
Microvalve Bioprinting of MSC-Chondrocyte Co-Cultures

 ,
,  ,
, 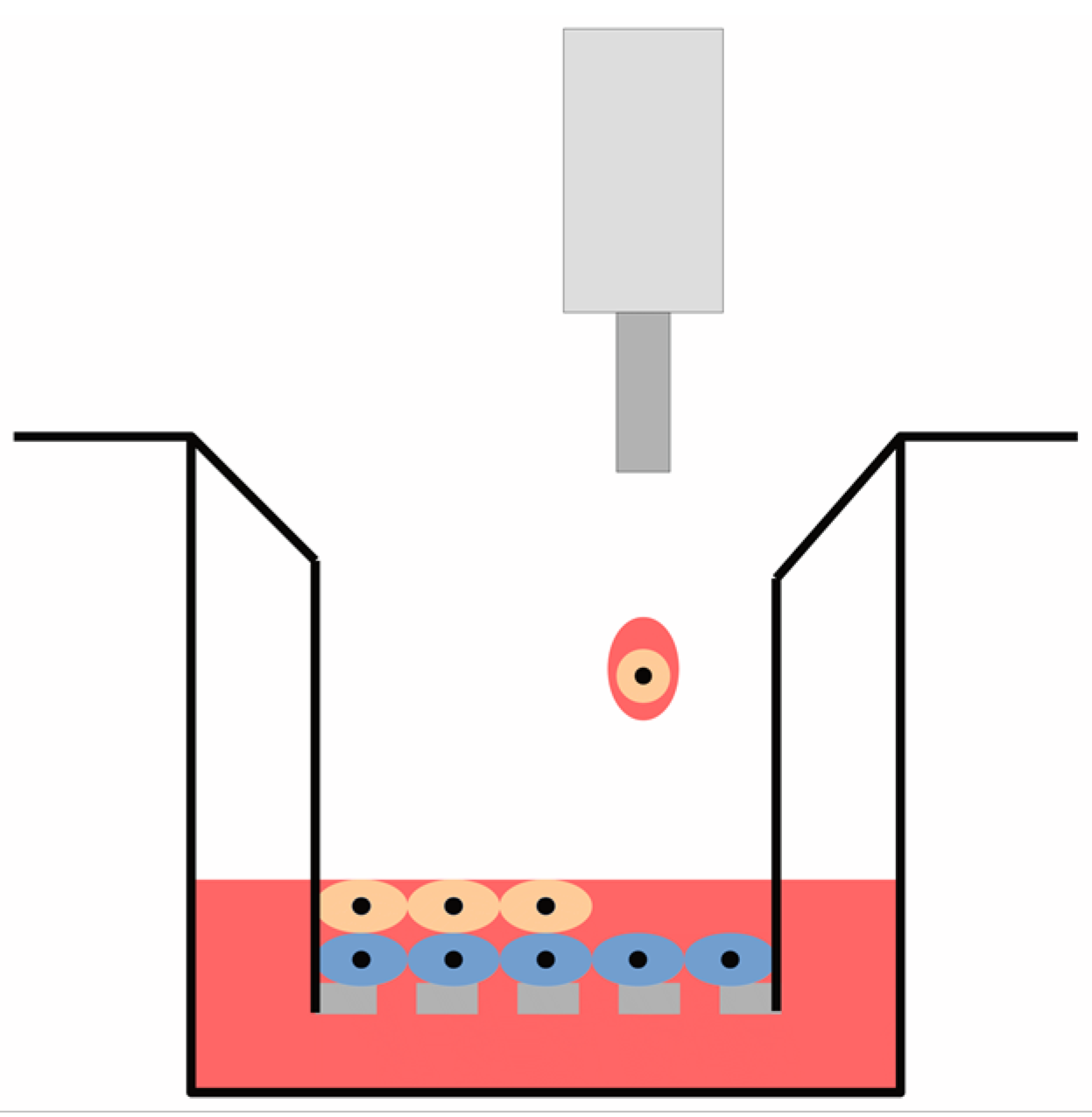{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Model Construction
2.3. Printing Configuration
