
Platelet Activation Mechanisms and Consequences of Immune Thrombocytopenia

 , ,
, ,  and
and {kind=link}
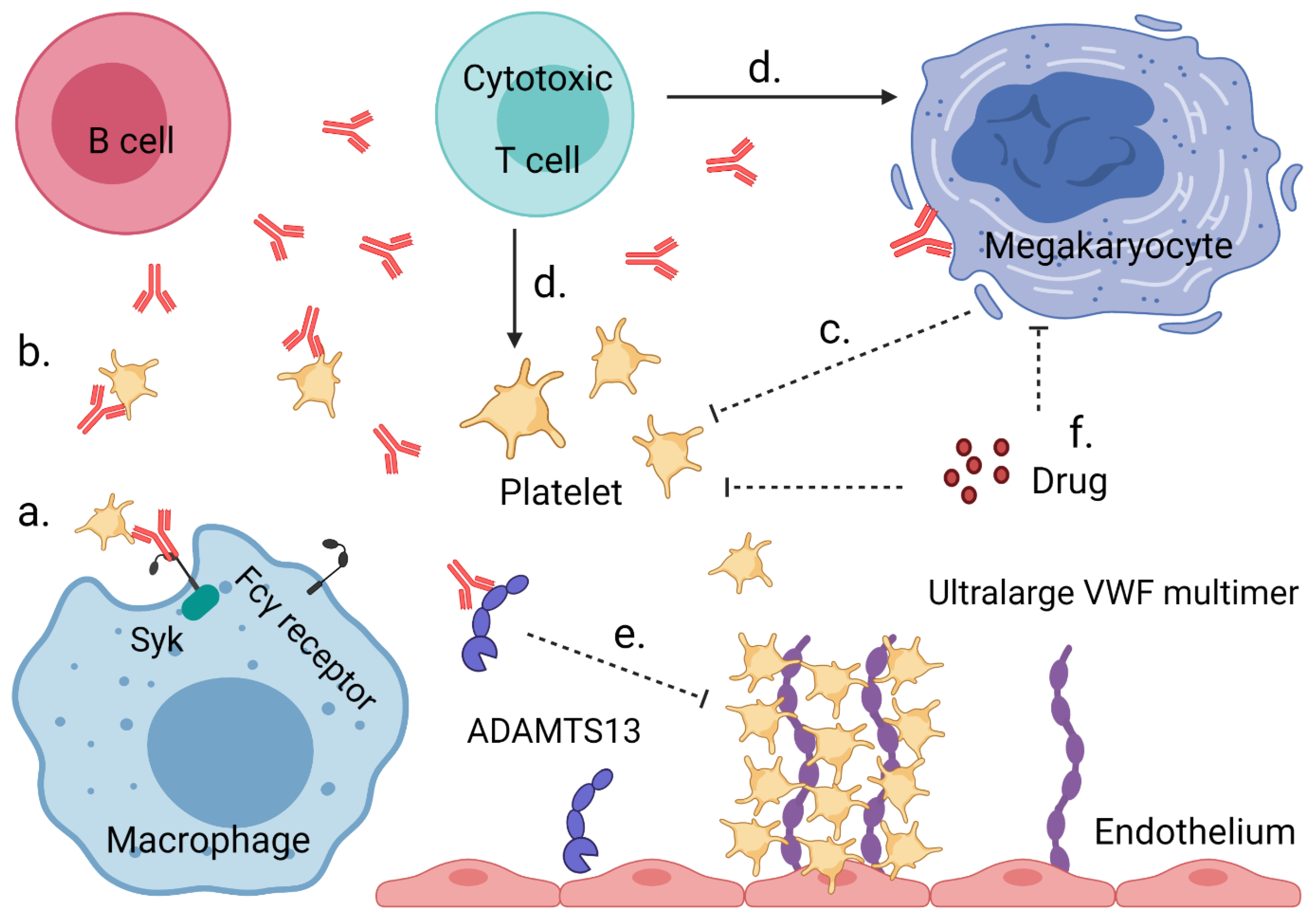{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Platelets in Idiopathic Thrombocytopenic Purpura (ITP)
3. Platelets in Systemic Lupus Erythematosus (SLE)
4. Platelets in Antiphospholipid Syndrome (APS)
5. Platelets in Drug-Induced Thrombocytopenia (DITP)
6. Platelets in Heparin-Induced Thrombocytopenia (HIT)
7. Platelets in Vaccine-Induced Thrombosis with Thrombocytopenia (VITT)
8. Platelets in Thrombotic Thrombocytopenia Purpura (TTP)
9. Platelets in Hemolysis, Elevated Liver Enzymes and Low Platelet (HELLP) Syndrome
10. Conclusions and Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaushansky, K.; Lok, S.; Holly, R.D.; Broudy, V.C.; Lin, N.; Bailey, M.C.; Forstrom, J.W.; Buddle, M.M.; Oort, P.J.; Hagen, F.S.; et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 1994, 369, 568–571. [Google Scholar] [CrossRef]
- Greenberg, E.M.; Kaled, E.S. Thrombocytopenia. Crit Care Nurs. Clin N. Am. 2013, 25, 427–434. [Google Scholar] [CrossRef]
- Grodzielski, M.; Goette, N.P.; Glembotsky, A.C.; Constanza Baroni Pietto, M.; Méndez-Huergo, S.P.; Pierdominici, M.S.; Montero, V.S.; Rabinovich, G.A.; Molinas, F.C.; Heller, P.G.; et al. Multiple concomitant mechanisms contribute to low platelet count in patients with immune thrombocytopenia. Sci. Rep. 2019, 9, 2208. [Google Scholar] [CrossRef]
- Van der Meijden, P.E.; Heemskerk, J.W. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P. Arterial thrombosis: Insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Mastenbroek, T.G.; van Geffen, J.P.; Heemskerk, J.W.; Cosemans, J.M. Acute and persistent platelet and coagulant activities in atherothrombosis. J. Thromb. Haemost. 2015, 13, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.S. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Witt, S.; Verdoold, R.; Cosemans, J.M.; Heemskerk, J.W. Insights into platelet-based control of coagulation. Thromb. Res. 2014, 133, S139–S148. [Google Scholar] [CrossRef]
- Nagy, M.; Heemskerk, J.W.; Swieringa, F. Use of microfluidics to assess the platelet-based control of coagulation. Platelets 2017, 28, 441–448. [Google Scholar] [CrossRef]
- Fernández, D.I.; Kuijpers, M.J.; Heemskerk, J.W. Platelet calcium signaling by G-protein coupled and ITAM-linked receptors regulating anoctamin-6 and procoagulant activity. Platelets 2021, 32, 863–871. [Google Scholar] [CrossRef]
- Swieringa, F.; Kuijpers, M.J.; Heemskerk, J.W.; van der Meijden, P.E. Targeting platelet receptor function in thrombus formation: The risk of bleeding. Blood Rev. 2014, 28, 9–21. [Google Scholar] [CrossRef]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef]
- National Heart Lung and Blood Institute of NIH USA. Thrombocytopenia. Available online: https://www.nhlbi.nih.gov/health-topics/thrombocytopenia (accessed on 1 August 2021).
- Tinazzi, E.; Osti, N.; Beri, R.; Argentino, G.; Veneri, D.; Dima, F.; Bason, C.; Jadav, G.; Dolcino, M.; Puccetti, A. Pathogenesis of immune thrombocytopenia in common variable immunodeficiency. Autoimmun. Rev. 2020, 19, 102616. [Google Scholar] [CrossRef]
- Neunert, C.; Lim, W.; Crowther, M.; Cohen, A.; Solberg, L.; Crowther, M.A. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011, 117, 4190–4207. [Google Scholar] [CrossRef] [Green Version]
