
Endothelial Dysfunction through Oxidatively Generated Epigenetic Mark in Respiratory Viral Infections

 , ,
, ,  and
and 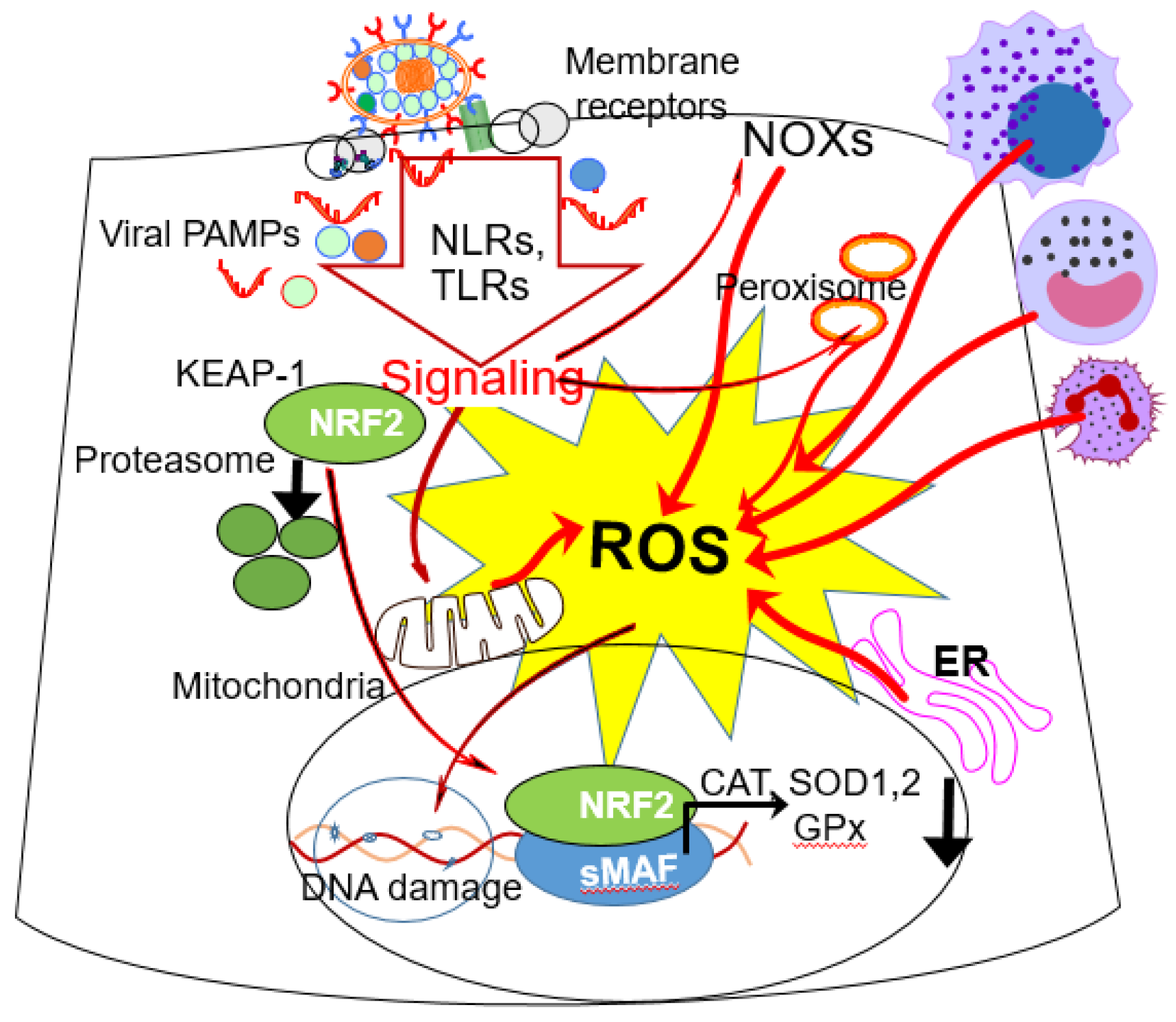{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Endothelial Cells
2.1. The Pulmonary Endothelial Cell
2.2. Permeability of the Alveolar Endothelium during Respiratory Viral Infections
3. Respiratory Viruses and Their Effects on the Bronchial Endothelium
3.1. Respiratory Syncytial Virus
3.2. Influenza Virus and SARS-CoV-2
4. RSV, H1N1 and SARS-CoV-2 Infections and Oxidative Stress
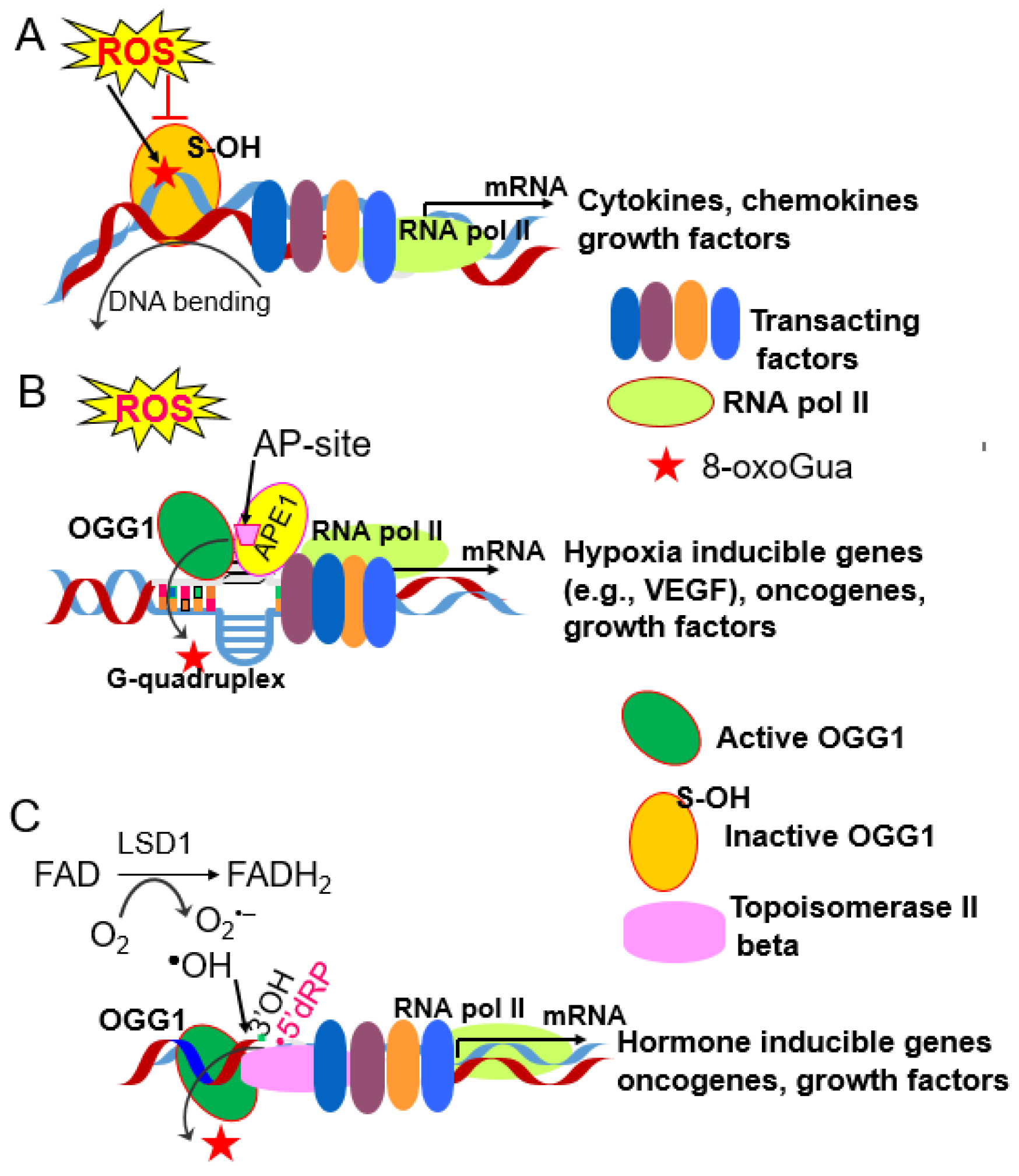
5. Gene Expression Driving Pulmonary Pathologies via Dysregulation of Bronchial Endothelial Cells
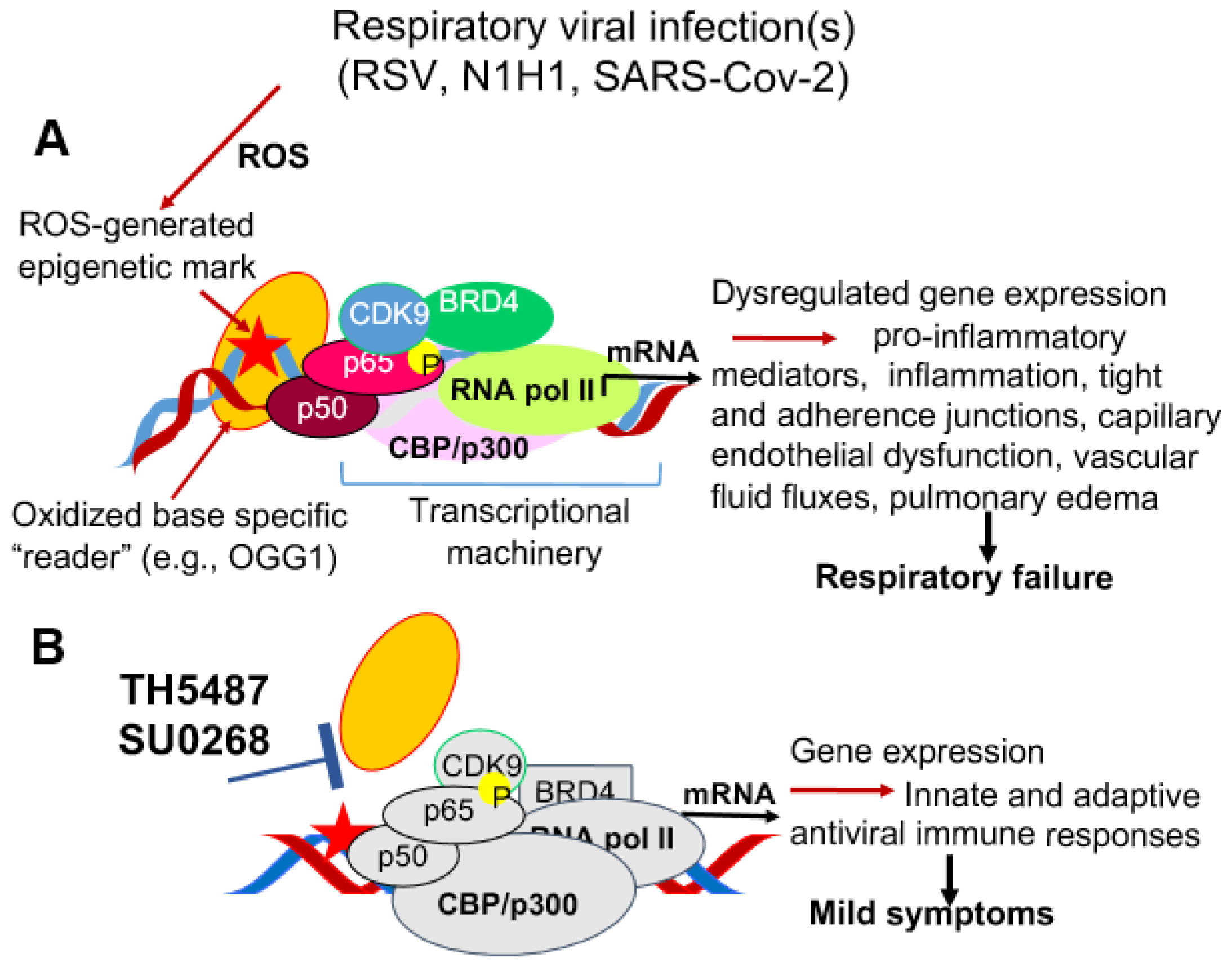
6. Unifying Approach to Ameliorate Endothelial Dysfunction via DNA Occupancy of Transcription Factors
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pober, J.S.; Sessa, W. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Herrero, R.; Sanchez, G.; Lorente, J.A. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 2018, 6, 32. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fosse, J.H.; Haraldsen, G.; Falk, K.; Edelmann, R. Endothelial Cells in Emerging Viral Infections. Front. Cardiovasc. Med. 2021, 8, 619690. [Google Scholar] [CrossRef]
- Handel, A.; Yates, A.; Pilyugin, S.S.; Antia, R. Sharing the burden: Antigen transport and firebreaks in immune responses. J. R. Soc. Interface 2008, 6, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spray, D.C.; Hanstein, R.; Lopez-Quintero, S.V.; Jr, R.F.S.; Suadicani, S.O.; Thi, M.M. Gap junctions and Bystander effects: Good Samaritans and executioners. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 2, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavigne, G.M.; Russell, H.; Sherry, B.; Ke, R. Autocrine and paracrine interferon signalling as ‘ring vaccination’ and ‘contact tracing’ strategies to suppress virus infection in a host. Proc. R. Soc. B: Boil. Sci. 2021, 288, 20203002. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.-B. Endothelial progenitor cells in angiogenesis. Sheng li xue bao: [Acta Physiol. Sin.] 2005, 57, 1–6. [Google Scholar]
- Schleimer, R.P.; A Sterbinsky, S.; Kaiser, J.; A Bickel, C.; A Klunk, D.; Tomioka, K.; Newman, W.; Luscinskas, F.W.; A Gimbrone, M.; McIntyre, B.W. IL-4 induces adherence of human eosinophils and basophils but not neutrophils to endothelium. Association with expression of VCAM-1. J. Immunol. 1992, 148. [Google Scholar]
- Montefort, S.; Feather, I.H.; Wilson, S.J.; Haskard, D.O.; Lee, T.H.; Holgate, S.T.; Howarth, P.H. The Expression of Leukocyte-Endothelial Adhesion Molecules Is Increased in Perennial Allergic Rhinitis. Am. J. Respir. Cell Mol. Biol. 1992, 7, 393–398. [Google Scholar] [CrossRef]
- Schleimer, R.P.; Bochner, B.S. Endothelial leukocyte adhesion molecule-1 and intercellular adhesion molecule-1 mediate the adhesion of eosinophils to endothelial cells in vitro and are expressed by endothelium in allergic cutaneous inflammation in vivo. J. Immunol. 1991, 147, 380–381. [Google Scholar]
- Kyan-Aung, U.; O Haskard, D.; Poston, R.N.; Thornhill, M.; Lee, T. Endothelial leukocyte adhesion molecule-1 and intercellular adhesion molecule-1 mediate the adhesion of eosinophils to endothelial cells in vitro and are expressed by endothelium in allergic cutaneous inflammation in vivo. J. Immunol. 1991, 146, 521–528. [Google Scholar]
- Stefanović, L.; Bogić, M.; Balaban, J.; Mitrović, O.; Stosović, R.; Andrejević, S. [The role of endothelial cells in allergic inflammation reactions]. Srp. Arh. za Celok. Lek. 1998, 126. [Google Scholar]
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir. Res. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, J.; Americh, L.; Gayon, R.; Julien, D.; Mansat, M.; Mansat, P.; Anract, P.; Cantagrel, A.; Cattan, P.; Reimund, J.-M.; et al. A comparative study of endothelial cell markers expressed in chronically inflamed human tissues: MECA-79, Duffy antigen receptor for chemokines, von Willebrand factor, CD31, CD34, CD105 and CD146. J. Pathol. 2005, 206, 260–268. [Google Scholar] [CrossRef]