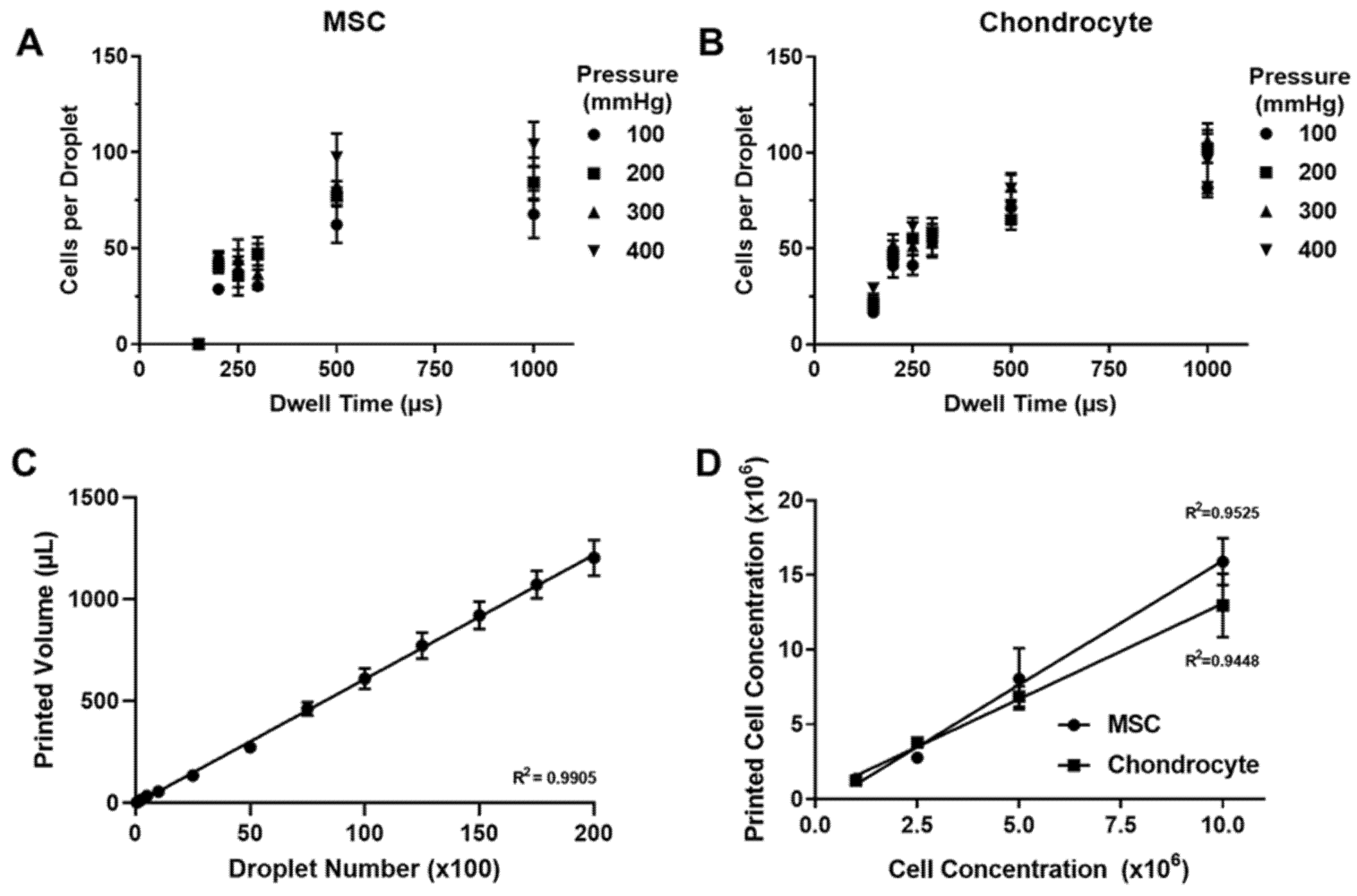
2.4. Characterisation of Printing Performance
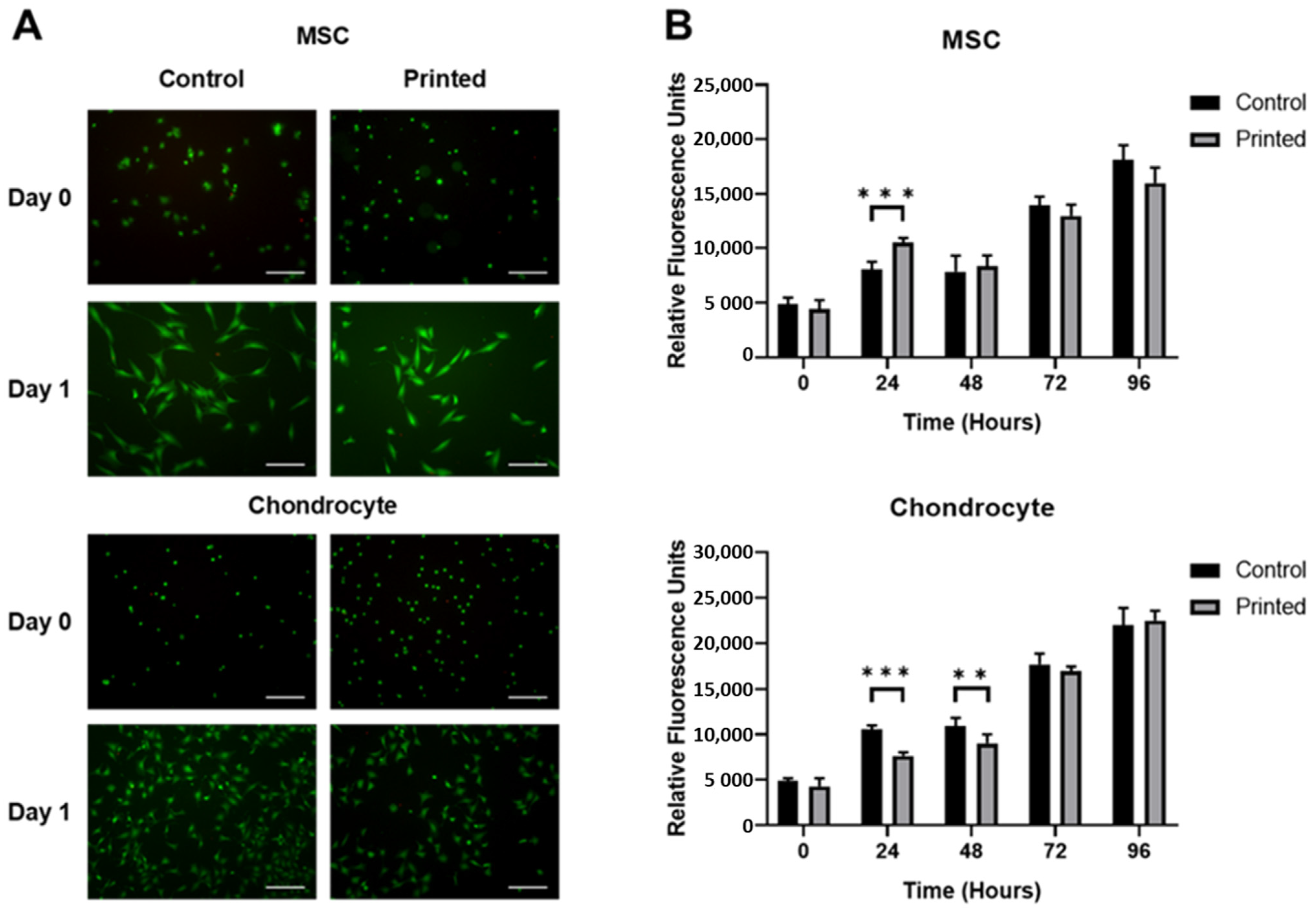
2.5. Cell Viability Assay
2.6. Metabolic Activity Assay
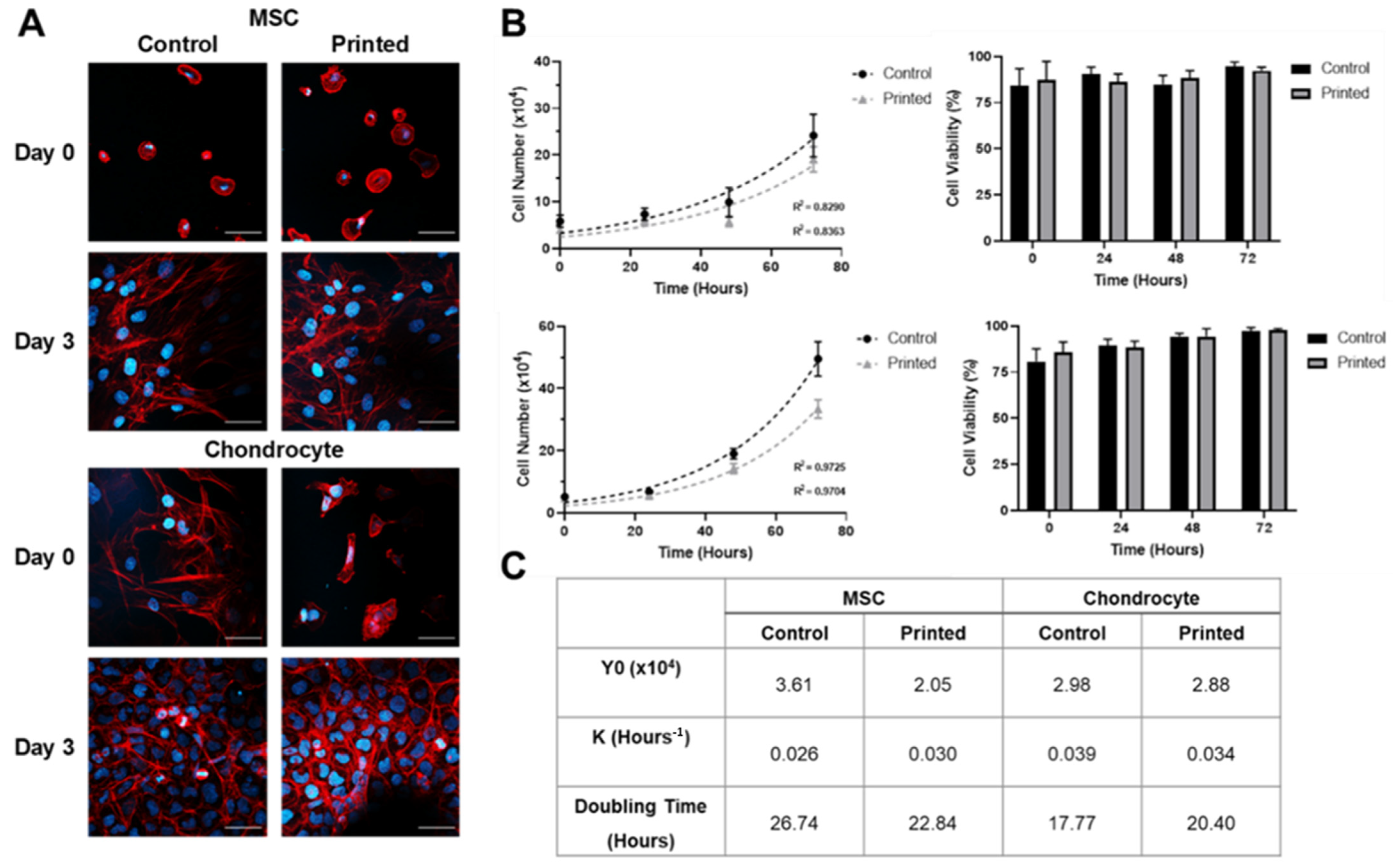