- Efficace, F.; Mandelli, F.; Fazi, P.; Santoro, C.; Gaidano, G.; Cottone, F.; Borchiellini, A.; Carpenedo, M.; Simula, M.P.; Di Giacomo, V.; et al. Health-related quality of life and burden of fatigue in patients with primary immune thrombocytopenia by phase of disease. Am. J. Hematol. 2016, 91, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Rodeghiero, F. Is ITP a thrombophilic disorder? Am. J. Hematol. 2016, 91, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufferey, A.; Kapur, R.; Semple, J.W. Pathogenesis and therapeutic mechanisms in immune thrombocytopenia (ITP). J. Clin. Med. 2017, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Solanilla, A.; Pasquet, J.M.; Viallard, J.F.; Contin, C.; Grosset, C.; Déchanet-Merville, J.; Dupouy, M.; Landry, M.; Belloc, F.; Nurden, P.; et al. Platelet-associated CD154 in immune thrombocytopenic purpura. Blood 2005, 105, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.; Ghanima, W. Immune thrombocytopenia. N. Engl. J. Med. 2019, 381, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Marini, I.; Bakchoul, T. Pathophysiology of autoimmune thrombocytopenia: Current insight with a focus on thrombopoiesis. Hämostaseologie 2019, 39, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Uzun, G.; Bakchoul, T. Primary immune thrombocytopenia: Novel insights into pathophysiology and disease management. J. Clin. Med. 2021, 10, 789. [Google Scholar] [CrossRef]
- Provan, D.; Arnold, D.M.; Bussel, J.B.; Chong, B.H.; Cooper, N.; Gernsheimer, T.; Ghanima, W.; Godeau, B.; Gonzalez-Lopez, T.J.; Grainger, J.; et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019, 3, 3780–3817. [Google Scholar] [CrossRef] [Green Version]
- McMillan, R. Autoantibodies and autoantigens in chronic immune thrombocytopenic purpura. Semin. Hematol. 2000, 37, 239–248. [Google Scholar] [CrossRef]
- Podolanczuk, A.; Lazarus, A.H.; Crow, A.R.; Grossbard, E.; Bussel, J.B. Of mice and men: An open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 2009, 113, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Crow, A.R.; Song, S.; Siragam, V.; Arus, A.H. Mechanisms of action of intravenous immunoglobulin in the treatment of immune thrombocytopenia. Pediatr. Blood Cancer 2006, 47, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R. Pathophysiology and therapeutic options in primary immune thrombocytopenia. Blood Transfus. 2011, 9, 262. [Google Scholar] [PubMed]
- Urbanus, R.T.; van der Wal, D.E.; Koekman, C.A.; Huisman, A.; van den Heuvel, D.J.; Gerritsen, H.C.; Deckmyn, H.; Akkerman, J.W.; Schutgens, R.E.; Gitz, E. Patient autoantibodies induce platelet destruction signals via raft-associated glycoprotein Ibα and Fc RIIa in immune thrombocytopenia. Haematologica 2013, 98, e70–e72. [Google Scholar] [CrossRef] [Green Version]
- Arman, M.; Krauel, K. Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J. Thromb. Haemost. 2015, 13, 893–908. [Google Scholar] [CrossRef]
- Zeng, D.F.; Chen, F.; Wang, S.; Chen, S.L.; Xu, Y.; Shen, M.Q.; Du, C.H.; Wang, C.; Kong, P.Y.; Cheng, T.M.; et al. Autoantibody against integrin αv β3 contributes to thrombocytopenia by blocking the migration and adhesion of megakaryocytes. J. Thromb. Haemost. 2018, 16, 1843–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, R.; Wang, L.; Tomer, A.; Nichol, J.; Pistillo, J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004, 103, 1364–1369. [Google Scholar] [CrossRef] [Green Version]
- Iraqi, M.; Perdomo, J.; Yan, F.; Choi, P.Y.; Chong, B.H. Immune thrombocytopenia: Antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica 2015, 100, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Leytin, V.; Mykhaylov, S.; Starkey, A.F.; Allen, D.J.; Lau, H.; Ni, H.; Semple, J.W.; Lazarus, A.H.; Freedman, J. Intravenous immunoglobulin inhibits anti-glycoprotein IIb-induced platelet apoptosis in a murine model of immune thrombocytopenia. Br. J. Haematol. 2006, 133, 78–82. [Google Scholar] [CrossRef]
