
Redox Sensitive Cysteine Residues as Crucial Regulators of Wild-Type and Mutant p53 Isoforms





Abstract
:1. Introduction
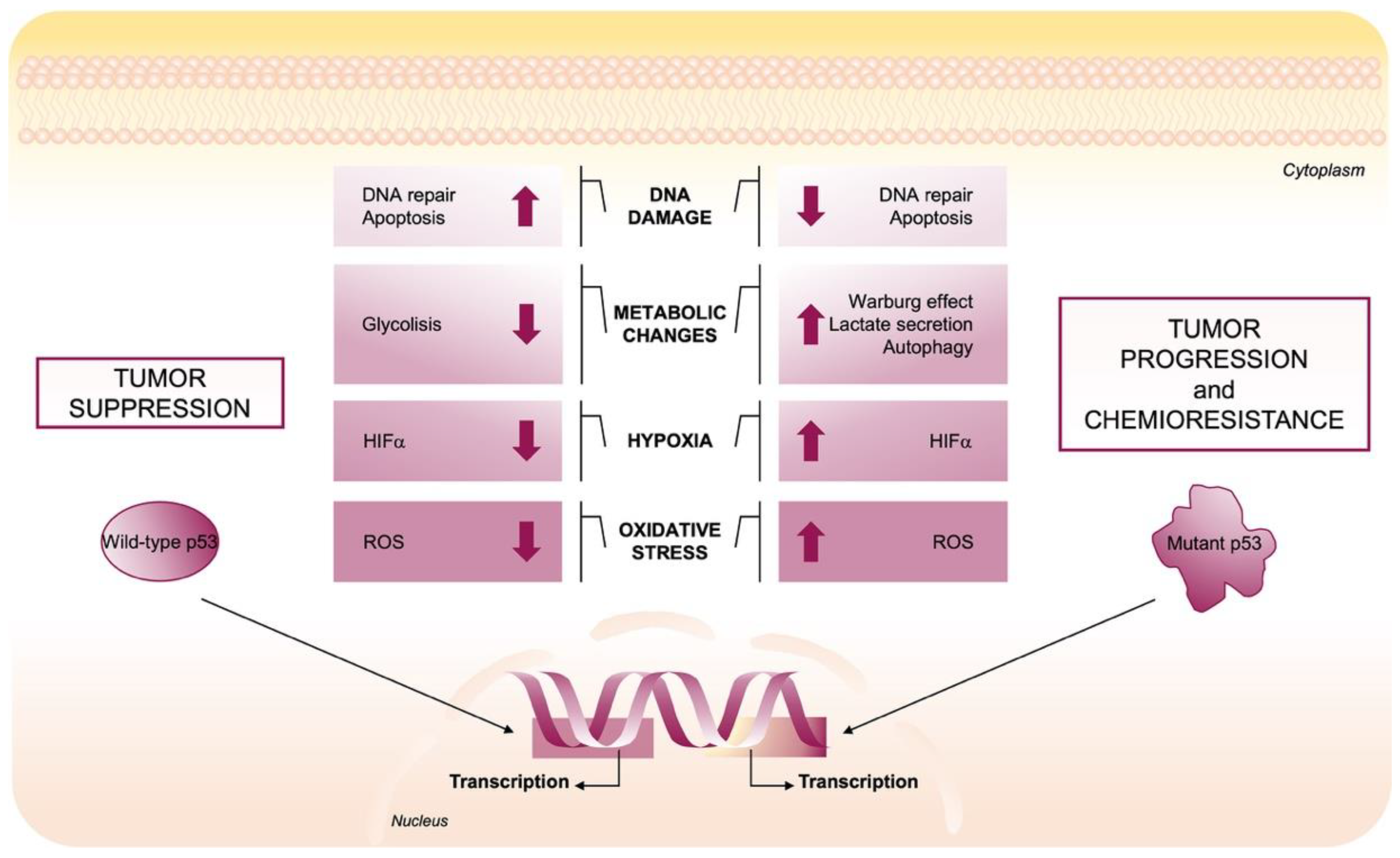
2. Structure and Function of Wild-Type p53 and of Its Mutant Counterpart
2.1. Wild-Type p53 Regulates Redox Balance
2.2. Mutant p53 Gain-of-Function Structure and Roles in Cancer
2.3. Mutant p53-Induced Oncogenic Mechanisms to Promote ROS Production
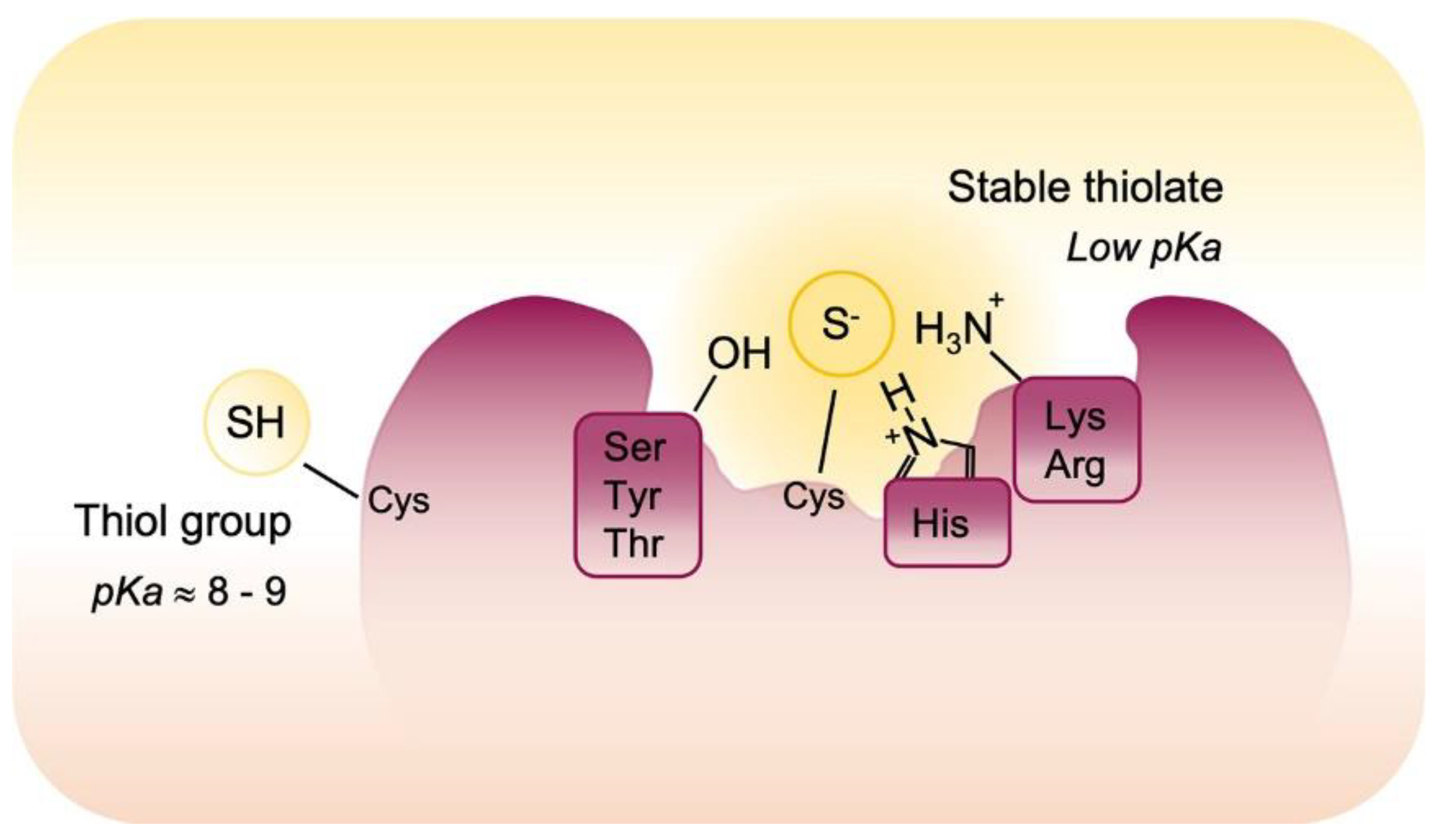
3. Oxidative Post-Translational Modifications of Proteins Cysteines
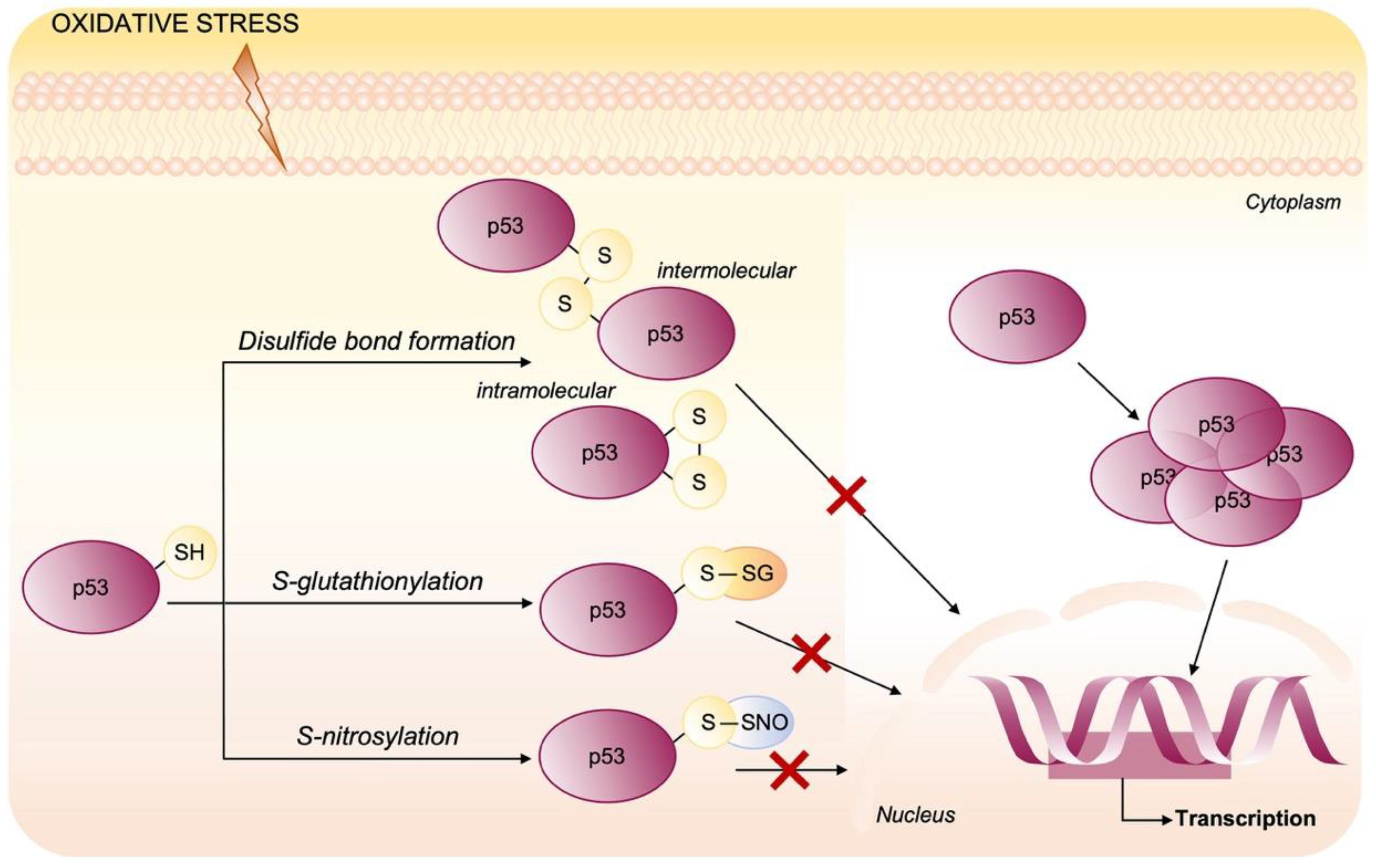
Oxidative Post-Translational Modifications of p53
4. Targeting Cysteines as a Strategy to Reactivate Mutant p53
