
Molecular Basis of Accelerated Aging with Immune Dysfunction-Mediated Inflammation (Inflamm-Aging) in Patients with Systemic Sclerosis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
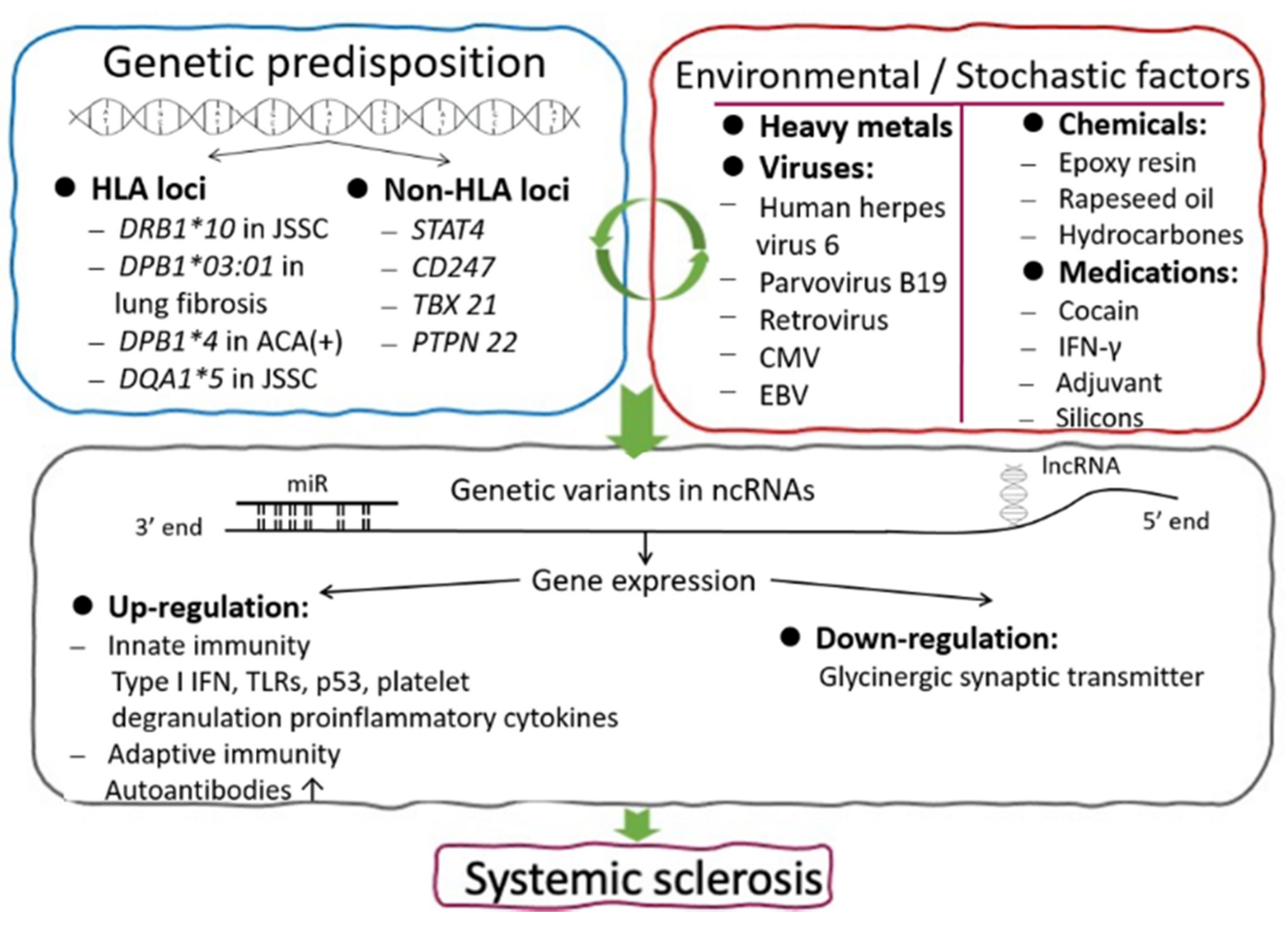
2. The Roles of Genetics, Environmental Risk Factors, Stochastic Processes, and Epigenetics in the Pathogenesis of Patients with Systemic Sclerosis
2.1. Genetic Predisposition in Patients with SSc
2.1.1. The Role of Major Histocompatibility Complex (MHC) Class II
2.1.2. The Role of Non-HLA Genes
2.2. Environmental Risk Factors and Stochastic Processes in Patients with SSc
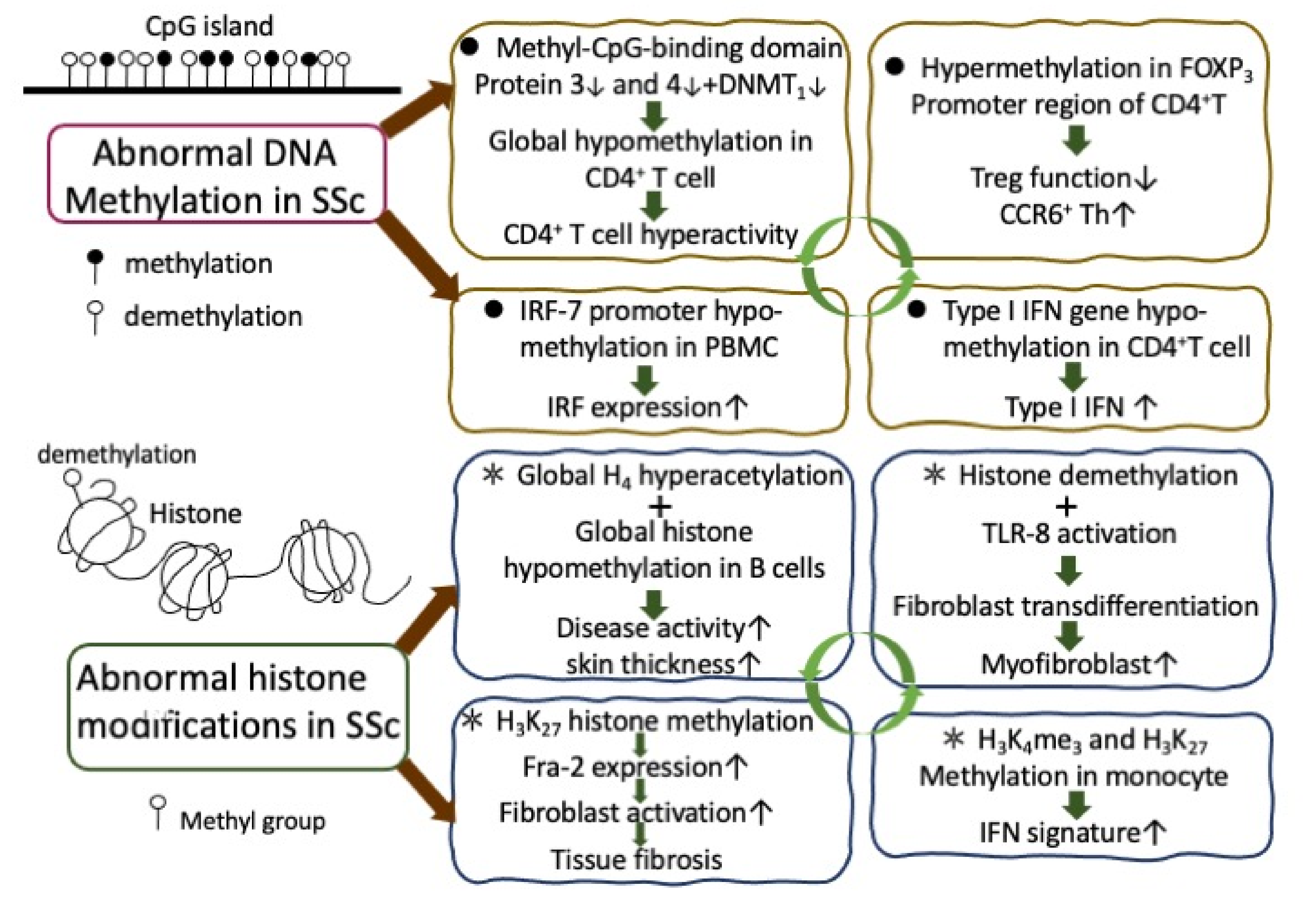
2.3. Aberrant Epigenetic Expression in Patients with SSc
2.3.1. Abnormal DNA Methylation and Histone Modifications in the Immune-Related Cells of SSc Patients
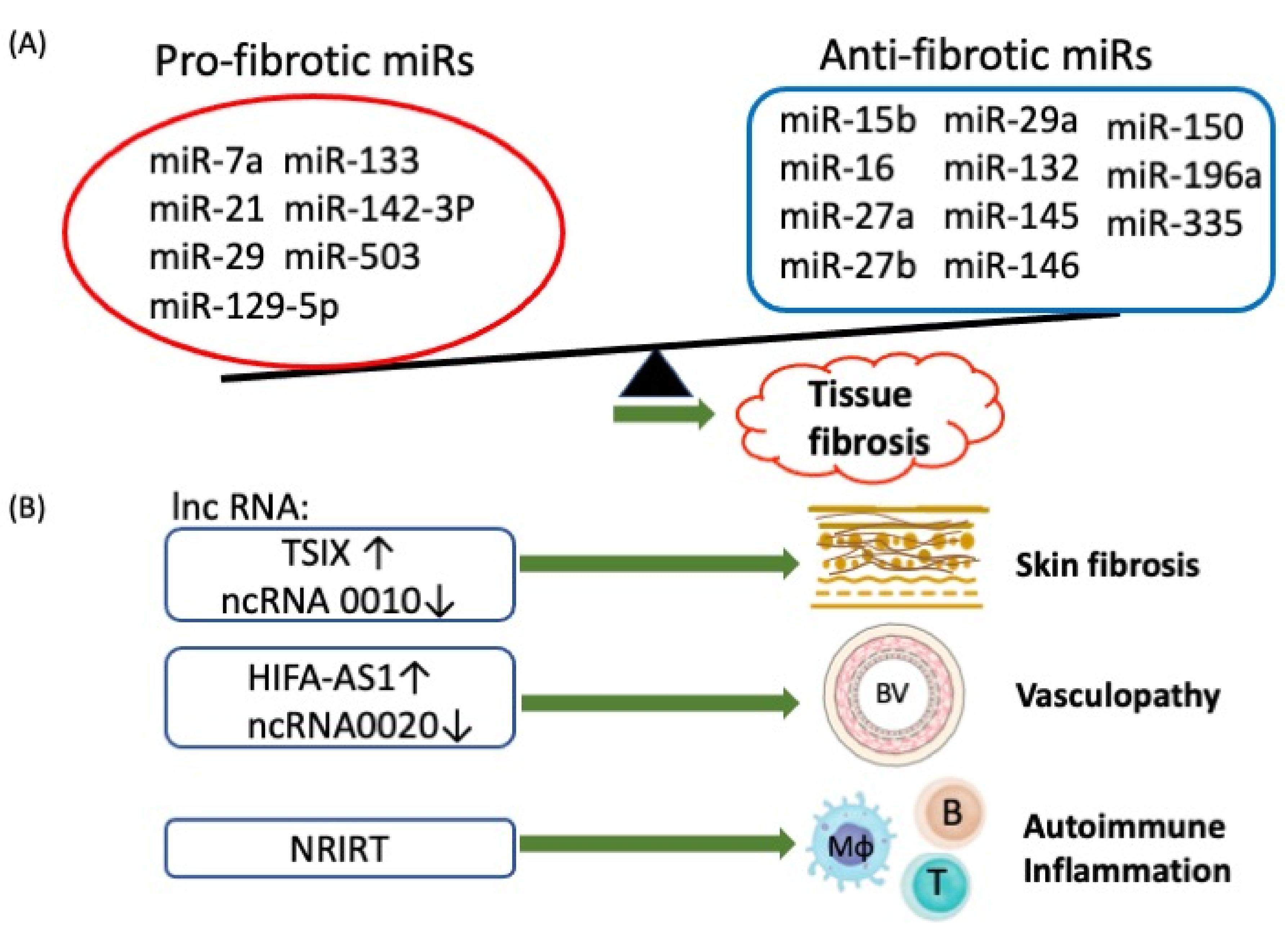
2.3.2. Aberrant Non-coding RNA (ncRNA) Expression in Patients with SSc
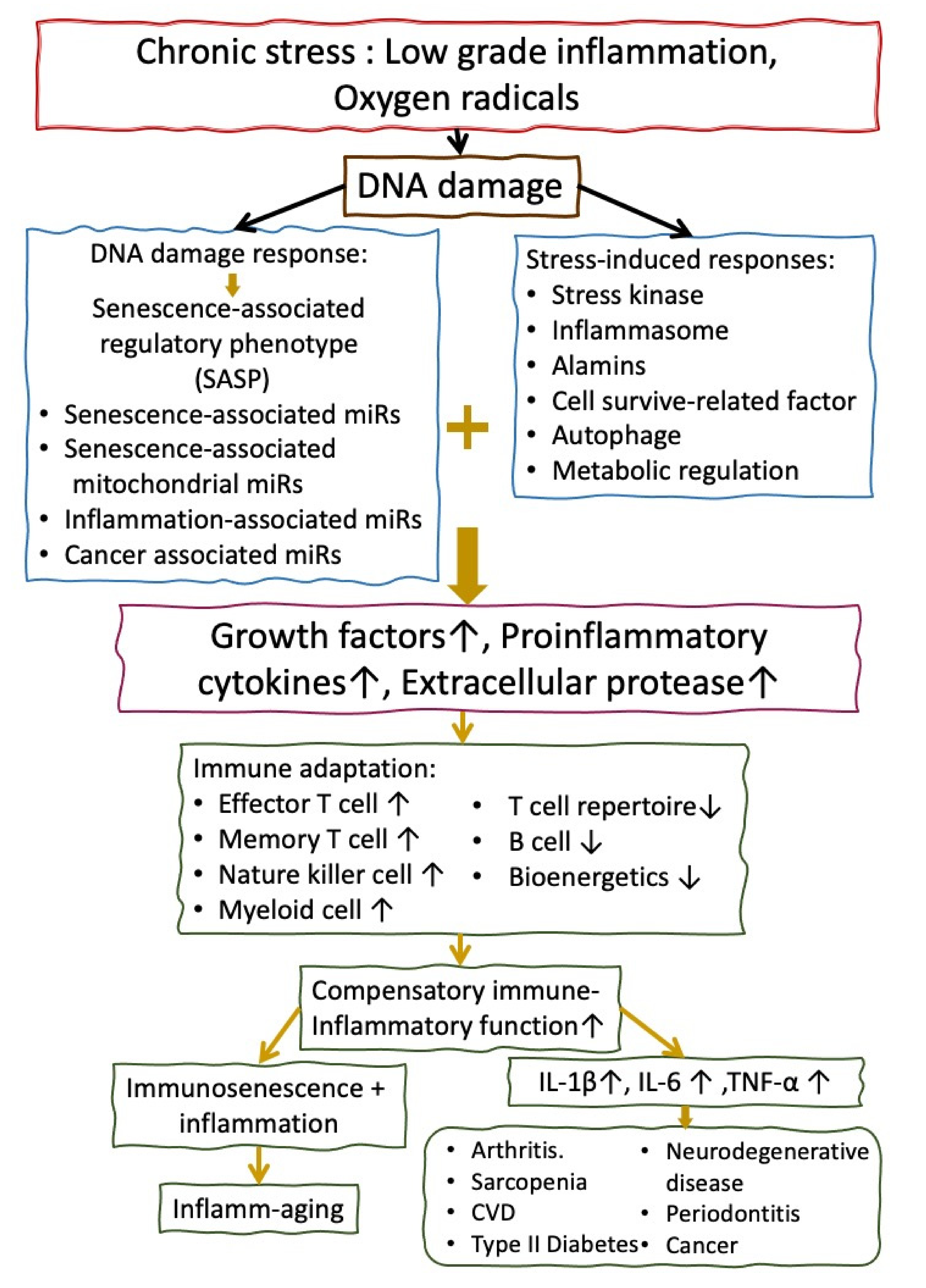
3. The Molecular Basis of Inflamm-aging (Immune Senescence) in the Elderly People
4. The Molecular Basis of Accelerated Inflamm-aging in Patients with SSc
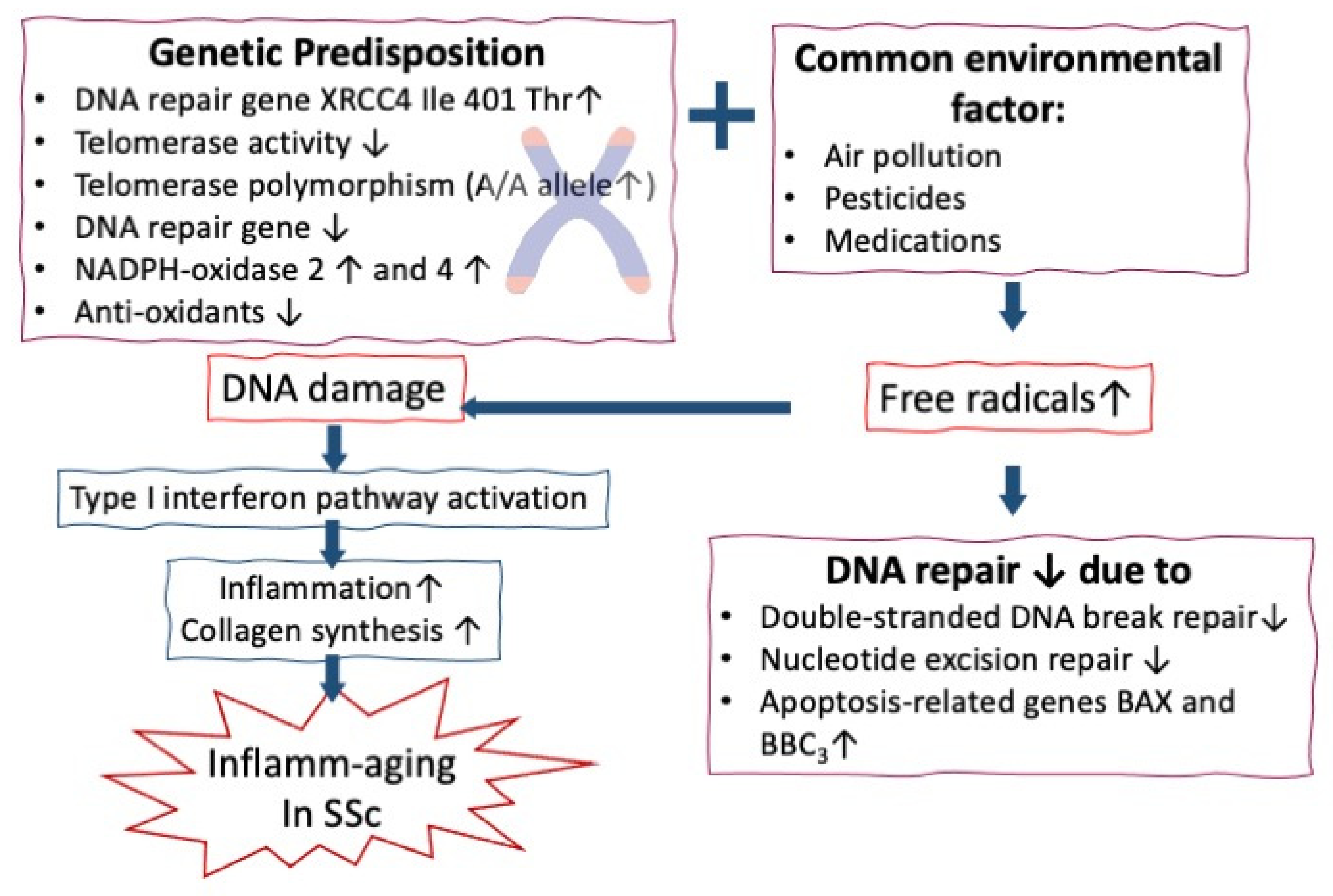
4.1. Role of Increased Chromosome Instability, DNA Damage, and Telomere Shortening in Accerelted Infalamm-aging in SSc
4.2. Roles of Increased Oxidative Stress in Accelerating Inflamm-aging of Patients with SSc
4.3. Immunopathological/Inflammatory Basis of Accelerated Inflamm-aging in Patient with SSc
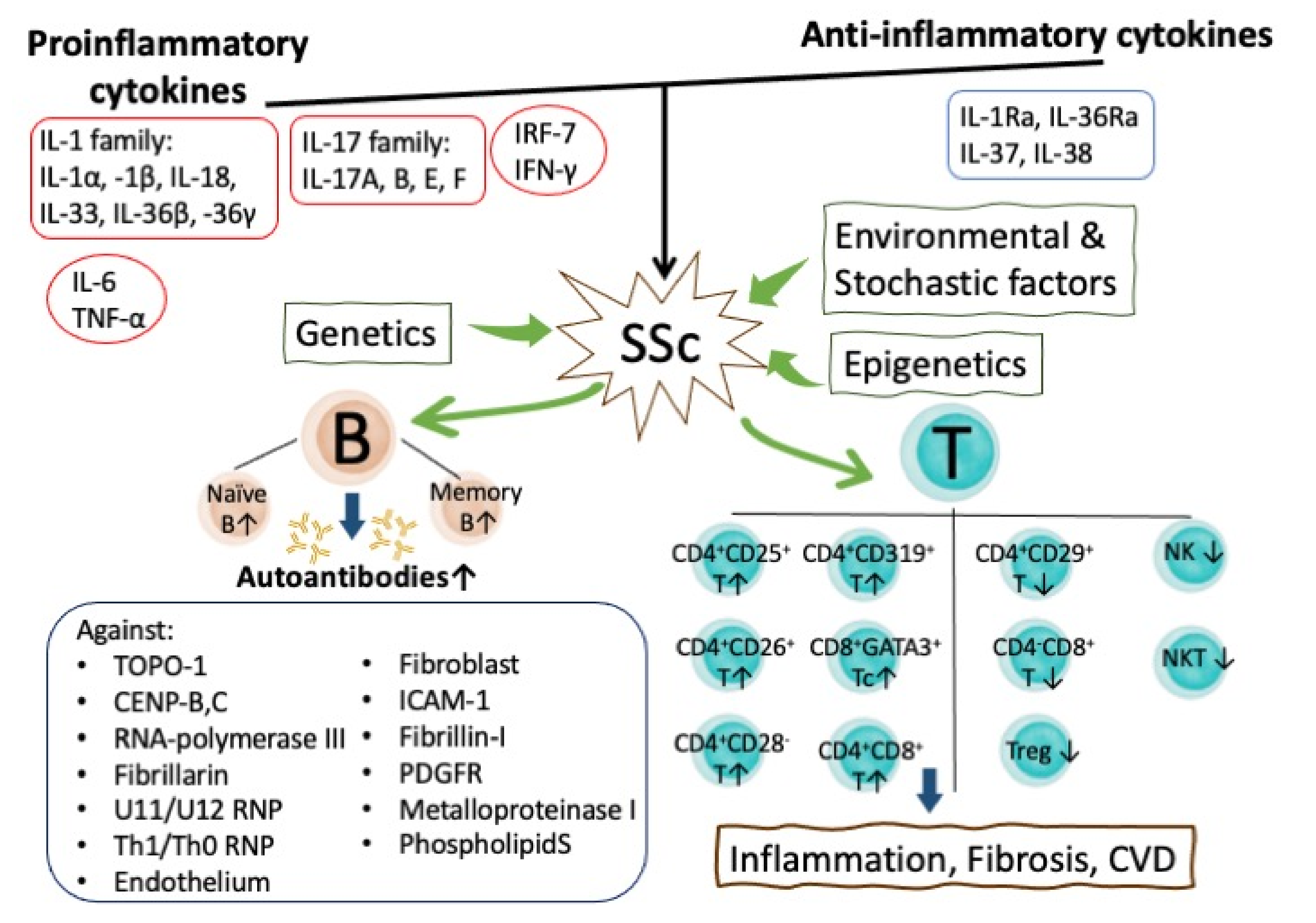
4.3.1. Aberrant Pro-inflammatory Cytokine Expression in Mediating Inflamm-Aging in SSc
4.3.2. Autoantibody-mediated Inflamm-aging in SSc Patients
5. Abnormal Immunobiological Basis of Accelerated Inflamm-aging in SSc Patients
5.1. The Role of Abnormal B Lymphocyte Homeostasis and Functions in the Inflamm-aging of SSc Patients
5.2. The Roles of Abnormal T Lymphocyte Subsets in the Accelerated Inflamm-aging of SSc Patients
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Ciechomska, M.; van Laar, J.; O’Reilly, S. Current frontiers in systemic sclerosis pathogenesis. Exp. Dermatol. 2015, 24, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Mitoma, C.; Mitoma, H.; Tsuji, G.; Chiba, T.; Nakahara, T.; Uchi, H.; Kadono, T. Pathogenesis of systemic sclerosis-current concept and emerging treatments. Immunol. Res. 2017, 65, 790–797. [Google Scholar] [CrossRef]
- Zuo, X.; Zhang, L.; Luo, H.; Li, Y.; Zhu, H. Systematic approach to understanding the pathogenesis of systemic sclerosis. Clin. Genet. 2017, 92, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, M.; Lepri, G.; Damiani, A.; Barsotti, S.; Di Battista, M.; Codullo, V.; Della Rossa, A.; Guiducci, S.; Allanore, Y. One year in review 2020: Systemic sclerosis. Clin. Exp. Rheumatol 2020, 38 (Suppl. 125), 3–17. [Google Scholar]
- Yamamoto, T. Scleroderma--pathophysiology. Eur. J. Dermatol. 2009, 19, 14–24. [Google Scholar] [CrossRef]
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef]
- Yoshizaki, A. Pathogenic roles of B lymphocytes in systemic sclerosis. Immunol. Lett. 2018, 195, 76–82. [Google Scholar] [CrossRef]
- Varga, J.; Abraham, D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J. Clin. Invest. 2007, 117, 557–567. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Hsieh, S.C.; Wu, T.H.; Li, K.J.; Shen, C.Y.; Liao, H.T.; Wu, C.H.; Kuo, Y.M.; Lu, C.S.; Yu, C.L. Pathogenic Roles of Autoantibodies and Aberrant Epigenetic Regulation of Immune and Connective Tissue Cells in the Tissue Fibrosis of Patients with Systemic Sclerosis. Int. J. Mol. Sci 2020, 21, 3069. [Google Scholar] [CrossRef]