- Alvarez Roman, M.T.; Fernandez Bello, I.; Arias-Salgado, E.G.; Rivas Pollmar, M.I.; Jimenez Yuste, V.; Martin Salces, M.; Butta, N.V. Effects of thrombopoietin receptor agonists on procoagulant state in patients with immune thrombocytopenia. Thromb. Haemost. 2014, 112, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Goette, N.P.; Glembotsky, A.C.; Lev, P.R.; Grodzielski, M.; Contrufo, G.; Pierdominici, M.S.; Espasandin, Y.R.; Riveros, D.; García, A.J.; Molinas, F.C.; et al. Platelet apoptosis in adult immune thrombocytopenia: Insights into the mechanism of damage triggered by auto-antibodies. PLoS ONE 2016, 11, e0160563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, S.; Bonnotte, B. Emerging therapies in immune thrombocytopenia. J. Clin. Med. 2021, 10, 1004. [Google Scholar] [CrossRef]
- Cantoni, S.; Carpenedo, M.; Nichelatti, M.; Sica, L.; Rossini, S.; Milella, M.; Popescu, C.; Cairoli, R. Clinical relevance of antiplatelet antibodies and the hepatic clearance of platelets in patients with immune thrombocytopenia. Blood 2016, 128, 2183–2185. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; van der Wal, D.E.; Zhu, G.; Xu, M.; Yougbare, I.; Ma, L.; Vadasz, B.; Carrim, N.; Grozovsky, R.; Ruan, M.; et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat. Commun. 2015, 6, 7737. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fu, J.; Ling, Y.; Yago, T.; McDaniel, J.M.; Song, J.; Bai, X.; Kondo, Y.; Qin, Y.; Hoover, C.; et al. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proc. Natl. Acad. Sci. USA 2017, 114, 8360–8365. [Google Scholar] [CrossRef] [Green Version]
- Deppermann, C.; Kratofil, R.M.; Peiseler, M.; David, B.A.; Zindel, J.; Castanheira, F.V.; van der Wal, F.; Carestia, A.; Jenne, C.N.; Marth, J.D.; et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 2020, 217, e20190723. [Google Scholar] [CrossRef]
- Quach, M.E.; Dragovich, M.A.; Chen, W.; Syed, A.K.; Cao, W.; Liang, X.; Deng, W.; De Meyer, S.F.; Zhu, G.; Peng, J.; et al. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood 2018, 131, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riswari, S.F.; Tunjungputri, R.N.; Kullaya, V.; Garishah, F.M.; Utari, G.S.R.; Farhanah, N.; Overheul, G.J.; Alisjahbana, B.; Gasem, M.H.; Urbanus, R.T.; et al. Desialylation of platelets induced by Von Willebrand Factor is a novel mechanism of platelet clearance in dengue. PLoS Pathog. 2019, 15, e1007500. [Google Scholar] [CrossRef]
- Najaoui, A.; Bakchoul, T.; Stoy, J.; Bein, G.; Rummel, M.J.; Santoso, S.; Sachs, U.J. Autoantibody-mediated complement activation on platelets is a common finding in patients with immune thrombocytopenic purpura (ITP). Eur. J. Haematol. 2012, 88, 167–174. [Google Scholar] [CrossRef]
- Hed, J. Role of complement in immune or idiopathic thrombocytopenic purpura. Acta Paediatr. Suppl. 1998, 424, 37–40. [Google Scholar] [CrossRef]
- Peerschke, E.I.; Andemariam, B.; Yin, W.; Bussel, J.B. Complement activation on platelets correlates with a decrease in circulating immature platelets in patients with immune thrombocytopenic purpura. Br. J. Haematol. 2010, 148, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, L.M.; Davis, J.M.; Buchli, R.; Vangundy, R.S.; Schwartz, K.A.; Gerlach, J.A. MHC class I-associated peptides identified from normal platelets and from individuals with idiopathic thrombocytopenic purpura. Hum. Immunol. 2005, 66, 874–883. [Google Scholar] [CrossRef]
- Olsson, B.; Andersson, P.O.; Jernas, M.; Jacobsson, S.; Carlsson, B.; Carlsson, L.M.; Wadenvik, H. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat. Med. 2003, 9, 1123–1124. [Google Scholar] [CrossRef]