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junior, P.L.D.S.; Câmara, D.A.D.; Porcacchia, A.S.; Fonseca, P.M.M.; Jorge, S.D.; Araldi, R.P.; Ferreira, A.K. The Roles of ROS in Cancer Heterogeneity and Therapy. Oxidative Med. Cell. Longev. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Mohan, G.M.; Shailender, G.; Malla, R.R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Fra, A.; Yoboue, E.D.; Sitia, R. Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol. Neurosci. 2017, 10, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, E.S.; Ceder, S.; Bykov, V.J.N.; Wiman, K.G. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 2019, 11, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B. Wild-Type p53: Tumors Can’t Stand It. Cell 2007, 128, 837–840. [Google Scholar] [CrossRef] [Green Version]
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med. 2016, 7, a026054. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [Green Version]
- Kamaraj, B.; Bogaerts, A. Structure and Function of p53-DNA Complexes with Inactivation and Rescue Mutations: A Molecular Dynamics Simulation Study. PLoS ONE 2015, 10, e0134638. [Google Scholar] [CrossRef]
- Hupp, T.R.; Meek, D.W.; Midgley, C.A.; Lane, D.P. Role of Cysteine Residues in Regulation of p53 Function Previous Studies of p53 Have Implicated Cysteine Residues in Site-Specific DNA Binding via zinc Coordination and Redox Regulation. Mol. Cell. Biol. 1995, 15, 3892–3903. Available online: https://journals.asm.org/journal/mcb (accessed on 9 November 2021).