2.7. Visualisation of Cell Morphology
2.8. Cell Proliferation Rate
2.9. Membrane Sectioning
2.10. Haematoxylin and Eosin Staining
2.11. Indirect Immunofluorescence Staining
2.12. Cell Tracker Staining
2.13. Model Cell Proliferation Assay
2.14. Statistical Analysis
3. Results
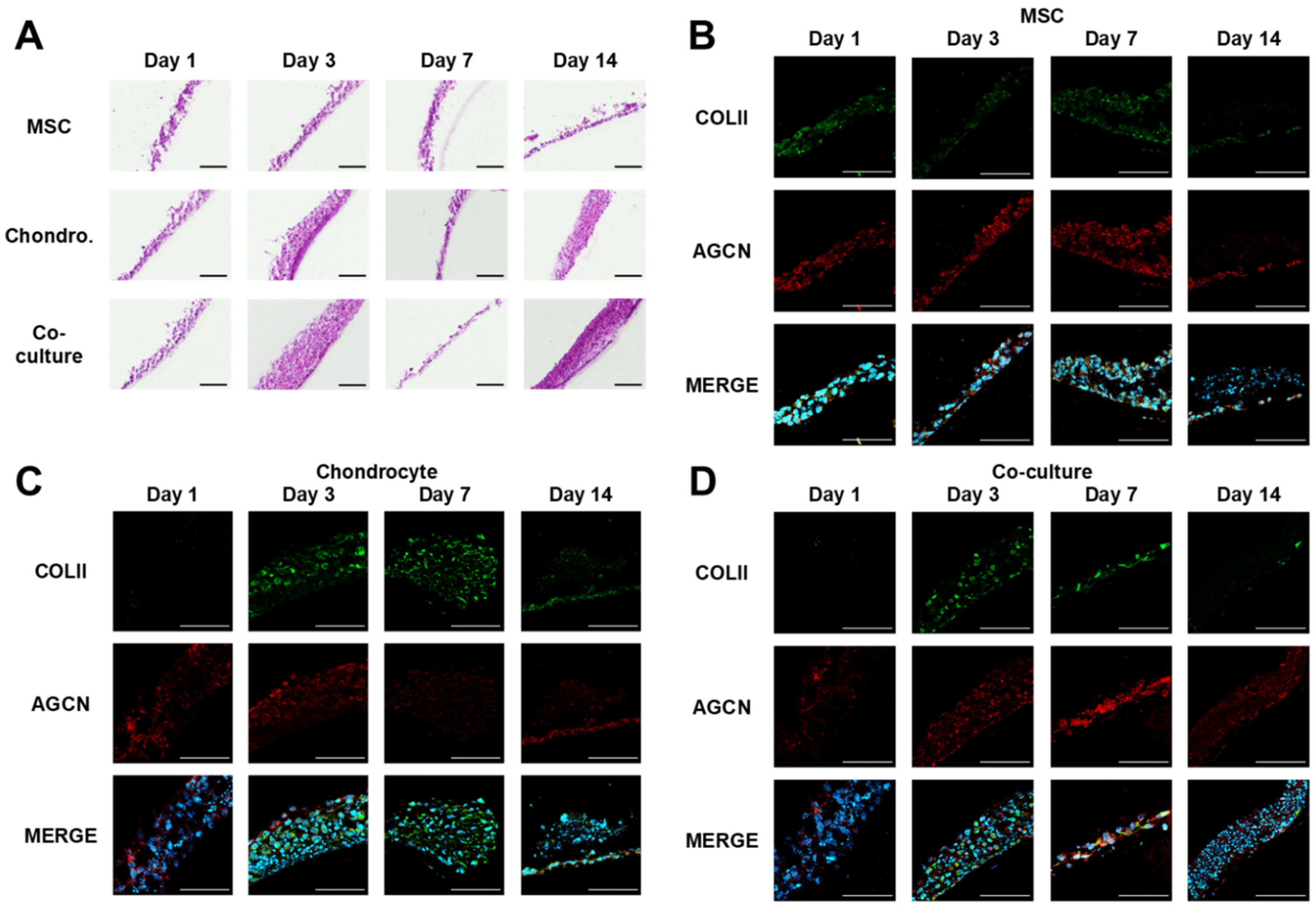
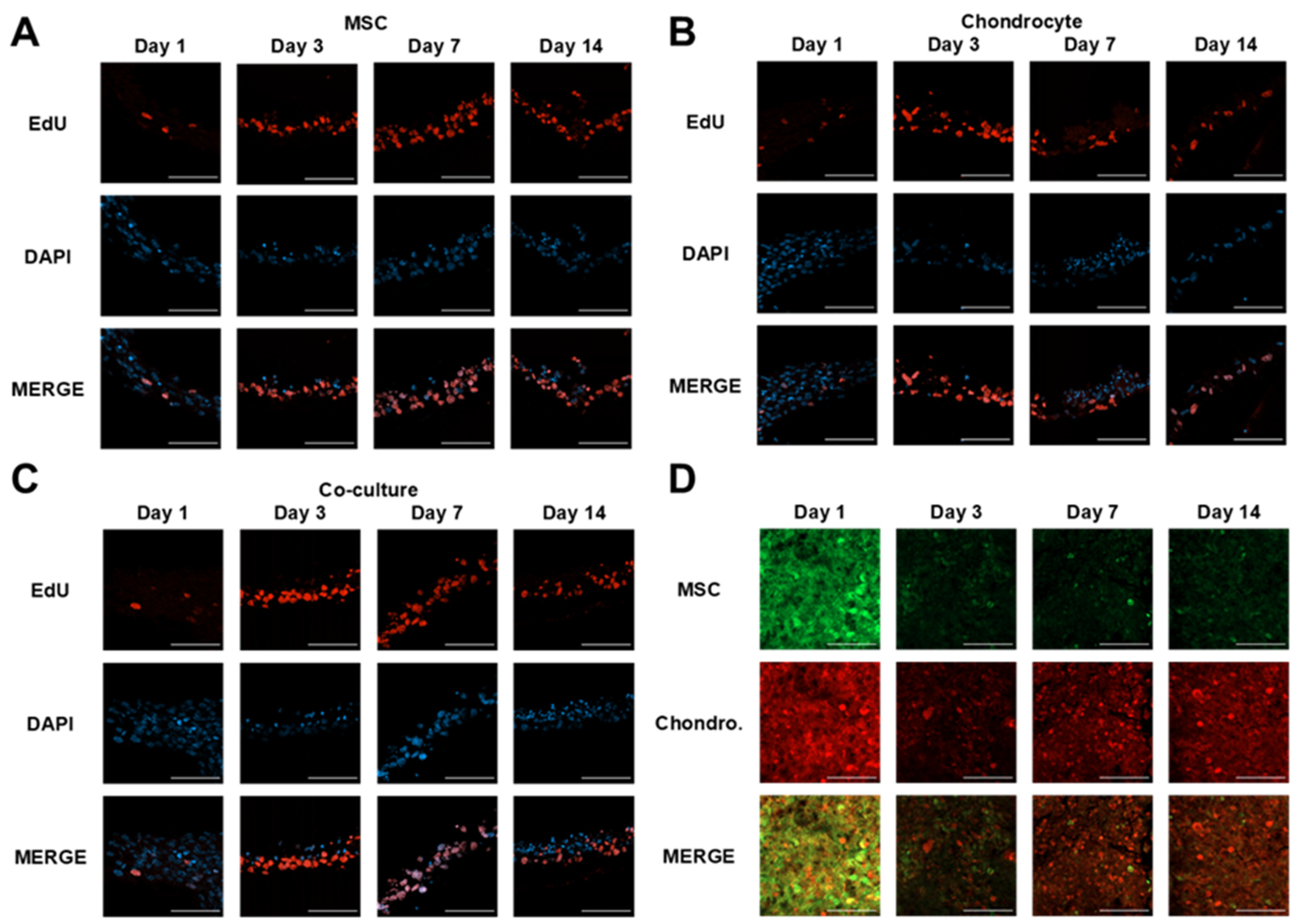
3.1. Microvalve Printing of MSCs and Chondrocytes
3.2. MSC and Chondrocyte Co-Cultures
4. Discussion
- Further validation of the physiological relevance of the model, using sets of donor-matched primary cells cultured as per ACI processes, and including further cell types relevant to the joint niche.
