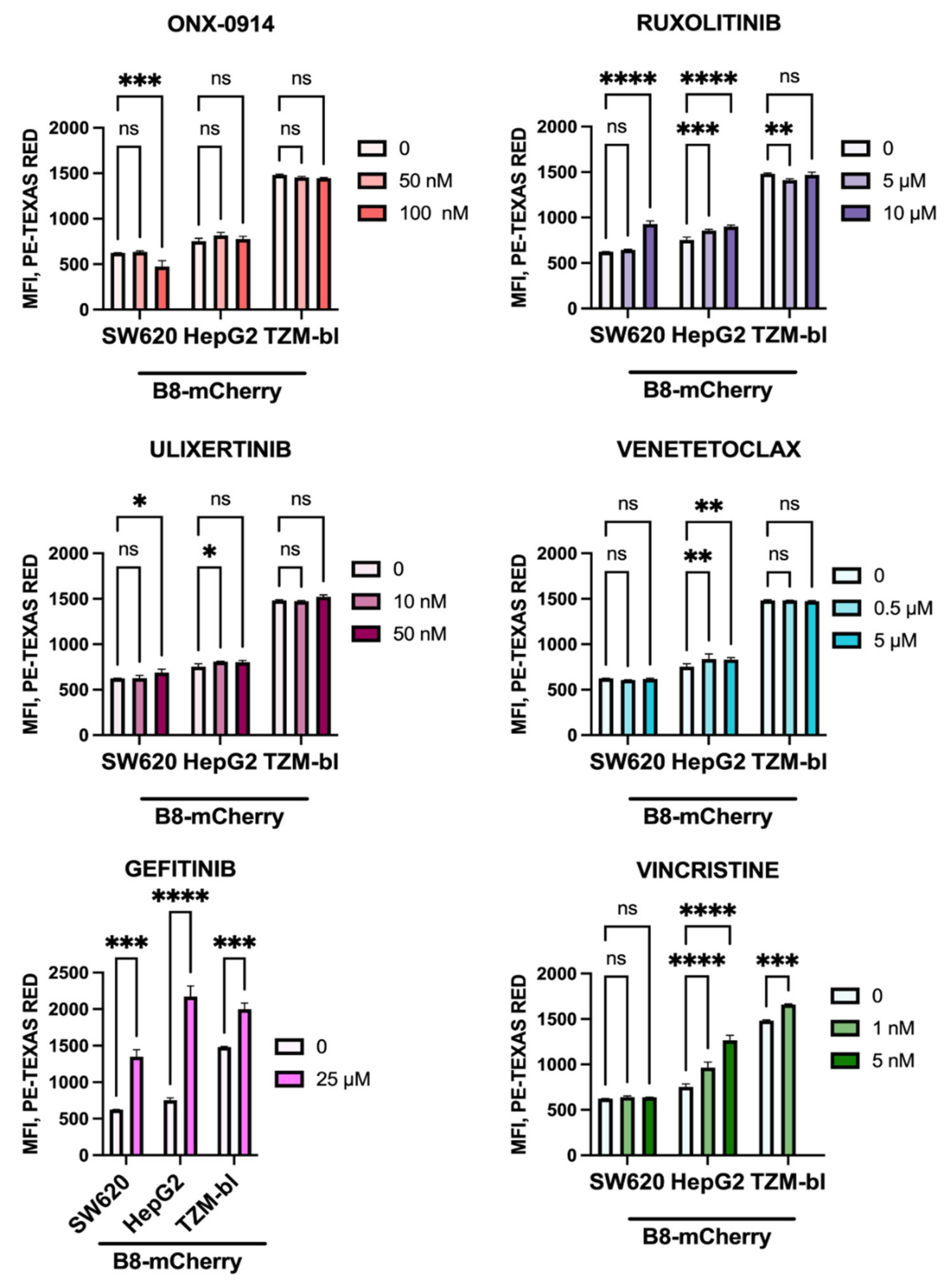
3.7. Anti-Cancer Drugs Ruxolitinib, Vincristine, and Gefitinib Stimulate the Expression of β5i-Containing Proteasomes in Modified Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kisselev, A.F.; Kaganovich, D.; Goldberg, A.L. Binding of Hydrophobic Peptides to Several Non-catalytic Sites Promotes Peptide Hydrolysis by All Active Sites of 20 S Proteasomes. J. Biol. Chem. 2002, 277, 22260–22270. [Google Scholar]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef] [Green Version]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Mccarthy, M.K.; Weinberg, J.B. The immunoproteasome and viral infection: A complex regulator of inflammation. Front. Microbiol. 2015, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-γ Induces Different Subunit Organizations and Functional Diversity of Proteasomes. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-C.; Seifert, U.; Kato, T.; Rice, C.M.; Feinstone, S.M.; Kloetzel, P.-M.; Rehermann, B. Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J. Clin. Investig. 2006, 116, 3006–3014. [Google Scholar] [CrossRef] [Green Version]
- Kotamraju, S.; Matalon, S.; Matsunaga, T.; Shang, T.; Hickman-Davis, J.M.; Kalyanaraman, B. Upregulation of immunoproteasomes by nitric oxide: Potential antioxidative mechanism in endothelial cells. Free Radic. Biol. Med. 2006, 40, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Guan, X.Q.; Kisselev, A.F.; Papasian, C.J.; Qureshi, A.A.; Morrison, D.C.; Van Way, C.W.; Vogel, S.N.; Qureshi, N. LPS-induced formation of immunoproteasomes: TNF-alpha and nitric oxide production are regulated by altered composition of proteasome-active sites. Cell Biochem. Biophys. 2011, 60, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khilji, M.S.; Verstappen, D.; Dahlby, T.; Burstein Prause, M.C.; Pihl, C.; Bresson, S.E.; Bryde, T.H.; Andersen, P.A.K.; Klindt, K.; Zivkovic, D.; et al. The intermediate proteasome is constitutively expressed in pancreatic beta cells and upregulated by stimulatory, low concentrations of interleukin 1β. PLoS ONE 2020, 15, e0222432. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.V.; Karpov, V.L. Biological consequences of structural and functional proteasome diversity. Heliyon 2018, 4, e00894. [Google Scholar] [CrossRef] [Green Version]
- De Verteuil, D.A.; Rouette, A.; Hardy, M.-P.; Lavallée, S.; Trofimov, A.; Gaucher, E.; Perreault, C. Immunoproteasomes Shape the Transcriptome and Regulate the Function of Dendritic Cells. J. Immunol. 2014, 193, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010, 432, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, S.P.; Collin, J.; Irina, N.; Anyfantis, G.; Kyung, B.K.; Lako, M.; Armstrong, L. A Putative Role for the Immunoproteasome in the Maintenance of Pluripotency in Human Embryonic Stem Cells. Stem Cells 2012, 30, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Moebius, J.; Van Den Broek, M.; Groettrup, M.; Basler, M. Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur. J. Immunol. 