- Artlett, C.M. Immunology of systemic sclerosis. Front. Biosci. 2005, 10, 1707–1719. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Role of cellular senescence and NOX4-mediated oxidative stress in systemic sclerosis pathogenesis. Curr. Rheumatol. Rep. 2015, 17, 473. [Google Scholar] [CrossRef] [Green Version]
- Vona, R.; Giovannetti, A.; Gambardella, L.; Malorni, W.; Pietraforte, D.; Straface, E. Oxidative stress in the pathogenesis of systemic scleroderma: An overview. J. Cell Mol. Med. 2018, 22, 3308–3314. [Google Scholar] [CrossRef]
- Abdulle, A.E.; Diercks, G.F.H.; Feelisch, M.; Mulder, D.J.; van Goor, H. The Role of Oxidative Stress in the Development of Systemic Sclerosis Related Vasculopathy. Front. Physiol. 2018, 9, 1177. [Google Scholar] [CrossRef] [Green Version]
- Ames, P.R.J.; Bucci, T.; Merashli, M.; Amaral, M.; Arcaro, A.; Gentile, F.; Nourooz-Zadeh, J.; DelgadoAlves, J. Oxidative/nitrative stress in the pathogenesis of systemic sclerosis: Are antioxidants beneficial? Free Radic. Res. 2018, 52, 1063–1082. [Google Scholar] [CrossRef]
- Svegliati, S.; Spadoni, T.; Moroncini, G.; Gabrielli, A. NADPH oxidase, oxidative stress and fibrosis in systemic sclerosis. Free Radic. Biol. Med. 2018, 125, 90–97. [Google Scholar] [CrossRef]
- Frasca, L.; Lande, R. Toll-like receptors in mediating pathogenesis in systemic sclerosis. Clin. Exp. Immunol. 2020, 201, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, A.; Straface, E.; Rosato, E.; Casciaro, M.; Pioggia, G.; Gangemi, S. Role of Alarmins in the Pathogenesis of Systemic Sclerosis. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Cypiene, A.; Laucevicius, A.; Venalis, A.; Dadoniene, J.; Ryliskyte, L.; Petrulioniene, Z.; Kovaite, M.; Gintautas, J. The impact of systemic sclerosis on arterial wall stiffness parameters and endothelial function. Clin. Rheumatol. 2008, 27, 1517–1522. [Google Scholar] [CrossRef]
- Machida, A.; Funaki, T.; Nishida, K.; Imai, R.I.; Nakaoka, Y.; Seki, S.I.; Baba, Y.I.; Kubo, T.; Yamasaki, N.; Kitaoka, H.; et al. Premature Onset Aortic Stenosis in Systemic Sclerosis: A Report of a Series of Cases. Intern. Med. 2020, 59, 3177–3181. [Google Scholar] [CrossRef]
- Stochmal, A.; Czuwara, J.; Trojanowska, M.; Rudnicka, L. Antinuclear Antibodies in Systemic Sclerosis: An Update. Clin. Rev. Allergy Immunol. 2020, 58, 40–51. [Google Scholar] [CrossRef]
- Emerit, I. Chromosomal breakage in systemic sclerosis and related disorders. Dermatologica 1976, 153, 145–156. [Google Scholar] [CrossRef]
- Dehbi, A.Z.; Radstake, T.R.; Broen, J.C. Accelerated telomere shortening in rheumatic diseases: Cause or consequence? Expert. Rev. Clin. Immunol. 2013, 9, 1193–1204. [Google Scholar] [CrossRef]
- Onishi, A.; Sugiyama, D.; Kumagai, S.; Morinobu, A. Cancer incidence in systemic sclerosis: Meta-analysis of population-based cohort studies. Arthritis Rheum. 2013, 65, 1913–1921. [Google Scholar] [CrossRef]
- Maria, A.T.J.; Partouche, L.; Goulabchand, R.; Riviere, S.; Rozier, P.; Bourgier, C.; Le Quellec, A.; Morel, J.; Noel, D.; Guilpain, P. Intriguing Relationships Between Cancer and Systemic Sclerosis: Role of the Immune System and Other Contributors. Front. Immunol 2018, 9, 3112. [Google Scholar] [CrossRef] [Green Version]
- Fragoulis, G.E.; Daoussis, D.; Pagkopoulou, E.; Garyfallos, A.; Kitas, G.D.; Dimitroulas, T. Cancer risk in systemic sclerosis: Identifying risk and managing high-risk patients. Expert Rev. Clin. Immunol. 2020, 16, 1105–1113. [Google Scholar] [CrossRef]