- Using higher-throughput proteomic analysis techniques, such as supernatant analysis, and positional transcriptomic techniques such as MALDI-imaging, in order to add further depth to the analysis and take advantage of the highly scalable printed culture format.
- Improvements to the cell culture platform that could enable further increases in cell culture density per well, the introduction of cytokines or other biomolecules, as well as address throughput limitations relating to the manual exchange of media during culture. A multi-microvalve platform could be developed to enable the simultaneous printing of multiple models. This has the potential to greatly increase printing throughput beyond approximately one model per second depending on the number of microvalves incorporated. Through increasing printing throughout, this modification would also reduce the total time that cells are held in suspension when printing significant numbers of models.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence. Osteoarthritis: Care and Management—Clinical Guideline CG177; National Institute for Health and Care Excellence: London, UK, 2014; Available online: https://www.nice.org.uk/guidance/cg177 (accessed on 26 November 2021).
- Maksymowych, W.P.; Russell, A.S.; Chiu, P.; Yan, A.; Jones, N.; Clare, T.; Lambert, R.G. Targeting tumour necrosis factor alleviates signs and symptoms of inflammatory osteoarthritis of the knee. Arthritis Res. Ther. 2012, 14, R206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvidge, J.; Bullement, A.; Hatswell, A.J. Cost effectiveness of characterised chondrocyte implantation for treatment of cartilage defects of the knee in the UK. PharmacoEconomics 2016, 34, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.B.; Wright, K.T.; Wales, J.; Kuiper, J.H.; McCarthy, H.; Gallacher, P.; Harrison, P.E.; Roberts, S. Efficacy and safety of autologous cell therapies for knee cartilage defects (autologous stem cells, chondrocytes or the two): Randomized controlled trial design. Regen. Med. 2017, 12, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Mollon, B.; Kandel, R.; Chahal, J.; Theodoropoulos, J. The clinical status of cartilage tissue regeneration in humans. Osteoarthr. Cartil. 2013, 21, 1824–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armoiry, X.; Cummins, E.; Connock, M.; Metcalfe, A.; Royle, P.; Johnston, R.; Rodrigues, J.; Waugh, N.; Mistry, H. Autologous chondrocyte implantation with chondrosphere for treating articular cartilage defects in the knee: An evidence review group perspective of a NICE single technology appraisal. PharmacoEconomics 2019, 37, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.J.; Cash, B.M. Matrix-induced autologous chondrocyte implantation with autologous bone grafting for osteochondral lesions of the femoral trochlea. Arthrosc. Tech. 2019, 8, e259–e266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foldager, C.B.; Gomoll, A.H.; Lind, M.; Spector, M. Cell seeding densities in autologous chondrocyte implantation techniques for cartilage repair. Cartilage 2012, 3, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wright, K.T.; Perry, J.; Tins, B.; Hopkins, T.; Hulme, C.; McCarthy, H.S.; Brown, A.; Richardson, J.B. Combined autologous chondrocyte and bone marrow mesenchymal stromal cell implantation in the knee: An 8-year follow up of two first-in-man cases. Cell Transplant. 2019, 28, 924–931. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.M.; Mytelka, D.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef]
- Johnson, C.I.; Argyle, D.J.; Clements, D.N. In vitro models for the study of osteoarthritis. Vet. J. 2016, 209, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurtrey, R.J. Analytic models of oxygen and nutrient diffusion, metabolism dynamics, and architecture optimization in three-dimensional tissue constructs with applications and insights in cerebral organoids. Tissue Eng. Part. C Methods 2016, 22, 221–249. [Google Scholar] [CrossRef] [Green Version]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The effect of 2D and 3D cell cultures on treatment response, EMT profile and stem cell features in head and neck cancer. Cancer Cell Int. 2019, 19, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Whalley, R.D.; Ferreira, A.M.; Dalgarno, K. High throughput physiological micro-models for in vitro pre-clinical drug testing: A review of engineering systems approaches. Prog. Biomed. Eng. 2020, 2, 022001. [Google Scholar] [CrossRef]