- Butler, J.S.; Loh, S.N. Structure, Function, and Aggregation of the Zinc-Free Form of the p53 DNA Binding Domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Loh, S.N. The missing Zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Formigari, A.; Gregianin, E.; Irato, P. The effect of zinc and the role of p53 in copper-induced cellular stress responses. J. Appl. Toxicol. 2013, 33, 527–536. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nature 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Maillet, A.; Pervaiz, S. Redox Regulation of p53, Redox Effectors Regulated by p53: A Subtle Balance. Antioxid. Redox Signal. 2012, 16, 1285–1294. [Google Scholar] [CrossRef]
- Hainaut, P.; Miiner, J. Redox Modulation of p53 Conformation and Sequence-specific DNA Binding in Vitro. Cancer Res. 1993, 53, 4469–4473. [Google Scholar]
- Martin, A.C.; Facchiano, A.; Cuff, A.L.; Hernandez-Boussard, T.; Olivier, M.; Hainaut, P.; Thornton, J.M. Integrating mutation data and structural analysis of the TP53 tumor-suppressor protein. Hum. Mutat. 2002, 19, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2017, 25, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V. Stress-Responsive Sestrins Link p53 with Redox Regulation and Mammalian Target of Rapamycin Signaling. Antioxid. Redox Signal. 2011, 15, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, H.; Shi, H.; Du, Y.; Yu, J.; Gu, S.; Chen, X.; Liu, K.J.; Hu, C.-A.A. PUMA Overexpression Induces Reactive Oxygen Species Generation and Proteasome-Mediated Stathmin Degradation in Colorectal Cancer Cells. Cancer Res. 2005, 65, 1647–1654. [Google Scholar] [CrossRef] [Green Version]
- Drane, P.; Bravard, A.; Bouvard, V.; May, E. Reciprocal down-regulation of p53 and SOD2 gene expression–implication in p53 mediated apoptosis. Oncogene 2001, 20, 430–439. [Google Scholar] [CrossRef]
- Hussain, S.P.; Amstad, P.; He, P.; Robles, A.; Lupold, S.; Kaneko, I.; Ichimiya, M.; Sengupta, S.; Mechanic, L.; Okamura, S.; et al. p53-Induced Up-Regulation of MnSOD and GPx but not Catalase Increases Oxidative Stress and Apoptosis. Cancer Res. 2004, 64, 2350–2356. [Google Scholar] [CrossRef] [Green Version]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, E.J.; Chumakov, P. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, E.H.; Casterline, B.W.; Burke, D.J.; Leto, T.L. Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-β-mediated migration of human lung and breast epithelial cells. Br. J. Cancer 2014, 110, 2569–2582. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Li, S.; Swaroop, M.; Guan, K.; Oberley, L.W.; Sun, Y. Transcriptional Activation of the Human Glutathione Peroxidase Promoter by p53. J. Biol. Chem. 1999, 274, 12061–12066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.Y.; Kim, H.-B.; Piao, C.; Lee, K.H.; Hyun, J.W.; Chang, I.-Y.; You, H.J. The critical role of catalase in prooxidant and antioxidant function of p53. Cell Death Differ. 2012, 20, 117–129. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant p53 blocks SESN1/AMPK/PGC-1α/UCP2 axis increasing mitochondrial O2−· production in cancer cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Fujiwara, T.; Kadowaki, Y.; Fukazawa, T.; Waku, T.; Itoshima, T.; Yamatsuji, T.; Nishizaki, M.; Roth, A.J.; Tanaka, N. Overexpression of the wild-type p53 gene inhibits NF-κB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene 2000, 19, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, P.S.; Beghelli, S.; Zamboni, G.; Scarpa, A. Genetic abnormalities in pancreatic cancer. Mol. Cancer 2003, 2, 7. [Google Scholar] [CrossRef]