- Sandhofer, M.; Fritz, J.; Altmann, H. Scleroderma, an ageing process? I. Clinical and immunological aspects (author’s transl). Aktuelle Gerontol 1977, 7, 645–651. [Google Scholar]
- Goto, M. Werner’s syndrome: From clinics to genetics. Clin. Exp. Rheumatol. 2000, 18, 760–766. [Google Scholar]
- De Martinis, M.; Ciccarelli, F.; Sirufo, M.M.; Ginaldi, L. An overview of environmental risk factors in systemic sclerosis. Expert Rev. Clin. Immunol. 2016, 12, 465–478. [Google Scholar] [CrossRef]
- McCormic, Z.D.; Khuder, S.S.; Aryal, B.K.; Ames, A.L.; Khuder, S.A. Occupational silica exposure as a risk factor for scleroderma: A meta-analysis. Int. Arch. Occup. Environ. Health 2010, 83, 763–769. [Google Scholar] [CrossRef]
- Angiolilli, C.; Marut, W.; van der Kroef, M.; Chouri, E.; Reedquist, K.A.; Radstake, T. New insights into the genetics and epigenetics of systemic sclerosis. Nat. Rev. Rheumatol. 2018, 14, 657–673. [Google Scholar] [CrossRef]
- Feghali-Bostwick, C.; Medsger, T.A., Jr.; Wright, T.M. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003, 48, 1956–1963. [Google Scholar] [CrossRef]
- Aho, K.; Koskenvuo, M.; Tuominen, J.; Kaprio, J. Occurrence of rheumatoid arthritis in a nationwide series of twins. J. Rheumatol. 1986, 13, 899–902. [Google Scholar] [PubMed]
- Bammer, H.; Schaltenbrand, G.; Solcher, H. Examinations of twins in multiple sclerosis. Dtsch. Z. Nervenheilkd 1960, 181, 261–279. [Google Scholar]
- Bhaskar, L.; Nagaraju, G.P. Clinical and Immunogenetic Aspects of Systemic Lupus Erythematosus. Crit. Rev. Immunol. 2019, 39, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.G.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: Genetics, epigenetics, and environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef]
- Katsumoto, T.R.; Whitfield, M.L.; Connolly, M.K. The pathogenesis of systemic sclerosis. Annu. Rev. Pathol. 2011, 6, 509–537. [Google Scholar] [CrossRef]
- Gourh, P.; Safran, S.A.; Alexander, T.; Boyden, S.E.; Morgan, N.D.; Shah, A.A.; Mayes, M.D.; Doumatey, A.; Bentley, A.R.; Shriner, D.; et al. HLA and autoantibodies define scleroderma subtypes and risk in African and European Americans and suggest a role for molecular mimicry. Proc. Natl. Acad. Sci. USA 2020, 117, 552–562. [Google Scholar] [CrossRef] [Green Version]
- Chairta, P.; Nicolaou, P.; Christodoulou, K. Genomic and genetic studies of systemic sclerosis: A systematic review. Hum. Immunol. 2017, 78, 153–165. [Google Scholar] [CrossRef]
- Mayes, M.D.; Bossini-Castillo, L.; Gorlova, O.; Martin, J.E.; Zhou, X.; Chen, W.V.; Assassi, S.; Ying, J.; Tan, F.K.; Arnett, F.C.; et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am. J. Hum. Genet. 2014, 94, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Guo, X.; Yi, L.; Guo, G.; Tu, W.; Wu, W.; Yang, L.; Xiao, R.; Li, Y.; Chu, H.; et al. Association of HLA-DPB1 with scleroderma and its clinical features in Chinese population. PLoS ONE 2014, 9, e87363. [Google Scholar] [CrossRef] [PubMed]
- Vigone, B.; Santaniello, A.; Marchini, M.; Montanelli, G.; Caronni, M.; Severino, A.; Beretta, L. Role of class II human leucocyte antigens in the progression from early to definite systemic sclerosis. Rheumatology 2015, 54, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Lambert, N.C.; Distler, O.; Muller-Ladner, U.; Tylee, T.S.; Furst, D.E.; Nelson, J.L. HLA-DQA1*0501 is associated with diffuse systemic sclerosis in Caucasian men. Arthritis Rheum. 2000, 43, 2005–2010. [Google Scholar] [CrossRef]