- Zola, H.; Swart, B.; Nicholson, I.; Aasted, B.; Bensussan, A.; Boumsell, L.; Buckley, C.; Clark, G.; Drbal, K.; Engel, P.; et al. CD molecules 2005: Human cell differentiation molecules. Blood 2005, 106, 3123–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostmans, Y.; De Smedt, K.; Richert, B.; Komi, D.E.A.; Maurer, M.; Michel, O. Markers for the involvement of endothelial cells and the coagulation system in chronic urticaria: A systematic review. Allergy 2021, 76, 2998–3016. [Google Scholar] [CrossRef]
- Niethamer, T.K.; Stabler, C.T.; Leach, J.; A Zepp, J.; Morley, M.P.; Babu, A.; Zhou, S.; E Morrisey, E. Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, A.; Walters, M.S.; Shieh, J.-H.; Shen, L.-B.; Gomi, K.; Downey, R.J.; Crystal, R.G.; Moore, M.A.S. Extracellular vesicles from human airway basal cells respond to cigarette smoke extract and affect vascular endothelial cells. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Karnati, S.; Seimetz, M.; Kleefeldt, F.; Sonawane, A.; Madhusudhan, T.; Bachhuka, A.; Kosanovic, D.; Weissmann, N.; Krüger, K.; Ergün, S. Chronic Obstructive Pulmonary Disease and the Cardiovascular System: Vascular Repair and Regeneration as a Therapeutic Target. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms Regulating Endothelial Permeability. Pulm. Circ. 2014, 4, 535–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asosingh, K.; Weiss, K.; Queisser, K.; Wanner, N.; Yin, M.; Aronica, M.; Erzurum, S. Endothelial cells in the innate response to allergens and initiation of atopic asthma. J. Clin. Investig. 2018, 128, 3116–3128. [Google Scholar] [CrossRef] [Green Version]
- Shoda, T.; Futamura, K.; Orihara, K.; Emi-Sugie, M.; Saito, H.; Matsumoto, K.; Matsuda, A. Recent advances in understanding the roles of vascular endothelial cells in allergic inflammation. Allergol. Int. 2015, 65, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; McCann, K.L.; Imani, F. MAPK and heat shock protein 27 activation are associated with respiratory syncytial virus induction of human bronchial epithelial monolayer disruption. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L436–L445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, A.; Mavunda, K.; Krilov, L.R. Current State of Respiratory Syncytial Virus Disease and Management. Infect. Dis. Ther. 2021, 1–12. [Google Scholar] [CrossRef]
- Martin, G.S.; Brigham, K.L. Fluid Flux and Clearance in Acute Lung Injury. Compr. Physiol. 2012, 2, 2471–2480. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhang, Y.; Luxon, B.A.; Garofalo, R.P.; Casola, A.; Sinha, M.; Brasier, A.R. Identification of NF-κB-Dependent Gene Networks in Respiratory Syncytial Virus-Infected Cells. J. Virol. 2002, 76, 6800–6814. [Google Scholar] [CrossRef] [Green Version]
- Ganter, M.T.; Roux, J.; Miyazawa, B.; Howard, M.; Frank, J.A.; Su, G.; Sheppard, D.; Violette, S.M.; Weinreb, P.; Horan, G.S.; et al. Interleukin-1β Causes Acute Lung Injury via αvβ5 and αvβ6 Integrin–Dependent Mechanisms. Circ. Res. 2008, 102, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Pugin, J.; Ricou, B.; Steinberg, K.P.; Suter, P.M.; Martin, T.R. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am. J. Respir. Crit. Care Med. 1996, 153, 1850–1856. [Google Scholar] [CrossRef]
- Petrache, I.; Birukova, A.; Ramirez, S.I.; Garcia, J.G.N.; Verin, A.D. The Role of the Microtubules in Tumor Necrosis Factor-α–Induced Endothelial Cell Permeability. Am. J. Respir. Cell Mol. Biol. 2003, 28, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Bruewer, M.; Luegering, A.; Kucharzik, T.; Parkos, C.A.; Madara, J.L.; Hopkins, A.; Nusrat, A. Proinflammatory Cytokines Disrupt Epithelial Barrier Function by Apoptosis-Independent Mechanisms. J. Immunol. 2003, 171, 6164–6172. [Google Scholar] [CrossRef] [Green Version]
- Mazzon, E.; Cuzzocrea, S. Role of TNF-α in lung tight junction alteration in mouse model of acute lung inflammation. Respir. Res. 2007, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Hermanns, M.I.; Kasper, J.; Dubruel, P.; Pohl, C.; Uboldi, C.; Vermeersch, V.; Fuchs, S.; Unger, R.E.; Kirkpatrick, C.J. An impaired alveolar-capillary barrier in vitro: Effect of proinflammatory cytokines and consequences on nanocarrier interaction. J. R. Soc. Interface 2009, 7, S41–S54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zhang, Q.; Wang, M.; Zhao, S.; Ma, J.; Luo, N.; Li, N.; Li, Y.; Xu, G.; Li, J. Interferon-γ and tumor necrosis factor-α disrupt epithelial barrier function by altering lipid composition in membrane microdomains of tight junction. Clin. Immunol. 2008, 126, 67–80. [Google Scholar] [CrossRef]