2010, 40, 3439–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussong, S.A.; Roehrich, H.; Kapphahn, R.J.; Maldonado, M.; Pardue, M.T.; Ferrington, D.A. A novel role for the immunoproteasome in retinal function. Investig. Opthalmol. Vis. Sci. 2011, 52, 714–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Hwang, S.M.; Gomes, A.V. Identification of the immunoproteasome as a novel regulator of skeletal muscle differentiation. Mol. Cell. Biol. 2014, 34, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Morozov, A.V.; Karpov, V.L. Proteasomes and Several Aspects of Their Heterogeneity Relevant to Cancer. Front. Oncol. 2019, 9, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltsev, A.V.; Funikov, S.Y.; Burov, A.V.; Spasskaya, D.S.; Ignatyuk, V.A.; Astakhova, T.M.; Lyupina, Y.; Deikin, A.; Tutyaeva, V.; Bal, N.; et al. Immunoproteasome Inhibitor ONX-0914 Affects Long-Term Potentiation in Murine Hippocampus. J. Neuroimmune Pharmacol. 2021, 16, 7–11. [Google Scholar] [CrossRef]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Dooves, S.; Kooijman, L.; Chan, E.T.; Kirk, C.J.; Smith, V.D.; Koot, S.; Mamber, C.; Jansen, A.H.; et al. Reactive glia show increased immunoproteasome activity in Alzheimer’s disease. Brain 2013, 136, 1415–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, D.; Lee, M.J.; Baek, A.; Yeo, I.J.; Miller, Z.; Baek, Y.M.; Lee, S.; Kim, D.-E.; Hong, J.T.; Kim, K.B. LMP2 Inhibitors as a Potential Treatment for Alzheimer’s Disease. J. Med. Chem. 2020, 63, 3763–3783. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, G.; Huber, E.M.; Xin, B.-T.; van Rooden, E.J.; Al-Ayed, K.; Kim, K.-B.; Kisselev, A.F.; Driessen, C.; van der Stelt, M.; van der Marel, G.A.; et al. Structure-Based Design of β1i or β5i Specific Inhibitors of Human Immunoproteasomes. J. Med. Chem. 2014, 57, 6197–6209. [Google Scholar] [CrossRef]
- Lickliter, J.; Anderl, J.; Kirk, C.J.; Wang, J.; Bomba, D. KZR-616, a Selective Inhibitor of the Immunoproteasome, Shows a Promising Safety and Target Inhibition Profile in a Phase I, Double-Blind, Single (SAD) and Multiple Ascending Dose (MAD) Study in Healthy Volunteers. Arthritis Rheumatol. 2017, 69 (Suppl. 10), 3674–3675. [Google Scholar]
- Ettari, R.; Zappala, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef]
- Muchamuel, T.; Anderl, J.; Fan, R.A.; Johnson, H.; Kirk, C.; Lowe, E. FRI0296 Kzr-616, a selective inhibitor of the immunoproteasome, blocks the disease progression in multiple models of systemic lupus erythematosus (SLE). Ann. Rheum. Dis. 2018, 77, 685. [Google Scholar]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Eynde, B.J.V.D. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, B.; Stroobant, V.; Bousquet-Dubouch, M.P.; Colau, D.; Chapiro, J.; Parmentier, N.; Dalet, A.; Eynde, B.J.V.D. Analysis of the Processing of Seven Human Tumor Antigens by Intermediate Proteasomes. J. Immunol. 2012, 189, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Zanker, D.; Waithman, J.; Yewdell, J.W.; Chen, W. Mixed proteasomes function to increase viral peptide diversity and broaden antiviral CD8+ T cell responses. J. Immunol. 2013, 191, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Khilji, M.S.; Bresson, S.E.; Verstappen, D.; Pihl, C.; Andersen, P.A.K.; Agergaard, J.B.; Dahlby, T.; Bryde, T.H.; Klindt, K.; Nielsen, C.K.; et al. The inducible β5i proteasome subunit contributes to proinsulin degradation in GRP94-deficient β-cells and is overexpressed in type 2 diabetes pancreatic islets. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E892–E900. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Benham, A.M.; Plougastel, B.; Neefjes, J.; Trowsdale, J. Dynamics of proteasome distribution in living cells. EMBO J. 1997, 16, 6087–6094. [Google Scholar] [CrossRef] [PubMed]