- Stevens, A.M.; Kanaan, S.B.; Torok, K.S.; Medsger, T.A.; Mayes, M.D.; Reveille, J.D.; Klein-Gitelman, M.; Reed, A.M.; Lee, T.; Li, S.C.; et al. Brief Report: HLA-DRB1, DQA1, and DQB1 in Juvenile-Onset Systemic Sclerosis. Arthritis Rheumatol. 2016, 68, 2772–2777. [Google Scholar] [CrossRef]
- Lopez-Isac, E.; Acosta-Herrera, M.; Kerick, M.; Assassi, S.; Satpathy, A.T.; Granja, J.; Mumbach, M.R.; Beretta, L.; Simeon, C.P.; Carreira, P.; et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 2019, 10, 4955. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Chou, C.; Lima, M.; Zhou, D.; Zhou, X. Systemic Sclerosis is a Complex Disease Associated Mainly with Immune Regulatory and Inflammatory Genes. Open Rheumatol. J. 2014, 8, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Ota, Y.; Kuwana, M. Updates on genetics in systemic sclerosis. Inflamm. Regen. 2021, 41, 17. [Google Scholar] [CrossRef]
- Beretta, L.; Barturen, G.; Vigone, B.; Bellocchi, C.; Hunzelmann, N.; De Langhe, E.; Cervera, R.; Gerosa, M.; Kovacs, L.; Ortega Castro, R.; et al. Genome-wide whole blood transcriptome profiling in a large European cohort of systemic sclerosis patients. Ann. Rheum. Dis. 2020, 79, 1218–1226. [Google Scholar] [CrossRef]
- Sun, Y.H.; Xie, M.; Wu, S.D.; Zhang, J.; Huang, C.Z. Identification and Interaction Analysis of Key Genes and MicroRNAs in Systemic Sclerosis by Bioinformatics Approaches. Curr. Med. Sci. 2019, 39, 645–652. [Google Scholar] [CrossRef]
- Jafarinejad-Farsangi, S.; Gharibdoost, F.; Farazmand, A.; Kavosi, H.; Jamshidi, A.; Karimizadeh, E.; Noorbakhsh, F.; Mahmoudi, M. MicroRNA-21 and microRNA-29a modulate the expression of collagen in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity 2019, 52, 108–116. [Google Scholar] [CrossRef]
- Erasmus, L.D. Scleroderma in goldminers on the Witwatersrand with particular reference to pulmonary manifestations. S. Afr. J. Lab. Clin. Med. 1957, 3, 209–231. [Google Scholar] [PubMed]
- Sluis-Cremer, G.K.; Hessel, P.A.; Nizdo, E.H.; Churchill, A.R.; Zeiss, E.A. Silica, silicosis, and progressive systemic sclerosis. Br. J. Ind. Med. 1985, 42, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.M.; Udnoon, S.; Watkins, J.; Walker, A.E.; Darke, C.S. Immunological mechanisms in the pathogenesis of vinyl chloride disease. Br. Med. J. 1976, 1, 936–938. [Google Scholar] [CrossRef] [Green Version]
- Walder, G. Solvents and Scleroderma. Lancet 1965, 2, 436–437. [Google Scholar] [CrossRef]
- Yamakage, A.; Ishikawa, H.; Saito, Y.; Hattori, A. Occupational scleroderma-like disorder occurring in men engaged in the polymerization of epoxy resins. Dermatologica 1980, 161, 33–44. [Google Scholar] [CrossRef]
- Diaz de Rojas, F.; Castro Garcia, M.; Abaitua Borda, I.; Alonso Gordo, J.M.; Posada de la Paz, M.; Kilbourne, E.M.; Tabuenca Oliver, J.M. The association of oil ingestion with toxic oil syndrome in two convents. Am. J. Epidemiol. 1987, 125, 907–911. [Google Scholar] [CrossRef]
- Finch, W.R.; Rodnan, G.P.; Buckingham, R.B.; Prince, R.K.; Winkelstein, A. Bleomycin-induced scleroderma. J. Rheumatol. 1980, 7, 651–659. [Google Scholar]
- Marie, I. Systemic sclerosis and exposure to heavy metals. Autoimmun. Rev. 2019, 18, 62–72. [Google Scholar] [CrossRef]
- Roberts-Thomson, P.J.; Walker, J.G. Stochastic processes in the aetiopathogenesis of scleroderma. Intern. Med. J. 2012, 42, 235–242. [Google Scholar] [CrossRef]