- Nagy, M.; Mastenbroek, T.G.; Mattheij, N.J.; De Witt, S.; Clemetson, K.J.; Kirschner, J.; Schulz, A.; Braun, A.; Cosemans, J.M.; Zieger, B.; et al. Variable impairment of platelet functions in patients with severe, genetically linked immune deficiencies. Haematologica 2018, 103, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.P.; Poncz, M. Inherited thrombocytopenias. In Platelets, 2nd ed; Michelson, A.D., Ed.; Academic Press: New York, NY, USA, 2007; pp. 985–998. [Google Scholar]
- Scherlinger, M.; Guillotin, V.; Truchetet, M.E.; Contin-Bordes, C.; Sisirak, V.; Duffau, P.; Lazaro, E.; Richez, C.; Blanco, P. Systemic lupus erythematosus and systemic sclerosis: All roads lead to platelets. Autoimmun. Rev. 2018, 17, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Abdel Galil, S.M.; Edrees, A.M.; Ajeeb, A.K.; Aldoobi, G.S.; El-Boshy, M.; Hussain, W. Prognostic significance of platelet count in SLE patients. Platelets 2017, 28, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Soh, M.S.; Ahn, Y.H.; Um, Y.J.; Jung, J.Y.; Suh, C.H.; Kim, H.A. Thrombocytopenia in systemic lupus erythematosus: Clinical manifestations, treatment, and prognosis in 230 patients. Medicine 2016, 95, e2818. [Google Scholar] [CrossRef]
- Drenkard, C.; Villa, A.R.; Alarcon-Segovia, D.; Perez-Vazquez, M.E. Influence of the antiphospholipid syndrome in the survival of patients with systemic lupus erythematosus. J. Rheumatol. 1994, 21, 1067–1072. [Google Scholar] [PubMed]
- Fayyaz, A.; Igoe, A.; Kurien, B.T.; Danda, D.; James, J.A.; Stafford, H.A.; Scofield, R.H. Haematological manifestations of lupus. Lupus Sci. Med. 2015, 2, e000078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Berlacher, M.D.; Vieth, J.A.; Heflin, B.C.; Gay, S.R.; Antczak, A.J.; Tasma, B.E.; Boardman, H.J.; Singh, N.; Montel, A.H.; Kahaleh, M.B.; et al. FcγRIIa ligation induces platelet hypersensitivity to thrombotic stimuli. Am. J. Pathol. 2013, 182, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, N.; Allaeys, I.; Marcoux, G.; Machlus, K.R.; Mailhot, B.; Zufferey, A.; Levesque, T.; Becker, Y.; Tessandier, N.; Melki, I.; et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc. Natl. Acad. Sci. USA 2018, 115, e1550–e1559. [Google Scholar] [CrossRef] [Green Version]
- Linge, P.; Fortin, P.R.; Lood, C.; Bengtsson, A.A.; Boilard, E. The non-haemostatic role of platelets in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2018, 14, 195. [Google Scholar] [CrossRef]
- Lee, Y.H.; Nath, S.K. Systemic lupus erythematosus susceptibility loci defined by genome scan meta-analysis. Hum. Genet. 2005, 118, 434–443. [Google Scholar] [CrossRef]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; de Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Garcia, D.; Erkan, D. Diagnosis and management of the antiphospholipid syndrome. N. Engl. J. Med. 2018, 378, 2010–2021. [Google Scholar] [CrossRef]
- Artim-Esen, B.; Diz-Kucukkaya, R.; Inanc, M. The significance and management of thrombocytopenia in antiphospholipid syndrome. Curr. Rheumatol. Rep. 2015, 17, 14. [Google Scholar] [CrossRef]