- Hickman, J.A.; Graeser, R.; De Hoogt, R.; Vidic, S.; Brito, C.; Gutekunst, M.; Van Der Kuip, H.; IMI PREDECT Consortium. Three-dimensional models of cancer for pharmacology and cancer cell biology: Capturing tumor complexity in vitro/ex vivo. Biotechnol. J. 2014, 9, 1115–1128. [Google Scholar] [CrossRef]
- Joshi, P.; Datar, A.; Yu, K.-N.; Kang, S.-Y.; Lee, M.-Y. High-content imaging assays on a miniaturized 3D cell culture platform. Toxicol. In Vitro 2018, 50, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Langhans, S.A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, G.; Vinciguerra, D.; Balasso, A.; Nicolas, V.; Goudin, N.; Garfa-Traore, M.; Fehér, A.; Dinnyes, A.; Nicolas, J.; Couvreur, P.; et al. Light sheet fluorescence microscopy versus confocal microscopy: In quest of a suitable tool to assess drug and nanomedicine penetration into multicellular tumor spheroids. Eur. J. Pharm. Biopharm. 2019, 142, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Guillotin, B.; Souquet, A.; Catros, S.; Duocastella, M.; Pippenger, B.; Bellance, S.; Bareille, R.; Rémy, M.; Bordenave, L.; Amédée, J.; et al. Laser assisted bioprinting of engineered tissue with high cell density and microscale organization. Biomaterials 2010, 31, 7250–7256. [Google Scholar] [CrossRef]
- Michael, S.; Sorg, H.; Peck, C.-T.; Koch, L.; Deiwick, A.; Chichkov, B.; Vogt, P.M.; Reimers, K. Tissue engineered skin substitutes created by laser-assisted bioprinting form skin-like structures in the dorsal skin fold chamber in mice. PLoS ONE 2013, 8, e57741. [Google Scholar] [CrossRef] [PubMed]
- Koch, L.; Deiwick, A.; Franke, A.; Schwanke, K.; Haverich, A.; Zweigerdt, R.; Chichkov, B.N. Laser bioprinting of human induced pluripotent stem cells—The effect of printing and biomaterials on cell survival, pluripotency, and differentiation. Biofabrication 2018, 10, 035005. [Google Scholar] [CrossRef]
- Yeo, M.; Ha, J.; Lee, H.; Kim, G. Fabrication of hASCs-laden structures using extrusion-based cell printing supplemented with an electric field. Acta Biomater. 2016, 38, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Paxton, N.; Smolan, W.; Böck, T.; Melchels, F.; Groll, J.; Jungst, T. Proposal to assess printability of bioinks for extrusion-based bioprinting and evaluation of rheological properties governing bioprintability. Biofabrication 2017, 9, 044107. [Google Scholar] [CrossRef]
- Miri, A.K.; Mirzaee, I.; Hassan, S.; Oskui, S.M.; Nieto, D.; Khademhosseini, A.; Zhang, Y.S. Effective bioprinting resolution in tissue model fabrication. Lab Chip 2019, 19, 2019–2037. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Yoon, S.; Kwon, K.Y.; Now, H.; Kim, Y.K.; Kim, W.-J.; Yoo, J.-Y.; Jung, S. Freeform micropatterning of living cells into cell culture medium using direct inkjet printing. Sci. Rep. 2017, 7, 14610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorber, B.; Hsiao, W.-K.; Hutchings, I.M.; Martin, K.R. Adult rat retinal ganglion cells and glia can be printed by piezoelectric inkjet printing. Biofabrication 2014, 6, 015001. [Google Scholar] [CrossRef]
- Chahal, D.; Ahmadi, A.; Cheung, K.C. Improving piezoelectric cell printing accuracy and reliability through neutral buoyancy of suspensions. Biotechnol. Bioeng. 2012, 109, 2932–2940. [Google Scholar] [CrossRef]
- Ribeiro, R.D.C.; Pal, D.; Ferreira, A.M.; Gentile, P.; Benning, M.; Dalgarno, K.; Ferreira-Duarte, A. Reactive jet impingement bioprinting of high cell density gels for bone microtissue fabrication. Biofabrication 2019, 11, 015014. [Google Scholar] [CrossRef] [PubMed]
- Faulkner-Jones, A.; Fyfe, C.; Cornelissen, D.-J.; Gardner, J.; King, J.; Courtney, A.; Shu, W. Bioprinting of human pluripotent stem cells and their directed differentiation into hepatocyte-like cells for the generation of mini-livers in 3D. Biofabrication 2015, 7, 044102. [Google Scholar] [CrossRef]
- Min, D.; Lee, W.; Bae, I.-H.; Lee, T.R.; Croce, P.; Yoo, S.-S. Bioprinting of biomimetic skin containing melanocytes. Exp. Dermatol. 2018, 27, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.; Singh, G.; Trasatti, J.P.; Bjornsson, C.; Xu, X.; Tran, T.N.; Yoo, S.-S.; Dai, G.; Karande, P. Design and fabrication of human skin by three-dimensional bioprinting. Tissue Eng. Part. C Methods 2014, 20, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, L.; Umehara, Y.; Jud, C.; Blank, F.; Fink, A.; Rothen-Rutishauser, B. Engineering an in vitro air-blood barrier by 3D bioprinting. Sci. Rep. 2015, 5, srep07974. [Google Scholar] [CrossRef] [Green Version]
- Faulkner-Jones, A.; Greenhough, S.; King, J.; Gardner, J.; Courtney, A.; Shu, W. Development of a valve-based cell printer for the formation of human embryonic stem cell spheroid aggregates. Biofabrication 2013, 5, 015013. [Google Scholar] [CrossRef]