- Freed-Pastor, W.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. P53 and Prognosis: New Insights and Further Complexity. Cell 2005, 120, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschauer, L.; Muller, P.A. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem. Soc. Trans. 2016, 44, 460–466. [Google Scholar] [CrossRef]
- He, C.; Li, L.; Guan, X.; Xiong, L.; Miao, X. Mutant p53 Gain of Function and Chemoresistance: The Role of Mutant p53 in Response to Clinical Chemotherapy. Chemotherapy 2016, 62, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Pozza, E.D.; Nadal-Serrano, M.; Oliver, J.O.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Reséndiz, I.; Gallardo-Pérez, J.C.; López-Macay, A.; Robledo-Cadena, D.X.; Villa, E.G.; Gariglio, P.; Saavedra, E.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S. Mutant p53R248Qdownregulates oxidative phosphorylation and upregulates glycolysis under normoxia and hypoxia in human cervix cancer cells. J. Cell. Physiol. 2018, 234, 5524–5536. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Kamat, C.D.; Green, D.E.; Warnke, L.; Thorpe, J.E.; Ceriello, A.; Ihnat, M.A. Mutant p53 facilitates pro-angiogenic, hyperproliferative phenotype in response to chronic relative hypoxia. Cancer Lett. 2007, 249, 209–219. [Google Scholar] [CrossRef]
- Butera, G.; Pacchiana, R.; Mullappilly, N.; Margiotta, M.; Bruno, S.; Conti, P.; Riganti, C.; Donadelli, M. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. BBA-Mol. Cell Res. 2018, 1865, 1914–1923. [Google Scholar] [CrossRef]
- Sánchez-Álvarez, M.; Strippoli, R.; Donadelli, M.; Bazhin, A.V.; Cordani, M. Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer. Cancers 2019, 11, 1415. [Google Scholar] [CrossRef] [Green Version]
- Bossi, G.; Lapi, E.; Strano, S.; Rinaldo, C.; Blandino, G.; Sacchi, A. Mutant p53 gain of function: Reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 2005, 25, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Torrens-Mas, M.; Cordani, M.; Mullappilly, N.; Pacchiana, R.; Riganti, C.; Palmieri, M.; Pons, D.; Roca, P.; Oliver, J.; Donadelli, M. Mutant p53 induces SIRT3/MnSOD axis to moderate ROS production in melanoma cells. Arch. Biochem. Biophys. 2019, 679, 108219. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [Green Version]
- Butturini, E.; De Prati, A.C.; Mariotto, S. Redox Regulation of STAT1 and STAT3 Signaling. Int. J. Mol. Sci. 2020, 21, 7034. [Google Scholar] [CrossRef] [PubMed]
- Lermant, A.; Murdoch, C.E. Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants 2019, 8, 315. [Google Scholar] [CrossRef] [Green Version]
- Dalle–Donne, I.; Milzani, A.D.G.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular Mechanisms and Potential Clinical Significance ofS-Glutathionylation. Antioxid. Redox Signal. 2008, 10, 445–474. [Google Scholar] [CrossRef]
- Butturini, E.; Gotte, G.; Dell’Orco, D.; Chiavegato, G.; Marino, V.; Canetti, D.; Cozzolino, F.; Monti, M.; Pucci, P.; Mariotto, S. Intermolecular disulfide bond influences unphosphorylated STAT3 dimerization and function. Biochem. J. 2016, 473, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Butturini, E.; Cozzolino, F.; Boriero, D.; de Prati, A.C.; Monti, M.; Rossin, M.; Canetti, D.; Cellini, B.; Pucci, P.; Mariotto, S. S-glutathionylation exerts opposing roles in the regulation of STAT1 and STAT3 signaling in reactive microglia. Free Radic. Biol. Med. 2018, 117, 191–201. [Google Scholar] [CrossRef]