- Al-Sadi, R. Mechanism of cytokine modulation of epithelial tight junction barrier. Front. Biosci. 2009, 14, 2765–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koss, M.; Pfeiffer, G.R.; Wang, Y.; Thomas, S.T.; Yerukhimovich, M.; Gaarde, W.A.; Doerschuk, C.M.; Wang, Q. Ezrin/Radixin/Moesin Proteins Are Phosphorylated by TNF-α and Modulate Permeability Increases in Human Pulmonary Microvascular Endothelial Cells. J. Immunol. 2006, 176, 1218–1227. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, J.A.; Ridley, A.J. Roles of Rho/ROCK and MLCK in TNF-α-induced changes in endothelial morphology and permeability. J. Cell. Physiol. 2007, 213, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Le, T.Q.; Kurihara, N.; Chida, J.; Cisse, Y.; Yano, M.; Kido, H. Influenza Virus–Cytokine-Protease Cycle in the Pathogenesis of Vascular Hyperpermeability in Severe Influenza. J. Infect. Dis. 2010, 202, 991–1001. [Google Scholar] [CrossRef] [Green Version]
- Ahdieh, M.; Vandenbos, T.; Youakim, A. Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-γ. Am. J. Physiol. Physiol. 2001, 281, C2029–C2038. [Google Scholar] [CrossRef] [Green Version]
- Dawre, S.; Maru, S. Human respiratory viral infections: Current status and future prospects of nanotechnology-based approaches for prophylaxis and treatment. Life Sci. 2021, 278, 119561. [Google Scholar] [CrossRef]
- Chatzis, O.; Darbre, S.; Pasquier, J.; Meylan, P.; Manuel, O.; Aubert, J.D.; Beck-Popovic, M.; Masouridi-Levrat, S.; Ansari, M.; Kaiser, L.; et al. Burden of severe RSV disease among immunocompromised children and adults: A 10 year retrospective study. BMC Infect. Dis. 2018, 18, 1–9. [Google Scholar] [CrossRef]
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.A.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.; et al. ICTV Virus Taxonomy Profile: Pneumoviridae. J. Gen. Virol. 2017, 98, 2912–2913. [Google Scholar] [CrossRef]
- Brasier, A.R. RSV Reprograms the CDK9•BRD4 Chromatin Remodeling Complex to Couple Innate Inflammation to Airway Remodeling. Viruses 2020, 12, 472. [Google Scholar] [CrossRef] [Green Version]
- Welliver, T.P.; Garofalo, R.P.; Hosakote, Y.; Hintz, K.H.; Avendano, L.; Sanchez, K.; Velozo, L.; Jafri, H.; Chavez-Bueno, S.; Ogra, P.L.; et al. Severe Human Lower Respiratory Tract Illness Caused by Respiratory Syncytial Virus and Influenza Virus Is Characterized by the Absence of Pulmonary Cytotoxic Lymphocyte Responses. J. Infect. Dis. 2007, 195, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Garofalo, R.P.; Cron, S.G.; Hosakote, Y.M.; Atmar, R.L.; Macias, C.G.; Piedra, P.A. Immunopathogenesis of Respiratory Syncytial Virus Bronchiolitis. J. Infect. Dis. 2007, 195, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Zlateva, K.T.; Van Ranst, M. DETECTION OF SUBGROUP B RESPIRATORY SYNCYTIAL VIRUS IN THE CEREBROSPINAL FLUID OF A PATIENT WITH RESPIRATORY SYNCYTIAL VIRUS PNEUMONIA. Pediatr. Infect. Dis. J. 2004, 23, 1065–1066. [Google Scholar] [CrossRef]
- E Bowles, N.; Ni, J.; Kearney, D.L.; Pauschinger, M.; Schultheiss, H.-P.; McCarthy, R.; Hare, J.; Bricker, J.; Bowles, K.R.; A Towbin, J. Detection of viruses in myocardial tissues by polymerase chain reaction: Evidence of adenovirus as a common cause of myocarditis in children and adults. J. Am. Coll. Cardiol. 2003, 42, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Nadal, D.; Wunderli, W.; Meurmann, O.; Briner, J.; Hirsig, J. Isolation of Respiratory Syncytial Virus from Liver Tissue and Extrahepatic Biliary Atresia Material. Scand. J. Infect. Dis. 1990, 22, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Gkentzi, D.; Dimitriou, G.; Karatza, A. Non-pulmonary manifestations of respiratory syncytial virus infection. J. Thorac. Dis. 2018, 10, S3815–S3818. [Google Scholar] [CrossRef] [PubMed]