- Hasselaar, P.; Derksen, R.H.; Oosting, J.D.; Blokzijl, L.; de Groot, P.G. Synergistic effect of low doses of tumor necrosis factor and sera from patients with systemic lupus erythematosus on the expression of procoagulant activity by cultured endothelial cells. Thromb. Haemost. 1989, 62, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Vreede, A.P.; Bockenstedt, P.L.; McCune, W.J.; Knight, J.S. Cryptic conspirators: A conversation about thrombocytopenia and antiphospholipid syndrome. Curr. Opin. Rheumatol. 2019, 31, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yang, M.; Wang, K.; Sun, J.; Song, L.; Diao, X.; Jiang, Z.; Cheng, G.; Wang, X. Excessive activation of the TLR9/TGF-β1/PDGF-B pathway in the peripheral blood of patients with systemic lupus erythematosus. Arthritis Res. Therap. 2017, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Urbanus, R.T.; Pennings, M.T.; Derksen, R.H.; de Groot, P. Platelet activation by dimeric b2-glycoprotein I requires signaling via both glycoprotein Iba and apolipoprotein E receptor 2′. J. Thromb. Haemost. 2008, 6, 1405–1412. [Google Scholar] [CrossRef]
- Hollerbach, A.; Müller-Calleja, N.; Ritter, S.; Häuser, F.; Canisius, A.; Orning, C.; Jurk, K.; Lackner, K.J. Platelet activation by antiphospholipid antibodies depends on epitope specificity and is prevented by mTOR inhibitors. Thromb. Haemost. 2019, 119, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Chayoua, W.; Nicolson, P.L.; Meijers, J.C.; Kardeby, C.; Garcia-Quintanilla, L.; Devreese, K.M.; de Laat, B.; Watson, S.P.; de Groot, P.G. Antiprothrombin antibodies induce platelet activation: A possible explanation for anti-FXa therapy failure in patients with antiphospholipid syndrome? J. Thromb. Haemost. 2021, 19, 1776–1782. [Google Scholar] [CrossRef]
- Aster, R.H. Drug-induced thrombocytopenia. In Platelets, 2nd ed.; Michelson, A.D., Ed.; Academic Press: New York, NY, USA, 2007; pp. 887–902. [Google Scholar]
- Asvadi, P.; Ahmadi, Z.; Chong, B.H. Drug-induced thrombocytopenia: Localization of the binding site of GPIX-specific quinine-dependent antibodies. Blood 2003, 102, 1670–1677. [Google Scholar] [CrossRef] [Green Version]
- Baaten, C.C.; Moenen, F.C.; Henskens, Y.M.; Swieringa, F.; Wetzels, R.J.; van Oerle, R.; Heijnen, H.F.; ten Cate, H.; Holloway, G.P.; Beckers, E.A.; et al. Impaired mitochondrial activity explains platelet dysfunction in thrombocytopenic cancer patients undergoing chemotherapy. Haematologica 2018, 103, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Majeed, A.; Powell, R. Heparin induced thrombocytopenia: Diagnosis and management update. Postgrad. Med. J. 2007, 83, 575–582. [Google Scholar] [CrossRef]
- Mayo, K.H.; Barker, S.; Kuranda, M.J.; Hunt, A.J.; Myers, J.A.; Maione, T.E. Molten globule monomer to condensed dimer: Role of disulfide bonds in platelet factor-4 folding and subunit association. Biochemistry 1992, 31, 12255–12265. [Google Scholar] [CrossRef]
- Baroletti, S.A.; Goldhaber, S.Z. Heparin-induced thrombocytopenia. Circulation 2006, 114, e355–e356. [Google Scholar] [CrossRef] [Green Version]
- Rauova, L.; Poncz, M.; McKenzie, S.E.; Reilly, M.P.; Arepally, G.; Weisel, J.W.; Nagaswami, C.; Cines, D.B.; Sachais, B.S. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005, 105, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A.; Selleng, K.; Warkentin, T.E. Autoimmune heparin-induced thrombocytopenia. J. Thromb. Haemost. 2017, 15, 2099–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warkentin, T.E.; Sheppard, J.I.; Moore, J.C.; Cook, R.J.; Kelton, J.G. Studies of the immune response in heparin-induced thrombocytopenia. Blood 2009, 113, 4963–4969. [Google Scholar] [CrossRef]
- Rollin, J.; Pouplard, C.; Sung, H.C.; Leroux, D.; Saada, A.; Gouilleux-Gruart, V.; Thibault, G.; Gruel, Y. Increased risk of thrombosis in FcγRIIA 131RR patients with HIT due to defective control of platelet activation by plasma IgG2. Blood 2015, 125, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Medvedev, N.; Delcea, M.; Greinacher, A. Anti-platelet factor 4/polyanion antibodies mediate a new mechanism of autoimmunity. Nat. Commun. 2017, 8, 14945. [Google Scholar] [CrossRef] [PubMed]