- Lu, Q. The critical importance of epigenetics in autoimmunity. J. Autoimmun. 2013, 41, 1–5. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Zhang, X. CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochem. Biophys. Res. Commun. 2009, 383, 421–425. [Google Scholar] [CrossRef]
- Tajima, S.; Suetake, I.; Takeshita, K.; Nakagawa, A.; Kimura, H. Domain Structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA Methyltransferases. Adv. Exp. Med. Biol. 2016, 945, 63–86. [Google Scholar]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Budden, D.M.; Hurley, D.G.; Cursons, J.; Markham, J.F.; Davis, M.J.; Crampin, E.J. Predicting expression: The complementary power of histone modification and transcription factor binding data. Epigenetics Chromatin 2014, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Lei, W.; Luo, Y.; Lei, W.; Luo, Y.; Yan, K.; Zhao, S.; Li, Y.; Qiu, X.; Zhou, Y.; Long, H.; et al. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand. J. Rheumatol. 2009, 38, 369–374. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Wang, Q.; Sun, X.H.; Liu, R.Z.; Shu, Y.; Kanekura, T.; Huang, J.H.; Li, Y.P.; Wang, J.C.; Zhao, M.; et al. DNA hypermethylation of the forkhead box protein 3 (FOXP3) promoter in CD4+ T cells of patients with systemic sclerosis. Br. J. Dermatol. 2014, 171, 39–47. [Google Scholar] [CrossRef]
- Almanzar, G.; Klein, M.; Schmalzing, M.; Hilligardt, D.; El Hajj, N.; Kneitz, H.; Wild, V.; Rosenwald, A.; Benoit, S.; Hamm, H.; et al. Disease Manifestation and Inflammatory Activity as Modulators of Th17/Treg Balance and RORC/FoxP3 Methylation in Systemic Sclerosis. Int. Arch. Allergy Immunol. 2016, 171, 141–154. [Google Scholar] [CrossRef]
- Coit, P.; Schollaert, K.L.; Mirizio, E.M.; Torok, K.S.; Sawalha, A.H. DNA methylation patterns in juvenile systemic sclerosis and localized scleroderma. Clin. Immunol. 2021, 228, 108756. [Google Scholar] [CrossRef]
- Baker Frost, D.; da Silveira, W.; Hazard, E.S.; Atanelishvili, I.; Wilson, R.C.; Flume, J.; Day, K.L.; Oates, J.C.; Bogatkevich, G.S.; Feghali-Bostwick, C.; et al. Differential DNA Methylation Landscape in Skin Fibroblasts from African Americans with Systemic Sclerosis. Genes 2021, 12, 129. [Google Scholar] [CrossRef]
- Rezaei, R.; Mahmoudi, M.; Gharibdoost, F.; Kavosi, H.; Dashti, N.; Imeni, V.; Jamshidi, A.; Aslani, S.; Mostafaei, S.; Vodjgani, M. IRF7 gene expression profile and methylation of its promoter region in patients with systemic sclerosis. Int. J. Rheum Dis. 2017, 20, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Pu, W.; Wang, L.; Jiang, S.; Zhou, X.; Tu, W.; Yu, L.; Zhang, J.; Guo, S.; Liu, Q.; et al. Genome-Wide DNA Methylation Analysis in Systemic Sclerosis Reveals Hypomethylation of IFN-Associated Genes in CD4(+) and CD8(+) T Cells. J. Invest. Dermatol. 2018, 138, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Pu, W.; Guo, S.; Jin, L.; He, D.; Wang, J. Genome-Wide DNA Methylation Profiles Reveal Common Epigenetic Patterns of Interferon-Related Genes in Multiple Autoimmune Diseases. Front. Genet. 2019, 10, 223. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Luo, Y.; Yin, Y.; Wang, Q.; Li, Y.; Kanekura, T.; Wang, J.; Liang, G.; Zhao, M.; et al. Aberrant histone modification in peripheral blood B cells from patients with systemic sclerosis. Clin. Immunol. 2013, 149, 46–54. [Google Scholar] [CrossRef]
- Kramer, M.; Dees, C.; Huang, J.; Schlottmann, I.; Palumbo-Zerr, K.; Zerr, P.; Gelse, K.; Beyer, C.; Distler, A.; Marquez, V.E.; et al. Inhibition of H3K27 histone trimethylation activates fibroblasts and induces fibrosis. Ann. Rheum. Dis. 2013, 72, 614–620. [Google Scholar] [CrossRef]