- Wu, Y.; Kennedy, P.; Bonazza, N.; Yu, Y.; Dhawan, A.; Ozbolat, I. Three-dimensional bioprinting of articular cartilage: A systematic review. Cartilage 2021, 12, 76–92. [Google Scholar] [CrossRef]
- Bammesberger, S.; Ernst, A.; Losleben, N.; Tanguy, L.; Zengerle, R.; Koltay, P. Quantitative characterization of non-contact microdispensing technologies for the sub-microliter range. Drug Discov. Today 2013, 18, 435–446. [Google Scholar] [CrossRef]
- Kwon, K.-S. Experimental analysis of waveform effects on satellite and ligament behavior via in situ measurement of the drop-on-demand drop formation curve and the instantaneous jetting speed curve. J. Micromech. Microeng. 2010, 20, 115005. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Derby, B. Experimental study of the parameters for stable drop-on-demand inkjet performance. Phys. Fluids 2019, 31, 32004. [Google Scholar] [CrossRef] [Green Version]
- Sip, C.G.; Bhattacharjee, N.; Folch, A. Microfluidic transwell inserts for generation of tissue culture-friendly gradients in well plates. Lab Chip 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Laschke, M.W.; Menger, M.D. Life is 3D: Boosting spheroid function for tissue engineering. Trends Biotechnol. 2017, 35, 133–144. [Google Scholar] [CrossRef] [PubMed]
- McMurtrey, R.J. Roles of diffusion dynamics in stem cell signaling and three-dimensional tissue development. Stem Cells Dev. 2017, 26, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.; Kim, D.; Moon, J. Influence of fluid physical properties on ink-jet printability. Langmuir 2009, 25, 2629–2635. [Google Scholar] [CrossRef]
- Wu, C.-H.; Hwang, W.-S. The effect of the echo-time of a bipolar pulse waveform on molten metallic droplet formation by squeeze mode piezoelectric inkjet printing. Microelectron. Reliab. 2015, 55, 630–636. [Google Scholar] [CrossRef]
- Ng, W.L.; Lee, J.M.; Yeong, W.Y.; Naing, M.W. Microvalve-based bioprinting—Process, bio-inks and applications. Biomater. Sci. 2017, 5, 632–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessemans, L.; Jully, V.; De Raikem, C.; Albanese, M.; Moniotte, N.; Silversmet, P.; Lemoine, D. Automated gravimetric calibration to optimize the accuracy and precision of TECAN freedom EVO liquid handler. J. Lab. Autom. 2016, 21, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Leijten, J.; Georgi, N.; Post, J.; van Blitterswijk, C.; Karperien, M. Trophic effects of mesenchymal stem cells increase chondrocyte proliferation and matrix formation. Tissue Eng. Part A 2011, 17, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Meretoja, V.; Dahlin, R.L.; Kasper, F.K.; Mikos, A.G. Enhanced chondrogenesis in co-cultures with articular chondrocytes and mesenchymal stem cells. Biomaterials 2012, 33, 6362–6369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sports Health Multidiscip. Approach 2009, 1, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Roughley, P.J.; Mort, J.S. The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 2014, 1, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldi, A.; Cinti, C.; Santi, S.; Bosco, D.; Vitale, M.; Zamai, L.; Gobbi, P.; Betts, C.; Falcieri, E. The behaviour of nuclear domains in the course of apoptosis. Histochem. Cell Biol. 1994, 102, 221–231. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Lee, A.J.; Barabino, G.A. Coculture-driven mesenchymal stem cell-differentiated articular chondrocyte-like cells support neocartilage development. Stem Cells Transl. Med. 2012, 1, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Duan, L.; Liang, Y.; Zhu, W.; Xiong, J.; Wang, D. Human umbilical cord blood-derived mesenchymal stem cells contribute to chondrogenesis in coculture with chondrocytes. BioMed Res. Int. 2016, 2016, 3827057. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dudman, J.; Ferreira, A.M.; Gentile, P.; Wang, X.; Dalgarno, K. Microvalve Bioprinting of MSC-Chondrocyte Co-Cultures. Cells 2021, 10, 3329. https://doi.org/10.3390/cells10123329
Dudman J, Ferreira AM, Gentile P, Wang X, Dalgarno K. Microvalve Bioprinting of MSC-Chondrocyte Co-Cultures. Cells. 2021; 10(12):3329. https://doi.org/10.3390/cells10123329
Chicago/Turabian StyleDudman, Joseph, Ana Marina Ferreira, Piergiorgio Gentile, Xiao Wang, and Kenneth Dalgarno. 2021. "Microvalve Bioprinting of MSC-Chondrocyte Co-Cultures" Cells 10, no. 12: 3329. https://doi.org/10.3390/cells10123329
APA StyleDudman, J., Ferreira, A. M., Gentile, P., Wang, X., & Dalgarno, K. (2021). Microvalve Bioprinting of MSC-Chondrocyte Co-Cultures. Cells, 10(12), 3329. https://doi.org/10.3390/cells10123329