- Mordakhanova, E.R.; Nevzorova, T.A.; Synbulatova, G.E.; Rauova, L.; Weisel, J.W.; Litvinov, R.I. Platelet activation in heparin-Induced thrombocytopenia is followed by platelet death via complex apoptotic and non-apoptotic pathways. Intern. J. Mol. Sci. 2020, 21, 2556. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.P.; Sinha, U.; André, P.; Taylor, S.M.; Pak, Y.; Deguzman, F.R.; Nanda, N.; Pandey, A.; Stolla, M.; Bergmeier, W.; et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood 2011, 117, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Arepally, G.M.; Mayer, I.M. Antibodies from patients with heparin-induced thrombocytopenia stimulate monocytic cells to express tissue factor and secrete interleukin-8. Blood 2001, 98, 1252–1254. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A. Heparin-induced thrombocytopenia. N. Engl. J. Med. 2015, 373, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Gimotty, P.A.; Crowther, M.A.; Warkentin, T.E. Predictive value of the 4Ts scoring system for heparin-induced thrombocytopenia: A systematic review and meta-analysis. Blood 2012, 120, 4160–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, A.; Winteroll, S. Laboratory diagnosis of heparin-induced thrombocytopenia and monitoring of alternative anticoagulants. Clin. Diagn. Lab. Immunol. 2003, 10, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Warkentin, T.E.; Greinacher, A. Management of heparin-induced thrombocytopenia. Curr. Opin. Hematol. 2016, 23, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Colarossi, G.; Maffulli, N.; Trivellas, A.; Schnoring, H.; Hatam, N.; Tingart, M.; Migliorini, F. Superior outcomes with argatroban for heparin-induced thrombocytopenia: A bayesian network meta-analysis. Intern. J. Clin. Pharm. 2021, 43, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Nilius, H.; Kaufmann, J.; Cuker, A.; Nagler, M. Comparative effectiveness and safety of anticoagulants for the treatment of heparin-induced thrombocytopenia. Am. J. Hematol. 2021, 96, 805–815. [Google Scholar] [CrossRef]
- Schultz, N.H.; Sorvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.H.; Skattor, T.H.; Tjonnfjord, G.E.; et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Cines, D.B.; Bussel, J.B. SARS-CoV-2 vaccine-induced immune thrombotic thrombocytopenia. N. Engl. J. Med. 2021, 384, 2254–2256. [Google Scholar] [CrossRef]
- Pavord, S.; Scully, M.; Hunt, B.J.; Lester, W.; Bagot, C.; Craven, B.; Rampotas, A.; Ambler, G.; Makris, M. Clinical features of vaccine-induced immune thrombocytopenia and thrombosis. N. Engl. J. Med. 2021, 385, 1680–1689. [Google Scholar] [CrossRef]
- Hwang, J.; Lee, S.B.; Lee, S.W.; Lee, M.H.; Koyanagi, A.; Jacob, L.; Tizaoui, K.; Yon, D.K.; Shin, J.I.; Smith, L. Comparison of vaccine-induced thrombotic events between ChAdOx1 nCoV-19 and Ad26.COV.2.S vaccines. J. Autoimmun. 2021, 122, 102681. [Google Scholar] [CrossRef]
- Ikenberg, B.; Demleitner, A.F.; Thiele, T.; Wiestler, B.; Gotze, K.; Mossmer, G.; Lingor, P. Cerebral venous sinus thrombosis after ChAdOx1 nCov-19 vaccination with a misleading first cerebral MRI scan. Stroke Vasc. Neurol. 2021. In print. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K.; Palankar, R.; Wesche, J.; Handtke, S.; Wolff, M.; Aurich, K.; Lalk, M.; Methling, K.; Volker, U.; et al. Insights in ChAdOx1 nCov-19 vaccine-induced immune thrombotic thrombocytopenia (VITT). Blood 2021. In print. [Google Scholar] [CrossRef] [PubMed]