- Eiland, L.S. Respiratory Syncytial Virus: Diagnosis, Treatment and Prevention. J. Pediatr. Pharmacol. Ther. 2009, 14, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ralston, S.L.; Lieberthal, A.S.; Meissner, H.C.; Alverson, B.K.; Baley, J.E.; Gadomski, A.M.; Johnson, D.W.; Light, M.J.; Maraqa, N.F.; Mendonca, E.A.; et al. Clinical Practice Guideline: The Diagnosis, Management, and Prevention of Bronchiolitis. Pediatrics 2014, 134, e1474–e1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, R.; Green, C.A.; Sande, C.J.; Drysdale, S.B. Respiratory syncytial virus: Diagnosis, prevention and management. Ther. Adv. Infect. Dis. 2019, 6. [Google Scholar] [CrossRef]
- Beijnen, E.M.S.; Van Haren, S.D. Vaccine-Induced CD8+ T Cell Responses in Children: A Review of Age-Specific Molecular Determinants Contributing to Antigen Cross-Presentation. Front. Immunol. 2020, 11, 607977. [Google Scholar] [CrossRef] [PubMed]
- Biagi, C.; Dondi, A.; Scarpini, S.; Rocca, A.; Vandini, S.; Poletti, G.; Lanari, M. Current State and Challenges in Developing Respiratory Syncytial Virus Vaccines. Vaccines 2020, 8, 672. [Google Scholar] [CrossRef]
- Walsh, E.E. Respiratory Syncytial Virus Infection. Clin. Chest Med. 2016, 38, 29–36. [Google Scholar] [CrossRef] [PubMed]
- E Johnson, J.; A Gonzales, R.; Olson, S.J.; Wright, P.F.; Graham, B.S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 2006, 20, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Norlander, A.E.; Peebles, R.S., Jr. Innate Type 2 Responses to Respiratory Syncytial Virus Infection. Viruses 2020, 12, 521. [Google Scholar] [CrossRef]
- Chang, C.-H.; Huang, Y.; Anderson, R. Activation of vascular endothelial cells by IL-1α released by epithelial cells infected with respiratory syncytial virus. Cell. Immunol. 2003, 221, 37–41. [Google Scholar] [CrossRef]
- Haeberle, H.A.; Kuziel, W.A.; Dieterich, H.-J.; Casola, A.; Gatalica, Z.; Garofalo, R.P. Inducible Expression of Inflammatory Chemokines in Respiratory Syncytial Virus-Infected Mice: Role of MIP-1α in Lung Pathology. J. Virol. 2001, 75, 878–890. [Google Scholar] [CrossRef] [Green Version]
- Arnold, R.; Nig, W.K. Respiratory Syncytial Virus Infection of Human Lung Endothelial Cells Enhances Selectively Intercellular Adhesion Molecule-1 Expression. J. Immunol. 2005, 174, 7359–7367. [Google Scholar] [CrossRef] [Green Version]
- Juliana, A.; Zonneveld, R.; Plötz, F.B.; van Meurs, M.; Wilschut, J. Neutrophil-endothelial interactions in respiratory syncytial virus bronchiolitis: An understudied aspect with a potential for prediction of severity of disease. J. Clin. Virol. 2019, 123, 104258. [Google Scholar] [CrossRef]
- Johansson, C.; Kirsebom, F.C.M. Neutrophils in respiratory viral infections. Mucosal Immunol. 2021, 1–13. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Tian, B.; Boldogh, I.; Garofalo, R.P.; Brasier, A.R. Respiratory Syncytial Virus Infection Induces a Reactive Oxygen Species-MSK1-Phospho-Ser-276 RelA Pathway Required for Cytokine Expression. J. Virol. 2009, 83, 10605–10615. [Google Scholar] [CrossRef] [Green Version]
- Brasier, A.R.; Boldogh, I. Targeting inducible epigenetic reprogramming pathways in chronic airway remodeling. Drugs Context 2019, 8, 1–10. [Google Scholar] [CrossRef]
- Muramoto, Y.; Takada, A.; Fujii, K.; Noda, T.; Iwatsuki-Horimoto, K.; Watanabe, S.; Horimoto, T.; Kida, H.; Kawaoka, Y. Hierarchy among Viral RNA (vRNA) Segments in Their Role in vRNA Incorporation into Influenza A Virions. J. Virol. 2006, 80, 2318–2325. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Q.; Wang, S.; Liu, S.; Hou, G.; Li, J.; Jiang, W.; Wang, K.; Peng, C.; Liu, D.; Guo, A.; et al. Diversity and distribution of type A influenza viruses: An updated panorama analysis based on protein sequences. Virol. J. 2019, 16, 1–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [CrossRef] [PubMed] [Green Version]
- Moore, K.A.; Ostrowsky, J.T.; Kraigsley, A.M.; Mehr, A.J.; Bresee, J.S.; Friede, M.H.; Gellin, B.G.; Golding, J.P.; Hart, P.J.; Moen, A.; et al. A Research and Development (R&D) roadmap for influenza vaccines: Looking toward the future. Vaccine 2021, 39, 6573–6584. [Google Scholar] [CrossRef] [PubMed]
- Poland, G.A.; Ovsyannikova, I.G.; Kennedy, R.B. The need for broadly protective COVID-19 vaccines: Beyond S-only approaches. Vaccine 2021, 39, 4239–4241. [Google Scholar] [CrossRef]