- Butturini, E.; Boriero, D.; de Prati, A.C.; Mariotto, S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 2019, 669, 22–30. [Google Scholar] [CrossRef]
- Soussi, T.; Mayb, P. Structural Aspects of the p53 Protein in Relation to Gene Evolution: A Second Look. J. Mol. Biol. 1996, 260, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a p53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Milner, J. A Structural Role for Metal Ions in the ‘Wild-type’ Conformation of the Tumor Suppressor Pro p53tein. Cancer Res. 1993, 53, 1739–1742. [Google Scholar] [PubMed]
- Stoner, C.S.; Pearson, G.D.; Koç, A.; Merwin, J.R.; Lopez, N.I.; Merrill, G.F. Effect of Thioredoxin Deletion and p53 Cysteine Replacement on Human p53 Activity in Wild-Type and Thioredoxin Reductase Null Yeast. Biochemistry 2009, 48, 9156–9169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotcher, J.; Clarke, D.J.; Mackay, C.L.; Hupp, T.; Sadlerc, P.J.; Langridge-Smith, P.R.R. Redox regulation of tumour suppressor protein p53: Identification of the sites of hydrogen peroxide oxidation and glutathionylation. Chem. Sci. 2013, 4, 1257–1269. Available online: https://pubs.rsc.org/en/content/articlelanding/2013/sc/c2sc21702c (accessed on 9 November 2021). [CrossRef]
- Held, J.; Danielson, S.R.; Behring, J.B.; Atsriku, C.; Britton, D.J.; Puckett, R.L.; Schilling, B.; Campisi, J.; Benz, C.C.; Gibson, B.W. Targeted Quantitation of Site-Specific Cysteine Oxidation in Endogenous Proteins Using a Differential Alkylation and Multiple Reaction Monitoring Mass Spectrometry Approach. Mol. Cell. Proteom. 2010, 9, 1400–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, S.; Ortiz, F.; Sun, X.Z.; Wu, H.-H.; Mason, A.; Momand, J. Copper uptake is required for pyrrolidine dithiocarbamate-mediated oxidation and protein level increase of p53 in cells. Biochem. J. 2002, 365, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Parks, D. Redox state regulates binding of p53 to sequence-specific DNA, but not to non-specific or mismatched DNA. Nucleic Acids Res. 1997, 25, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Buzek, J.; Latonen, L.; Kurki, S.; Peltonen, K.D.; Laiho, M. Redox state of tumor suppressor p53 regulates its sequence-specific DNA binding in DNA-damaged cells by cysteine 277. Nucleic Acids Res. 2002, 30, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is Inhibited by Glutathionylation of Cysteines Present in the Proximal DNA-Binding Domain During Oxidative Stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [Green Version]
- Calmels, S.; Hainaut, P.; Ohshima, H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res. 1997, 57, 3365–3369. [Google Scholar]
- Chazotte-Aubert, L.; Hainaut, P.; Ohshima, H. Nitric Oxide Nitrates Tyrosine Residues of Tumor-Suppressor p53 Protein in MCF-7 Cells. Biochem. Biophys. Res. Commun. 2000, 267, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C.S.; Whisenhunt, T.R.; Wesemann, D.R.; Harkins, E.L.; Van Meir, E.G.; Samanta, M. Inactivation of wild-type p53 protein function by reactive oxygen and nitrogen species in malignant glioma cells. Cancer Res. 2003, 63, 8670–8673. Available online: http://www.ncbi.nlm.nih.gov/pubmed/14695179 (accessed on 9 November 2021).