- Baldin, V.; Militello, M.; Thomas, Y.; Doucet, C.; Fic, W.; Boireau, S.; Jariel-Encontre, I.; Piechaczyk, M.; Bertrand, E.; Tazi, J.; et al. A novel role for PA28gamma-proteasome in nuclear speckle organization and SR protein trafficking. Mol. Biol. Cell 2008, 19, 1706–1716. [Google Scholar] [CrossRef] [Green Version]
- Kulichkova, V.A.; Artamonova, T.O.; Zaykova, J.J.; Ermolaeva, J.B.; Khodorkovskii, M.A.; Barlev, N.A.; Tomilin, A.N.; Tsimokha, A.S. Simultaneous EGFP and tag labeling of the β7 subunit for live imaging and affinity purification of functional human proteasomes. Mol. Biotechnol. 2015, 57, 36–44. [Google Scholar] [CrossRef]
- Schipper-Krom, S.; Sanz, A.S.; van Bodegraven, E.J.; Speijer, D.; Florea, B.I.; Ovaa, H.; Reits, E.A. Visualizing Proteasome Activity and Intracellular Localization Using Fluorescent Proteins and Activity-Based Probes. Front. Mol. Biosci. 2019, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funikov, S.Y.; Spasskaya, D.S.; Burov, A.V.; Teterina, E.V.; Ustyugov, A.A.; Karpov, V.L.; Morozov, A.V. Structures of the Mouse Central Nervous System Contain Different Quantities of Proteasome Gene Transcripts. Mol. Biol. 2021, 55, 54–63. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Dmitriev, A.A.; Kudryavtseva, A.V.; Shargunov, A.V.; Karpov, D.S.; Uroshlev, L.A.; Melnikova, N.V.; Blinov, V.M.; Poverennaya, E.V.; Archakov, A.I.; et al. PPLine: An Automated Pipeline for SNP, SAP, and Splice Variant Detection in the Context of Proteogenomics. J. Proteome Res. 2015, 14, 3729–3737. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2021, 1–7. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- De Jong, A.; Schuurman, K.G.; Rodenko, B.; Ovaa, H.; Berkers, C.R. Fluorescence-based proteasome activity profiling. Methods Mol. Biol. 2012, 803, 183–204. [Google Scholar] [CrossRef] [Green Version]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.-P. Label-Free Quantitative Proteomics Reveals the Dynamics of Proteasome Complexes Composition and Stoichiometry in a Wide Range of Human Cell Lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef]
- Koch, B.; Nijmeijer, B.; Kueblbeck, M.; Cai, Y.; Walther, N.; Ellenberg, J. Generation and validation of homozygous fluorescent knock-in cells using CRISPR-Cas9 genome editing. Nat. Protoc. 2018, 13, 1465–1487. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Arendt, C.S.; Hochstrasser, M. Distinct Elements in the Proteasomal β5 Subunit Propeptide Required for Autocatalytic Processing and Proteasome Assembly. J. Biol. Chem. 2016, 291, 1991–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, E.M.; Groll, M. A Nut for Every Bolt: Subunit-Selective Inhibitors of the Immunoproteasome and Their Therapeutic Potential. Cells 2021, 10, 1929. [Google Scholar] [CrossRef]
- Miles, E.L.; O’Gorman, C.; Zhao, J.; Samuel, M.; Walters, E.; Yi, Y.-J.; Sutovsky, M.; Prather, R.; Wells, K.D.; Sutovsky, P. Transgenic pig carrying green fluorescent proteasomes. Proc. Natl. Acad. Sci. USA 2013, 110, 6334–6339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heink, S.; Ludwig, D.; Kloetzel, P.M.; Kruger, E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA 2005, 102, 9241–9246. [Google Scholar] [CrossRef] [Green Version]