- Ru, Y.-X.; Li, Y.-C.; Zhao, Y.; Zhao, S.-X.; Yang, J.-P.; Zhang, H.-M.; Pang, T.-X. Multiple Organ Invasion by Viruses: Pathological Characteristics in Three Fatal Cases of the 2009 Pandemic Influenza A/H1N1. Ultrastruct. Pathol. 2011, 35, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.J.; Verkerk, M.S.A.; Van Der Bruggen, T.; Marx, J.J.M.; Van Asbeck, B.S.; Diepersloot, R.J.A. Iron chelation and hydroxyl radical scavenging reduce the inflammatory response of endothelial cells after infection with Chlamydia pneumoniae or influenza A. Eur. J. Clin. Investig. 2002, 32, 84–90. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. New Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Choreño-Parra, J.A.; Jiménez-Álvarez, L.A.; Ramírez-Martínez, G.; Cruz-Lagunas, A.; Thapa, M.; Fernández-López, L.A.; Carnalla-Cortés, M.; Choreño-Parra, E.M.; Mena-Hernández, L.; Sandoval-Vega, M.; et al. Expression of Surfactant Protein D Distinguishes Severe Pandemic Influenza A(H1N1) from Coronavirus Disease 2019. J. Infect. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- de Paula, C.B.V.; De Azevedo, M.L.V.; Nagashima, S.; Martins, A.P.C.; Malaquias, M.A.S.; Miggiolaro, A.F.R.D.S.; Júnior, J.D.S.M.; Avelino, G.; Carmo, L.A.P.D.; Carstens, L.B.; et al. IL-4/IL-13 remodeling pathway of COVID-19 lung injury. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Poor, H.D.; Ventetuolo, C.E.; Tolbert, T.; Chun, G.; Serrao, G.; Zeidman, A.; Dangayach, N.S.; Olin, J.; Kohli-Seth, R.; Powell, C.A. COVID-19 critical illness pathophysiology driven by diffuse pulmonary thrombi and pulmonary endothelial dysfunction responsive to thrombolysis. Clin. Transl. Med. 2020, 10. [Google Scholar] [CrossRef]
- Oxford, A.E.; Halla, F.; Robertson, E.B.; Morrison, B.E. Endothelial Cell Contributions to COVID-19. Pathogens 2020, 9, 785. [Google Scholar] [CrossRef]
- Bao, W.; Zhang, X.; Jin, Y.; Hao, H.; Yang, F.; Yin, D.; Chen, X.; Xue, Y.; Han, L.; Zhang, M. Factors Associated with the Expression of ACE2 in Human Lung Tissue: Pathological Evidence from Patients with Normal FEV1 and FEV1/FVC. J. Inflamm. Res. 2021, 14, 1677–1687. [Google Scholar] [CrossRef]
- Conde, J.N.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 2020, 11. [Google Scholar] [CrossRef]
- Vasquez-Bonilla, W.O.; Orozco, R.; Argueta, V.; Sierra, M.; Zambrano, L.I.; Muñoz-Lara, F.; López-Molina, D.S.; Arteaga-Livias, K.; Grimes, Z.; Bryce, C.; et al. A review of the main histopathological findings in coronavirus disease 2019. Hum. Pathol. 2020, 105, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Rotulo, G.A.; Casalini, E.; Brisca, G.; Piccotti, E.; Castagnola, E. Unexpected peak of bronchiolitis requiring oxygen therapy in February 2020: Could an undetected SARS-CoV2-RSV co-infection be the cause? Pediatr. Pulmonol. 2021, 56, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and Oxidative Stress. Biochem. (Moscow) 2020, 85, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Hu, W.; Yu, H.; Zhao, L.; Zhao, Y.; Zhao, X.; Xue, H.; Zhao, Y. Little to no expression of angiotensin-converting enzyme-2 on most human peripheral blood immune cells but highly expressed on tissue macrophages. Cytom. Part A 2020. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Ziegler, C.G.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.; Bals, J.; Hauser, B.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Guazzi, M.; Gomberg-Maitland, M.; Naeije, R. Impact of Pharmacologic Interventions—Treating Endothelial Dysfunction and Group 2 Pulmonary Hypertension. Prog. Cardiovasc. Dis. 2015, 57, 473–479. [Google Scholar] [CrossRef]
- Daiber, A.; Chlopicki, S. Revisiting pharmacology of oxidative stress and endothelial dysfunction in cardiovascular disease: Evidence for redox-based therapies. Free. Radic. Biol. Med. 2020, 157, 15–37. [Google Scholar] [CrossRef]
- Hosakote, Y.M.; Komaravelli, N.; Mautemps, N.; Liu, T.; Garofalo, R.P.; Casola, A. Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L991–L1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosakote, Y.M.; Jantzi, P.D.; Esham, D.L.; Spratt, H.; Kurosky, A.; Casola, A.; Garofalo, R.P. Viral-mediated Inhibition of Antioxidant Enzymes Contributes to the Pathogenesis of Severe Respiratory Syncytial Virus Bronchiolitis. Am. J. Respir. Crit. Care Med. 2011, 183, 1550–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, R.P.; Kolli, D.; Casola, A. Respiratory Syncytial Virus Infection: Mechanisms of Redox Control and Novel Therapeutic Opportunities. Antioxidants Redox Signal. 2013, 18, 186–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.-K.; Minakuchi, M.; Wuputra, K.; Ku, C.-C.; Pan, J.-B.; Kuo, K.-K.; Lin, Y.-C.; Saito, S.; Lin, C.-S.; Yokoyama, K.K. Redox control in the pathophysiology of influenza virus infection. BMC Microbiol. 2020, 20, 1–22. [Google Scholar] [CrossRef]