- Ciechomska, M.; O’Reilly, S.; Przyborski, S.; Oakley, F.; Bogunia-Kubik, K.; van Laar, J.M. Histone Demethylation and Toll-like Receptor 8-Dependent Cross-Talk in Monocytes Promotes Transdifferentiation of Fibroblasts in Systemic Sclerosis Via Fra-2. Arthritis Rheumatol. 2016, 68, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Van der Kroef, M.; Castellucci, M.; Mokry, M.; Cossu, M.; Garonzi, M.; Bossini-Castillo, L.M.; Chouri, E.; Wichers, C.G.K.; Beretta, L.; Trombetta, E.; et al. Histone modifications underlie monocyte dysregulation in patients with systemic sclerosis, underlining the treatment potential of epigenetic targeting. Ann. Rheum. Dis. 2019, 78, 529–538. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, Y.; Shu, Y.; Lu, Q.; Xiao, R. Epigenetic mechanisms: An emerging role in pathogenesis and its therapeutic potential in systemic sclerosis. Int. J. Biochem. Cell Biol. 2015, 67, 92–100. [Google Scholar] [CrossRef]
- Aslani, S.; Sobhani, S.; Gharibdoost, F.; Jamshidi, A.; Mahmoudi, M. Epigenetics and pathogenesis of systemic sclerosis; the ins and outs. Hum. Immunol. 2018, 79, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Walczyk, M.; Paradowska-Gorycka, A.; Olesinska, M. Epigenetics: The Future Direction in Systemic Sclerosis. Scand. J. Immunol. 2017, 86, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Salvioli, S.; Monti, D.; Lanzarini, C.; Conte, M.; Pirazzini, C.; Bacalini, M.G.; Garagnani, P.; Giuliani, C.; Fontanesi, E.; Ostan, R.; et al. Immune system, cell senescence, aging and longevity--inflamm-aging reappraised. Curr. Pharm. Des. 2013, 19, 1675–1679. [Google Scholar]
- Franceschi, C.; Bonafe, M.; Valensin, S. Human immunosenescence: The prevailing of innate immunity, the failing of clonotypic immunity, and the filling of immunological space. Vaccine 2000, 18, 1717–1720. [Google Scholar] [CrossRef]
- Larbi, A.; Franceschi, C.; Mazzatti, D.; Solana, R.; Wikby, A.; Pawelec, G. Aging of the immune system as a prognostic factor for human longevity. Physiology 2008, 23, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Monti, D.; Ostan, R.; Borelli, V.; Castellani, G.; Franceschi, C. Inflammaging and human longevity in the omics era. Mech. Ageing Dev. 2017, 165 (Pt B), 129–138. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.E. Stress, glucocorticoids and ageing of the immune system. Stress 2005, 8, 69–83. [Google Scholar] [CrossRef]
- Epel, E.S. Psychological and metabolic stress: A recipe for accelerated cellular aging? Hormones 2009, 8, 7–22. [Google Scholar] [CrossRef]
- Miller, G.E.; Murphy, M.L.; Cashman, R.; Ma, R.; Ma, J.; Arevalo, J.M.; Kobor, M.S.; Cole, S.W. Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav. Immun. 2014, 41, 191–199. [Google Scholar] [CrossRef] [Green Version]
- De Punder, K.; Heim, C.; Wadhwa, P.D.; Entringer, S. Stress and immunosenescence: The role of telomerase. Psychoneuroendocrinology 2019, 101, 87–100. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Monsurro, V.; Salvioli, S.; Capri, M.; Procopio, A.D.; Franceschi, C. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res. Rev. 2013, 12, 1056–1068. [Google Scholar] [CrossRef]
- Catana, C.S.; Calin, G.A. Berindan-Neagoe, I. Inflamma-miRs in Aging and Breast Cancer: Are They Reliable Players? Front. Med. 2015, 2, 85. [Google Scholar] [CrossRef] [Green Version]
- Teodori, L.; Petrignani, I.; Giuliani, A.; Prattichizzo, F.; Gurau, F.; Matacchione, G.; Olivieri, F.; Coppari, S.; Albertini, M.C. Inflamm-aging microRNAs may integrate signals from food and gut microbiota by modulating common signalling pathways. Mech. Ageing Dev. 2019, 182, 111127. [Google Scholar] [CrossRef]