- Papayannakos, C.; Daniel, R. Understanding lentiviral vector chromatin targeting: Working to reduce insertional mutagenic potential for gene therapy. Gene Ther. 2013, 20, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Niewerth, D.; Kaspers, G.J.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Anderl, J.; Blank, J.L.; van de Ven, P.M.; Zweegman, S.; Jansen, G.; et al. Interferon-γ-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J. Hematol. Oncol. 2014, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Moritz, K.E.; McCormack, N.M.; Abera, M.B.; Viollet, C.; Yauger, Y.J.; Sukumar, G.; Dalgard, C.L.; Burnett, B.G. The role of the immunoproteasome in interferon-γ-mediated microglial activation. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, H.Y.; He, X.J. Induction of immunoproteasomes in porcine kidney (PK)-15 cells by interferon-γ and tumor necrosis factor-α. J. Vet. Med. Sci. 2019, 81, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.; Murray, R.Z.; Mason, G.G.; Hendil, K.B.; Rivett, A.J. Association of immunoproteasomes with the endoplasmic reticulum. Biochem. J. 2000, 352, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Claud, E.C.; Much, M.W.; Chang, E.B. Stress granule formation mediates the inhibition of colonic Hsp70 translation by interferon-gamma and tumor necrosis factor-alpha. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G481–G492. [Google Scholar] [CrossRef] [Green Version]
- Turakhiya, A.; Meyer, S.R.; Marincola, G.; Böhm, S.; Vanselow, J.T.; Schlosser, A.; Hofmann, K.; Buchberger, A. ZFAND1 Recruits p97 and the 26S Proteasome to Promote the Clearance of Arsenite-Induced Stress Granules. Mol. Cell 2018, 70, 906–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, M.; Enenkel, C. Intracellular Dynamics of the Ubiquitin-Proteasome-System. F1000Research 2015, 4, 367. [Google Scholar] [CrossRef]
- Enenkel, C. Proteasome dynamics. Biochim. Biophys. Acta 2014, 1843, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okon, I.S.; Coughlan, K.A.; Zhang, M.; Wang, Q.; Zou, M.H. Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J. Biol. Chem. 2015, 290, 9101–9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.D.; Liu, Y.; Li, N.; Zhang, Y.; Jin, Q.; Hao, D.C.; Piao, H.-L.; Dai, Z.-R.; Ge, G.-B.; Yang, L. Induction of CYP1A1 increases gefitinib-induced oxidative stress and apoptosis in A549 cells. Toxicol. Vitr. 2017, 44, 36–43. [Google Scholar] [CrossRef]
- Bhuyan, A.A.M.; Wagner, T.; Cao, H.; Lang, F. Triggering of Suicidal Erythrocyte Death by Gefitinib. Cell. Physiol. Biochem. 2017, 41, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Johnston-Carey, H.K.; Pomatto, L.C.; Davies, K.J. The Immunoproteasome in oxidative stress, aging, and disease. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, A.; Zetterberg, M. The Immunoproteasome in Human Lens Epithelial Cells During Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5038–5045. [Google Scholar] [CrossRef] [Green Version]