- Brand, J.V.D.; Haagmans, B.; van Riel, D.; Osterhaus, A.; Kuiken, T. The Pathology and Pathogenesis of Experimental Severe Acute Respiratory Syndrome and Influenza in Animal Models. J. Comp. Pathol. 2014, 151, 83–112. [Google Scholar] [CrossRef] [Green Version]
- Heras, N.D.L.; Giménez, V.M.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Mamun, A.; Dominic, A.; Le, N.-T. SARS-CoV-2 Mediated Endothelial Dysfunction: The Potential Role of Chronic Oxidative Stress. Front. Physiol. 2021, 11, 605908. [Google Scholar] [CrossRef] [PubMed]
- Panfoli, I. Potential role of endothelial cell surface ectopic redox complexes in COVID-19 disease pathogenesis. Clin. Med. 2020, 20, e146–e147. [Google Scholar] [CrossRef]
- Komaravelli, N.; Kelley, J.P.; Garofalo, M.P.; Wu, H.; Casola, A.; Kolli, D. Role of dietary antioxidants in human metapneumovirus infection. Virus Res. 2015, 200, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, I.G.; De Brito, C.A.; Dos Reis, V.M.S.; Sato, M.N.; Pereira, N.Z. SARS-CoV-2 and Other Respiratory Viruses: What Does Oxidative Stress Have to Do with It? Oxidative Med. Cell. Longev. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Komaravelli, N.; Ansar, M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces NRF2 degradation through a promyelocytic leukemia protein - ring finger protein 4 dependent pathway. Free. Radic. Biol. Med. 2017, 113, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free. Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef] [PubMed]
- DeMaula, C.D.; Leutenegger, C.M.; Bonneau, K.R.; MacLachlan, N. The Role of Endothelial Cell-Derived Inflammatory and Vasoactive Mediators in the Pathogenesis of Bluetongue. Virology 2002, 296, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herseth, J.; Refsnes, M.; Låg, M.; Hetland, G.; Schwarze, P. IL-1β as a determinant in silica-induced cytokine responses in monocyte-endothelial cell co-cultures. Hum. Exp. Toxicol. 2008, 27, 387–399. [Google Scholar] [CrossRef]
- Lakshminarayanan, V.; Beno, D.W.A.; Costa, R.H.; Roebuck, K.A. Differential Regulation of Interleukin-8 and Intercellular Adhesion Molecule-1 by H2O2 and Tumor Necrosis Factor-α in Endothelial and Epithelial Cells. J. Biol. Chem. 1997, 272, 32910–32918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrou, G.I.; Hatziagapiou, K.; Vlahopoulos, S. Inflammation and tissue homeostasis: The NF-κB system in physiology and malignant progression. Mol. Biol. Rep. 2020, 47, 4047–4063. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.; Boldogh, I.; Casola, A.; Brasier, A.R. Nuclear factor-kappaB-dependent induction of interleukin-8 gene expression by tumor necrosis factor alpha: Evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation. Blood 1999, 94, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.A.; Cen, O.; Hengen, N.; Agan, J.; Moschovi, M.; Critselis, E.; Adamaki, M.; Bacopoulou, F.; Copland, J.A.; Boldogh, I.; et al. Dynamic aberrant NF-κB spurs tumorigenesis: A new model encompassing the microenvironment. Cytokine Growth Factor Rev. 2015, 26, 389–403. [Google Scholar] [CrossRef] [Green Version]
- E Hammond, M.; Lapointe, G.R.; Feucht, P.H.; Hilt, S.; A Gallegos, C.; A Gordon, C.; A Giedlin, M.; Mullenbach, G.; Tekamp-Olson, P. IL-8 induces neutrophil chemotaxis predominantly via type I IL-8 receptors. J. Immunol. 1995, 155, 1428–1433. [Google Scholar]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef]