- Althaus, K.; Moller, P.; Uzun, G.; Singh, A.; Beck, A.; Bettag, M.; Bosmuller, H.; Guthoff, M.; Dorn, F.; Petzold, G.C.; et al. Antibody-mediated procoagulant platelets in SARS-CoV-2-vaccination associated immune thrombotic thrombocytopenia. Haematologica 2021, 106, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K.; Mayerle, J.; Palankar, R.; Wesche, J.; Reiche, S.; Aebischer, A.; Warkentin, T.E.; Muenchhoff, M.; Hellmuth, J.C.; et al. Anti-platelet factor 4 antibodies causing VITT do not cross-react with SARS-CoV-2 spike protein. Blood 2021, 138, 1269–1277. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Lorenz, R.; Siess, W.; Weber, C. Vaccine-induced immune thrombotic thrombocytopenia (VITT): Targeting pathomechanisms with bruton tyrosine kinase inhibitors. Thromb. Haemost. 2021, 121, 1395–1399. [Google Scholar] [CrossRef]
- George, J.N. Thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2006, 354, 1927–1935. [Google Scholar] [CrossRef]
- Sadler, J.E. Pathophysiology of thrombotic thrombocytopenic purpura. Blood 2017, 130, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Kremer Hovinga, J.A.; Coppo, P.; Lämmle, B.; Moake, J.L.; Miyata, T.; Vanhoorelbeke, K. Thrombotic thrombocytopenic purpura. Nat. Rev. Dis. Primers 2017, 3, 17020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feys, H.B.; Roodt, J.; Vandeputte, N.; Pareyn, I.; Mottl, H.; Hou, S.; Lamprecht, S.; Van Rensburg, W.J.; Deckmyn, H.; Vanhoorelbeke, K. Inhibition of von Willebrand factor-platelet glycoprotein Ib interaction prevents and reverses symptoms of acute acquired thrombotic thrombocytopenic purpura in baboons. Blood 2012, 120, 3611–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846. [Google Scholar] [CrossRef] [Green Version]
- Kremer Hovinga, J.A.; George, J.N. Hereditary thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 381, 1653–1662. [Google Scholar] [CrossRef]
- Van Dorland, H.A.; Taleghani, M.M.; Sakai, K.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knobl, P.N.; von Krogh, A.S.; Schneppenheim, R.; Aebi-Huber, I.; et al. The international hereditary thrombotic thrombocytopenic purpura registry: Key findings at enrollment until 2017. Haematologica 2019, 104, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Masias, C.; Cataland, S.R. The role of ADAMTS13 testing in the diagnosis and management of thrombotic microangiopathies and thrombosis. Blood 2018, 132, 903–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bury, L.; Malara, A.; Momi, S.; Petito, E.; Balduini, A.; Gresele, P. Mechanisms of thrombocytopenia in platelet-type von Willebrand disease. Haematologica 2019, 104, 1473–1481. [Google Scholar] [CrossRef]
- Dupont, A.; Soukaseum, C.; Cheptou, M.; Adam, F.; Nipoti, T.; Lourenco-Rodrigues, M.D.; Legendre, P.; Proulle, V.; Rauch, A.; Kawecki, C.; et al. Relevance of platelet desialylation and thrombocytopenia in type 2B von Willebrand disease: Preclinical and clinical evidence. Haematologica 2019, 104, 2493–2500. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, S.; Lammle, B.; Cataland, S.R. Thrombotic thrombocytopenic purpura: Pathophysiology, diagnosis, and management. J. Clin. Med. 2021, 10, 536. [Google Scholar] [CrossRef]
- Verhenne, S.; Vandeputte, N.; Pareyn, I.; Izsvak, Z.; Rottensteiner, H.; Deckmyn, H.; De Meyer, S.F.; Vanhoorelbeke, K. Long-term prevention of congenital thrombotic thrombocytopenic purpura in ADAMTS13 knockout mice by sleeping beauty transposon-mediated gene therapy. Arter. Thromb. Vasc. Biol. 2017, 37, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abildgaard, U.; Heimdal, K. Pathogenesis of the syndrome of hemolysis, elevated liver enzymes, and low platelet count (HELLP): A review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 166, 117–123. [Google Scholar] [CrossRef]