- Terlecki-Zaniewicz, L.; Lammermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmullner, R.; Dellago, H.; Skalicky, S.; Pum, D.; et al. Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging 2018, 10, 1103–1132. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; Micolucci, L.; Cricca, M.; De Carolis, S.; Mensa, E.; Ceriello, A.; Procopio, A.D.; Bonafe, M.; Olivieri, F. Exosome-based immunomodulation during aging: A nano-perspective on inflamm-aging. Mech. Ageing Dev. 2017, 168, 44–53. [Google Scholar] [CrossRef]
- Rippo, M.R.; Olivieri, F.; Monsurro, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014, 56, 154–163. [Google Scholar] [CrossRef]
- Giuliani, A.; Prattichizzo, F.; Micolucci, L.; Ceriello, A.; Procopio, A.D.; Rippo, M.R. Mitochondrial (Dys) Function in Inflammaging: Do MitomiRs Influence the Energetic, Oxidative, and Inflammatory Status of Senescent Cells? Mediators. Inflamm. 2017, 2017, 2309034. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409. [Google Scholar] [CrossRef] [Green Version]
- Malaquin, N.; Martinez, A.; Rodier, F. Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. Exp. Gerontol. 2016, 82, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Wolff, D.J.; Needleman, B.W.; Wasserman, S.S.; Schwartz, S. Spontaneous and clastogen induced chromosomal breakage in scleroderma. J. Rheumatol. 1991, 18, 837–840. [Google Scholar]
- Takeuchi, F.; Nakano, K.; Yamada, H.; Kosuge, E.; Hirai, M.; Maeda, H.; Moroi, Y. Chromosome abnormalities in peripheral lymphocytes from patients with progressive systemic sclerosis. Rheumatol. Int. 1993, 12, 243–246. [Google Scholar] [CrossRef]
- Roberts-Thomson, P.J.; Male, D.A.; Walker, J.G.; Cox, S.R.; Shen, X.; Smith, M.D.; Ahern, M.J.; Turner, D.R. Genomic instability in scleroderma. Asian Pac. J. Allergy Immunol. 2004, 22, 153–158. [Google Scholar]
- Artlett, C.M.; Black, C.M.; Briggs, D.C.; Stevens, C.O.; Welsh, K.I. Telomere reduction in scleroderma patients: A possible cause for chromosomal instability. Br. J. Rheumatol. 1996, 35, 732–737. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, A.; Brouilette, S.W.; Lamb, K.; Radhakrishnan, K.; McGlynn, L.; Chee, M.M.; Parkinson, E.K.; Freeman, D.; Madhok, R.; Shiels, P.G. Association of increased telomere lengths in limited scleroderma, with a lack of age-related telomere erosion. Ann. Rheum. Dis. 2008, 67, 1780–1782. [Google Scholar] [CrossRef]
- Lakota, K.; Hanumanthu, V.S.; Agrawal, R.; Carns, M.; Armanios, M.; Varga, J. Short lymphocyte, but not granulocyte, telomere length in a subset of patients with systemic sclerosis. Ann. Rheum. Dis. 2019, 78, 1142–1144. [Google Scholar] [CrossRef]
- Sauer, I.; Ries, W.; Mittag, M.; Haustein, U.F. Biological age in patients with progressive scleroderma. Z. Gerontol. Geriatr. 1996, 29, 223–232. [Google Scholar]
- Ohtsuka, T.; Yamakage, A.; Yamazaki, S. The polymorphism of telomerase RNA component gene in patients with systemic sclerosis. Br. J. Dermatol 2002, 147, 250–254. [Google Scholar] [CrossRef]
- Tarhan, F.; Vural, F.; Kosova, B.; Aksu, K.; Cogulu, O.; Keser, G.; Gunduz, C.; Tombuloglu, M.; Oder, G.; Karaca, E.; et al. Telomerase activity in connective tissue diseases: Elevated in rheumatoid arthritis, but markedly decreased in systemic sclerosis. Rheumatol. Int. 2008, 28, 579–583. [Google Scholar] [CrossRef]