- Ma, Q.; Li, R.; Pan, W.; Huang, W.; Liu, B.; Xie, Y.; Wang, Z.; Li, C.; Jiang, H.; Huang, J.; et al. Phillyrin (KD-1) exerts anti-viral and anti-inflammatory activities against novel coronavirus (SARS-CoV-2) and human coronavirus 229E (HCoV-229E) by suppressing the nuclear factor kappa B (NF-κB) signaling pathway. Phytomedicine 2020, 78, 153296. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ahn, J.H.; Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Hong, K.S.; Jang, J.G.; Park, C.G.; et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Boldogh, S.; Garofalo, R.; Jamaluddin, M.; Brasier, A.R. Respiratory Syncytial Virus Influences NF-κB-Dependent Gene Expression through a Novel Pathway Involving MAP3K14/NIK Expression and Nuclear Complex Formation with NF-κB2. J. Virol. 2005, 79, 8948–8959. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, S.; Boldogh, I.; Brasier, A. Inside-Out Signaling Pathways from Nuclear Reactive Oxygen Species Control Pulmonary Innate Immunity. J. Innate Immun. 2016, 8, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Schmolke, M.; Viemann, D.; Roth, J.; Ludwig, S. Essential Impact of NF-κB Signaling on the H5N1 Influenza A Virus-Induced Transcriptome. J. Immunol. 2009, 183, 5180–5189. [Google Scholar] [CrossRef] [Green Version]
- Strengert, M.; Jennings, R.; Davanture, S.; Hayes, P.; Gabriel, G.; Knaus, U.G. Mucosal Reactive Oxygen Species Are Required for Antiviral Response: Role of Duox in Influenza A Virus Infection. Antioxidants Redox Signal. 2014, 20, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Nasi, A.; McArdle, S.; Gaudernack, G.; Westman, G.; Melief, C.; Rockberg, J.; Arens, R.; Kouretas, D.; Sjölin, J.; Mangsbo, S. Reactive oxygen species as an initiator of toxic innate immune responses in retort to SARS-CoV-2 in an ageing population, consider N-acetylcysteine as early therapeutic intervention. Toxicol. Rep. 2020, 7, 768–771. [Google Scholar] [CrossRef]
- Pasini, A.F.; Stranieri, C.; Cominacini, L.; Mozzini, C. Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-Cov-2 Complications. Antioxidants 2021, 10, 272. [Google Scholar] [CrossRef]
- Ansar, M.; Ivanciuc, T.; Garofalo, R.P.; Casola, A. Increased Lung Catalase Activity Confers Protection Against Experimental RSV Infection. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Olive, L.; Marx, W.; O'Neil, A.; Athan, E.; Carvalho, A.F.; Maes, M.; Walder, K.; et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 2020, 258, 118166. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Forcados, G.E.; Muhammad, A.; Oladipo, O.O.; Makama, S.; Meseko, C.A. Metabolic Implications of Oxidative Stress and Inflammatory Process in SARS-CoV-2 Pathogenesis: Therapeutic Potential of Natural Antioxidants. Front. Cell. Infect. Microbiol. 2021, 11, 654813. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P.; Birincioglu, M.; Rodriguez, H. Free radical-induced damage to DNA: Mechanisms and measurement1,2. Free Radic. Biol. Med. 2002, 32, 1102–1115. [Google Scholar] [CrossRef]
- Mitra, S.; Hazra, T.K.; Roy, R.; Ikeda, S.; Biswas, T.; Lock, J.; Boldogh, I.; Izumi, T. Complexities of DNA base excision repair in mammalian cells. Mol. Cells 1997, 7, 305–312. [Google Scholar]
- Izumi, T.; Schein, C.H.; Oezguen, N.; Feng, Y.; Braun§, W. Effects of Backbone Contacts 3‘ to the Abasic Site on the Cleavage and the Product Binding by Human Apurinic/Apyrimidinic Endonuclease (APE1). Biochemistry 2003, 43, 684–689. [Google Scholar] [CrossRef]
- Hegde, M.; Izumi, T.; Mitra, S. Oxidized Base Damage and Single-Strand Break Repair in Mammalian Genomes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, A.M.; Burrows, C.J. 8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis. DNA Repair 2017, 56, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. On the irrelevancy of hydroxyl radical to DNA damage from oxidative stress and implications for epigenetics. Chem. Soc. Rev. 2020, 49, 6524–6528. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, S.; Pramanik, S.; Harris, H.L.; Tarpley, M.; Sarkar, A.; Spagnol, G.; Sorgen, P.L.; Chowdhury, D.; Band, V.; Klinkebiel, D.; et al. Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc. Natl. Acad. Sci. USA 2020, 117, 11409–11420. [Google Scholar] [CrossRef]
- Pan, L.; Wang, H.; Luo, J.; Zeng, J.; Pi, J.; Liu, H.; Liu, C.; Ba, X.; Qu, X.; Xiang, Y.; et al. Epigenetic regulation of TIMP1 expression by 8-oxoguanine DNA glycosylase-1 binding to DNA:RNA hybrid. FASEB J. 2019, 33, 14159–14170. [Google Scholar] [CrossRef] [Green Version]
- Hao, W.; Qi, T.; Pan, L.; Wang, R.; Zhu, B.; Aguilera-Aguirre, L.; Radak, Z.; Hazra, T.K.; Vlahopoulos, S.; Bacsi, A.; et al. Effects of the stimuli-dependent enrichment of 8-oxoguanine DNA glycosylase1 on chromatinized DNA. Redox Biol. 2018, 18, 43–53. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 2017, 14, 669–678. [Google Scholar] [CrossRef]