- Koenig, M.; Roy, M.; Baccot, S.; Cuilleron, M.; de Filippis, J.P.; Cathébras, P. Thrombotic microangiopathy with liver, gut, and bone infarction (catastrophic antiphospholipid syndrome) associated with HELLP syndrome. Clin. Rheumatol. 2005, 24, 166–168. [Google Scholar] [CrossRef]
- Hulstein, J.J.; van Runnard Heimel, P.J.; Franx, A.; Lenting, P.J.; Bruinse, H.W.; Silence, K.; de Groot, P.G.; Fijnheer, R. Acute activation of the endothelium results in increased levels of active von Willebrand factor in hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome. J. Thromb. Haemost. 2006, 4, 2569–2575. [Google Scholar] [CrossRef]
- Lattuada, A.; Rossi, E.; Calzarossa, C.; Candolfi, R.; Mannucci, P.M. Mild to moderate reduction of a von Willebrand factor cleaving protease (ADAMTS-13) in pregnant women with HELLP microangiopathic syndrome. Haematologica 2003, 88, 1029–1034. [Google Scholar] [PubMed]
- Pourrat, O.; Coudroy, R.; Pierre, F. ADAMTS13 deficiency in severe postpartum HELLP syndrome. Br. J. Haematol. 2013, 163, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Azzam, H.A.; Abousamra, N.K.; Goda, H.; El-Shouky, R.; El-Gilany, A. The expression and concentration of CD40 ligand in normal pregnancy, preeclampsia, and hemolytic anemia, elevated liver enzymes and low platelet count (HELLP) syndrome. Blood Coagul Fibrinolys 2013, 24, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Jablonski, M.; Lepercq, J.; Blouin, J.; Benachi, A.; Hourmant, M.; Pirson, Y.; Dürrbach, A.; Grünfeld, J.P.; Knebelmann, B.; et al. Factor H, membrane cofactor protein, and factor I mutations in patients with hemolysis, elevated liver enzymes, and low platelet count syndrome. Blood 2008, 112, 4542–4545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ari, E.; Yilmaz, Y.; Gul, A.; Alahdab, Y.O.; Kedrah, A.E.; Macunluoglu, B.; Aydin, A.; Arikan, H.; Ozener, C. Human serum complement C3 and factor H in the syndrome of hemolysis, elevated liver enzymes, and low platelet count. Am. J. Reprod. Immunol. 2009, 62, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Lanir, N.; Thaler, I. HELLP syndrome associated with factor V R506Q mutation. Br. J. Haematol. 1996, 92, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Thulluru, H.K.; Mulders, J.; Michel, O.J.; Poutsma, A.; Windhorst, S.; Kleiverda, G.; Sie, D.; Lachmeijer, A.M.; Oudejans, C.B. HELLP babies link a novel lincRNA to the trophoblast cell cycle. J. Clin. Investig. 2012, 122, 4003–4011. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Urbanus, R.T.; ten Cate, H.; de Groot, P.G.; de Laat, B.; Heemskerk, J.W.M.; Roest, M. Platelet Activation Mechanisms and Consequences of Immune Thrombocytopenia. Cells 2021, 10, 3386. https://doi.org/10.3390/cells10123386
Sun S, Urbanus RT, ten Cate H, de Groot PG, de Laat B, Heemskerk JWM, Roest M. Platelet Activation Mechanisms and Consequences of Immune Thrombocytopenia. Cells. 2021; 10(12):3386. https://doi.org/10.3390/cells10123386
Chicago/Turabian StyleSun, Siyu, Rolf T. Urbanus, Hugo ten Cate, Philip G. de Groot, Bas de Laat, Johan W. M. Heemskerk, and Mark Roest. 2021. "Platelet Activation Mechanisms and Consequences of Immune Thrombocytopenia" Cells 10, no. 12: 3386. https://doi.org/10.3390/cells10123386
APA StyleSun, S., Urbanus, R. T., ten Cate, H., de Groot, P. G., de Laat, B., Heemskerk, J. W. M., & Roest, M. (2021). Platelet Activation Mechanisms and Consequences of Immune Thrombocytopenia. Cells, 10(12), 3386. https://doi.org/10.3390/cells10123386