- Brehmer, D.; Greff, Z.; Godl, K.; Blencke, S.; Kurtenbach, A.; Weber, M.; Müller, S.; Klebl, B.; Cotton, M.; Kéri, G.; et al. Cellular targets of gefitinib. Cancer Res. 2005, 65, 379–382. [Google Scholar]
- Verma, N.; Rai, A.K.; Kaushik, V.; Brünnert, D.; Chahar, K.R.; Pandey, J.; Goyal, P. Identification of gefitinib off-targets using a structure-based systems biology approach; their validation with reverse docking and retrospective data mining. Sci. Rep. 2016, 6, 33949. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martinez, A.; Ayala, R.; Posma, J.M.; Neves, A.L.; Gauguier, D.; Nicholson, J.K.; Dumas, M.E. MetaboSignal: A network-based approach for topological analysis of metabotype regulation via metabolic and signaling pathways. Bioinformatics 2017, 33, 773–775. [Google Scholar] [CrossRef] [Green Version]
- Motosugi, R.; Murata, S. Dynamic Regulation of Proteasome Expression. Front. Mol. Biosci. 2019, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishto, M.; Liepe, J.; Textoris-Taube, K.; Keller, C.; Henklein, P.; Weberruss, M.; Dahlmann, B.; Enenkel, C.; Voigt, A.; Kuckelkorn, U.; et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur. J. Immunol. 2014, 44, 3508–3521. [Google Scholar] [CrossRef]
- Winter, M.B.; La Greca, F.; Arastu-Kapur, S.; Caiazza, F.; Cimermancic, P.; Buchholz, T.J.; Anderl, J.L.; Ravalin, M.; Bohn, M.F.; Sali, A.; et al. Immunoproteasome functions explained by divergence in cleavage specificity and regulation. eLife 2017, 6, e27364. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Cytokine Stimulation) | Cell Lines | |||||||
|---|---|---|---|---|---|---|---|---|
| Relative MFI, % | SW620 | SW620B8-mCherry | TZM-bl | TZM-blB8-mCherry | HepG2 | HepG2B8-mCherry | U937 | U937B8-mCherry |
| No | 100 | 581 | 100 | 610 | 100 | 200 | 100 | 1928 |
| Yes | 123 | 2778 | 124 | 3199 | 158 | 626 | 109 | 2460 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burov, A.; Funikov, S.; Vagapova, E.; Dalina, A.; Rezvykh, A.; Shyrokova, E.; Lebedev, T.; Grigorieva, E.; Popenko, V.; Leonova, O.; et al. A Cell-Based Platform for the Investigation of Immunoproteasome Subunit β5i Expression and Biology of β5i-Containing Proteasomes. Cells 2021, 10, 3049. https://doi.org/10.3390/cells10113049
Burov A, Funikov S, Vagapova E, Dalina A, Rezvykh A, Shyrokova E, Lebedev T, Grigorieva E, Popenko V, Leonova O, et al. A Cell-Based Platform for the Investigation of Immunoproteasome Subunit β5i Expression and Biology of β5i-Containing Proteasomes. Cells. 2021; 10(11):3049. https://doi.org/10.3390/cells10113049
Chicago/Turabian StyleBurov, Alexander, Sergei Funikov, Elmira Vagapova, Alexandra Dalina, Alexander Rezvykh, Elena Shyrokova, Timofey Lebedev, Ekaterina Grigorieva, Vladimir Popenko, Olga Leonova, and et al. 2021. "A Cell-Based Platform for the Investigation of Immunoproteasome Subunit β5i Expression and Biology of β5i-Containing Proteasomes" Cells 10, no. 11: 3049. https://doi.org/10.3390/cells10113049
APA StyleBurov, A., Funikov, S., Vagapova, E., Dalina, A., Rezvykh, A., Shyrokova, E., Lebedev, T., Grigorieva, E., Popenko, V., Leonova, O., Spasskaya, D., Spirin, P., Prassolov, V., Karpov, V., & Morozov, A. (2021). A Cell-Based Platform for the Investigation of Immunoproteasome Subunit β5i Expression and Biology of β5i-Containing Proteasomes. Cells, 10(11), 3049. https://doi.org/10.3390/cells10113049