- Cobbs, C.S.; Samanta, M.; Harkins, L.E.; Gillespie, G.; Merrick, B.; MacMillan-Crow, L.A. Evidence for Peroxynitrite-Mediated Modifications to p53 in Human Gliomas: Possible Functional Consequences. Arch. Biochem. Biophys. 2001, 394, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, V.; Bayden, A.S.; Graves, P.R.; Kellogg, G.; Mikkelsen, R.B. Nitration of the Tumor Suppressor Protein p53 at Tyrosine 327 Promotes p53 Oligomerization and Activation. Biochemistry 2010, 49, 5331–5339. [Google Scholar] [CrossRef] [Green Version]
- Kaar, J.; Basse, N.; Joerger, A.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotcher, J.; Clarke, D.J.; Weidt, S.K.; Mackay, C.L.; Hupp, T.R.; Sadler, P.J.; Langridge-Smith, P.R.R. Identification of Two Reactive Cysteine Residues in the Tumor Suppressor Protein p53 Using Top-Down FTICR Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Rainwater, R.; Parks, D.; Anderson, E.M.; Tegtmeyer, P.; Mann, K. Role of cysteine residues in regulation of p53 function. Mol. Cell. Biol. 1995, 15, 3892–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méplan, C.; Richard, M.-J.; Hainaut, P. Metalloregulation of the tumor suppressor protein p53: Zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene 2000, 19, 5227–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.Z.; Vinci, C.; Makmura, L.; Han, S.; Tran, D.; Nguyen, J.; Hamann, M.; Grazziani, S.; Sheppard, S.; Gutova, M.; et al. Formation of Disulfide Bond in p53 Correlates with Inhibition of DNA Binding and Tetramerization. Antioxid. Redox Signal. 2003, 5, 655–665. [Google Scholar] [CrossRef]
- Zhang, Q.; Bergman, J.; Wiman, K.G.; Bykov, V.J. Role of Thiol Reactivity for Targeting Mutant p53. Cell Chem. Biol. 2018, 25, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Michael, A. Ueber die Addition von Natriumacetessig- und Natriummalonsaeureethern zu den Aethern ungesaettigter Saeuren. J. Prakt. Chem. 1887, 35, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2017, 18, 89–102. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2009, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.; Issaeva, N.; Selivanova, G.; Wiman, K.G. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: A statistical analysis of information in the National Cancer Institute database. Carcinogenesis 2002, 23, 2011–2018. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Bykov, V.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, R.L.; Sanfilippo, M.; Vella, V.; Pandini, G.; Vigneri, P.; Nicolosi, M.L.; Gianì, F.; Vigneri, R.; Frasca, F. Reactivation of p53 mutants by p53 reactivation and induction of massive apoptosis in thyroid cancer cells. Int. J. Cancer 2011, 130, 2259–2270. [Google Scholar] [CrossRef]
- Aryee, D.N.T.; Niedan, S.; Ban, J.; Schwentner, R.; Muehlbacher, K.; Kauer, M.; Kofler, R.; Kovar, H. Variability in functional p53 reactivation by PRIMA-1Met/APR-246 in Ewing sarcoma. Br. J. Cancer 2013, 109, 2696–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-L.; Zhou, J.; Chan, Z.-L.; Chooi, J.-Y.; Chen, Z.-R.; Chng, W.-J. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget 2015, 6, 36689–36699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yi, B.; Wang, C.; Chen, D.; Bae, S.; Wei, S.; Guo, R.-J.; Lu, C.; Nguyen, L.; Yang, W.-H.; et al. Silencing of CD24 Enhances the PRIMA-1–Induced Restoration of Mutant p53 in Prostate Cancer Cells. Clin. Cancer Res. 2015, 22, 2545–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zache, N.; Lambert, J.M.; Rökaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological Rescue of Mutant p53 Conformation and Function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Madka, V.; Zhang, Y.; Li, Q.; Mohammed, A.; Sindhwani, P.; Lightfoot, S.; Wu, X.-R.; Kopelovich, L.; Rao, C.V. p53-stabilizing Agent CP-31398 Prevents Growth and Invasion of Urothelial Cancer of the Bladder in Transgenic UPII-SV40T Mice. Neoplasia 2013, 15, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Rippin, T.M.; Bykov, V.N.; Freund, S.M.V.; Selivanova, G.; Wiman, K.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Madan, E.; Parker, T.M.; Bauer, M.; Dhiman, A.; Pelham, C.J.; Nagane, M.; Kuppusamy, M.L.; Holmes, M.; Holmes, T.R.; Shaik, K.; et al. The curcumin analog HO-3867 selectively kills cancer cells by converting mutant p53 protein to transcriptionally active wildtype p53. J. Biol. Chem. 2018, 293, 4262–4276. [Google Scholar] [CrossRef] [Green Version]