- Wang, R.; Hao, W.; Pan, L.; Boldogh, I.; Ba, X. The roles of base excision repair enzyme OGG1 in gene expression. Cell. Mol. Life Sci. 2018, 75, 3741–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, X.; Aguilera-Aguirre, L.; Rashid, Q.T.A.N.; Bacsi, A.; Radak, Z.; Sur, S.; Hosoki, K.; Hegde, M.L.; Boldogh, I. The Role of 8-Oxoguanine DNA Glycosylase-1 in Inflammation. Int. J. Mol. Sci. 2014, 15, 16975–16997. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.W.; Phang, T.; Edwards, M.G.; Geraci, M.W.; Gillespie, M.N. Promoter G-quadruplex sequences are targets for base oxidation and strand cleavage during hypoxia-induced transcription. Free. Radic. Biol. Med. 2012, 53, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastukh, V.; Roberts, J.T.; Clark, D.W.; Bardwell, G.C.; Patel, M.; Al-Mehdi, A.-B.; Borchert, G.M.; Gillespie, M.N. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1367–L1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemes, R.M.R.; Costa, A.J.; Bartolomeo, C.S.; Bassani, T.B.; Nishino, M.S.; Pereira, G.J.d.S.; Smaili, S.S.; Maciel, R.M.D.B.; Braconi, C.T.; da Cruz, E.F.; et al. 17β-estradiol reduces SARS-CoV-2 infection in vitro. Physiol. Rep. 2021, 9, e14707. [Google Scholar] [CrossRef] [PubMed]
- Erfinanda, L.; Ravindran, K.; Kohse, F.; Gallo, K.; Preissner, R.; Walther, T.; Kuebler, W.M. Oestrogen-mediated upregulation of the Mas receptor contributes to sex differences in acute lung injury and lung vascular barrier regulation. Eur. Respir. J. 2020, 57, 2000921. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA Oxidation as Triggered by H3K9me2 Demethylation Drives Estrogen-Induced Gene Expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuchegna, C.; Aceto, F.; Bertoni, A.; Romano, A.; Perillo, B.; Laccetti, P.; Gottesman, M.E.; Avvedimento, E.V.; Porcellini, A. Mechanism of retinoic acid-induced transcription: Histone code, DNA oxidation and formation of chromatin loops. Nucleic Acids Res. 2014, 42, 11040–11055. [Google Scholar] [CrossRef] [Green Version]
- Donley, N.; Jaruga, P.; Coskun, E.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Small Molecule Inhibitors of 8-Oxoguanine DNA Glycosylase-1 (OGG1). ACS Chem. Biol. 2015, 10, 2334–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, Y.; Auld, D.; Ji, D.; Beharry, A.A.; Kietrys, A.M.; Wilson, D.L.; Jimenez, M.; King, D.; Nguyen, Z.; Kool, E.T. Potent and Selective Inhibitors of 8-Oxoguanine DNA Glycosylase. J. Am. Chem. Soc. 2018, 140, 2105–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, Y.-K.; Kietrys, A.M.; Hebenbrock, M.; Lee, Y.; Wilson, D.L.; Kool, E. Dual Inhibitors of 8-Oxoguanine Surveillance by OGG1 and NUDT1. ACS Chem. Biol. 2019, 14, 2606–2615. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Lin, P.; Wu, Q.; Pu, Q.; Zhou, C.; Wang, B.; Gao, P.; Wang, Z.; Gao, A.; Overby, M.; et al. Small-Molecule Inhibitor of 8-Oxoguanine DNA Glycosylase 1 Regulates Inflammatory Responses during Pseudomonas aeruginosa Infection. J. Immunol. 2020, 205, 2231–2242. [Google Scholar] [CrossRef]
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Samson, L.D. A target to suppress inflammation. Science 2018, 362, 748–749. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlahopoulos, S.; Wang, K.; Xue, Y.; Zheng, X.; Boldogh, I.; Pan, L. Endothelial Dysfunction through Oxidatively Generated Epigenetic Mark in Respiratory Viral Infections. Cells 2021, 10, 3067. https://doi.org/10.3390/cells10113067
Vlahopoulos S, Wang K, Xue Y, Zheng X, Boldogh I, Pan L. Endothelial Dysfunction through Oxidatively Generated Epigenetic Mark in Respiratory Viral Infections. Cells. 2021; 10(11):3067. https://doi.org/10.3390/cells10113067
Chicago/Turabian StyleVlahopoulos, Spiros, Ke Wang, Yaoyao Xue, Xu Zheng, Istvan Boldogh, and Lang Pan. 2021. "Endothelial Dysfunction through Oxidatively Generated Epigenetic Mark in Respiratory Viral Infections" Cells 10, no. 11: 3067. https://doi.org/10.3390/cells10113067
APA StyleVlahopoulos, S., Wang, K., Xue, Y., Zheng, X., Boldogh, I., & Pan, L. (2021). Endothelial Dysfunction through Oxidatively Generated Epigenetic Mark in Respiratory Viral Infections. Cells, 10(11), 3067. https://doi.org/10.3390/cells10113067