- Selvendiran, K.; Ahmed, S.; Dayton, A.; Kuppusamy, M.L.; Tazi, M.; Bratasz, A.; Tong, L.; Rivera, B.K.; Kálai, T.; Hideg, K. Safe and targeted anticancer efficacy of a novel class of antioxidant-conjugated difluorodiarylidenyl piperidones: Differential cytotoxicity in healthy and cancer cells. Free Radic. Biol. Med. 2010, 48, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef]
- Bykov, V.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K. Reactivation of Mutant p53 and Induction of Apoptosis in Human Tumor Cells by Maleimide Analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.R.; Kelley, M.R.; Smith, M.L. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 14548–14553. [Google Scholar] [CrossRef] [Green Version]
- Synnott, N.C.; Bauer, M.; Madden, S.; Murray, A.; Klinger, R.; O’Donovan, N.; O’Connor, D.; Gallagher, W.; Crown, J.; Fersht, A.R.; et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018, 414, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Synnott, N.C.; Bauer, M.R.; Madden, S.F.; Murray, A.M.; Klinger, R.; O’Donovan, N.; O’Connor, D.; Gallagher, W.M.; Crown, J.; Fersht, A.R.; et al. Targeting mutant p53 with PK11007: A new approach for the treatment of patients with triple-negative breast cancer? J. Clin. Oncol. 2017, 35, e14099. [Google Scholar] [CrossRef]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, M.-Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Lambot, S.M.; Oullier, T.; le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Bjorklund, U.; Wiman, K. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Vakifahmetoglu, H.; Stridh, H.; Zhivotovsky, B.; Wiman, K.G. PRIMA-1MET induces mitochondrial apoptosis through activation of caspase-2. Oncogene 2008, 27, 6571–6580. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactivators—p53 Cys-Targeting | Mutations in p53 | Mechanism of Action | Ref. |
|---|---|---|---|
| APR-246 (PRIMA-1MET) Quinuclidinone | R175H; R273H; D259Y/K286E; K286E; S241F; R273C; P223L/V274F | Michael Addition | [84,86,87,88,89,90,91,92,93,94] |
| CP-31398 Styrylquinazoline | V173A; S241F; R249S; R273H | Michael Addition | [95,96,97,98] |
| HO-3867 Diarylidenyl piperidone curcumin analogue | Y163H; R175H; H193R; L194F; Y205F; P223L/V274F; C238Y; N239D; S241F; G245S; G245V; M246I; R248Q; R248W; R249S; R273H; C277F; R280K; E285K | Michael Addition | [99,100] |
| KSS-9 Piperlongumine derivative | R175H | Michael Addition | [101] |
| MIRA-1 Maleimide | R175H; P176Y/R248W; R248Q; R248W; R273H; R273H/P309S; R280K; R282W | Michael Addition | [102] |
| PK11007 Sulfonylpyrimidine | Y220; V143A | Nucleophilic aromatic substitution | [103] |
| STIMA-1 2-vinylquinazolin-4-(3H)-one | R175H; R273H | Michael Addition | [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butturini, E.; Butera, G.; Pacchiana, R.; Carcereri de Prati, A.; Mariotto, S.; Donadelli, M. Redox Sensitive Cysteine Residues as Crucial Regulators of Wild-Type and Mutant p53 Isoforms. Cells 2021, 10, 3149. https://doi.org/10.3390/cells10113149
Butturini E, Butera G, Pacchiana R, Carcereri de Prati A, Mariotto S, Donadelli M. Redox Sensitive Cysteine Residues as Crucial Regulators of Wild-Type and Mutant p53 Isoforms. Cells. 2021; 10(11):3149. https://doi.org/10.3390/cells10113149
Chicago/Turabian StyleButturini, Elena, Giovanna Butera, Raffaella Pacchiana, Alessandra Carcereri de Prati, Sofia Mariotto, and Massimo Donadelli. 2021. "Redox Sensitive Cysteine Residues as Crucial Regulators of Wild-Type and Mutant p53 Isoforms" Cells 10, no. 11: 3149. https://doi.org/10.3390/cells10113149
APA StyleButturini, E., Butera, G., Pacchiana, R., Carcereri de Prati, A., Mariotto, S., & Donadelli, M. (2021). Redox Sensitive Cysteine Residues as Crucial Regulators of Wild-Type and Mutant p53 Isoforms. Cells, 10(11), 3149. https://doi.org/10.3390/cells10113149