- Palomino, G.M.; Bassi, C.L.; Wastowski, I.J.; Xavier, D.J.; Lucisano-Valim, Y.M.; Crispim, J.C.; Rassi, D.M.; Marques-Neto, J.F.; Sakamoto-Hojo, E.T.; Moreau, P.; et al. Patients with systemic sclerosis present increased DNA damage differentially associated with DNA repair gene polymorphisms. J. Rheumatol. 2014, 41, 458–465. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Pappa, M.; Ntouros, P.A.; Nezos, A.; Mavragani, C.P.; Souliotis, V.L.; Sfikakis, P.P. Association Between DNA Damage Response, Fibrosis and Type I Interferon Signature in Systemic Sclerosis. Front. Immunol. 2020, 11, 582401. [Google Scholar] [CrossRef]
- Savas, E.; Aksoy, N.; Pehlivan, Y.; Sayiner, Z.A.; Ozturk, Z.A.; Tabur, S.; Orkmez, M.; Onat, A.M. Evaluation of oxidant and antioxidant status and relation with prolidase in systemic sclerosis. Wien. Klin. Wochenschr. 2014, 126, 341–346. [Google Scholar] [CrossRef]
- Sambo, P.; Jannino, L.; Candela, M.; Salvi, A.; Donini, M.; Dusi, S.; Luchetti, M.M.; Gabrielli, A. Monocytes of patients wiht systemic sclerosis (scleroderma spontaneously release in vitro increased amounts of superoxide anion. J. Invest. Dermatol. 1999, 112, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Allanore, Y.; Borderie, D.; Lemarechal, H.; Ekindjian, O.G.; Kahan, A. Acute and sustained effects of dihydropyridine-type calcium channel antagonists on oxidative stress in systemic sclerosis. Am. J. Med. 2004, 116, 595–600. [Google Scholar] [CrossRef]
- Servettaz, A.; Guilpain, P.; Goulvestre, C.; Chereau, C.; Hercend, C.; Nicco, C.; Guillevin, L.; Weill, B.; Mouthon, L.; Batteux, F. Radical oxygen species production induced by advanced oxidation protein products predicts clinical evolution and response to treatment in systemic sclerosis. Ann. Rheum. Dis. 2007, 66, 1202–1209. [Google Scholar] [CrossRef] [Green Version]
- Sambo, P.; Baroni, S.S.; Luchetti, M.; Paroncini, P.; Dusi, S.; Orlandini, G.; Gabrielli, A. Oxidative stress in scleroderma: Maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum. 2001, 44, 2653–2664. [Google Scholar] [CrossRef]
- Ogawa, F.; Shimizu, K.; Muroi, E.; Hara, T.; Sato, S. Increasing levels of serum antioxidant status, total antioxidant power, in systemic sclerosis. Clin. Rheumatol. 2011, 30, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.M.; Tanner, S.B.; Awad, J.A.; Roberts, L.J., II; Morrow, J.D. Evidence of free radical-mediated injury (isoprostane overproduction) in scleroderma. Arthritis Rheum. 1996, 39, 1146–1150. [Google Scholar] [CrossRef]
- Ogawa, F.; Shimizu, K.; Muroi, E.; Hara, T.; Hasegawa, M.; Takehara, K.; Sato, S. Serum levels of 8-isoprostane, a marker of oxidative stress, are elevated in patients with systemic sclerosis. Rheumatology 2006, 45, 815–818. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Ogawa, F.; Akiyama, Y.; Muroi, E.; Yoshizaki, A.; Iwata, Y.; Komura, K.; Bae, S.; Sato, S. Increased serum levels of N(epsilon)-(hexanoyl)lysine, a new marker of oxidative stress, in systemic sclerosis. J. Rheumatol. 2008, 35, 2214–2219. [Google Scholar] [CrossRef]
- Ogawa, F.; Shimizu, K.; Hara, T.; Muroi, E.; Hasegawa, M.; Takehara, K.; Sato, S. Serum levels of heat shock protein 70, a biomarker of cellular stress, are elevated in patients with systemic sclerosis: Association with fibrosis and vascular damage. Clin. Exp. Rheumatol. 2008, 26, 659–662. [Google Scholar]
- Boin, F.; Erre, G.L.; Posadino, A.M.; Cossu, A.; Giordo, R.; Spinetti, G.; Passiu, G.; Emanueli, C.; Pintus, G. Oxidative stress-dependent activation of collagen synthesis is induced in human pulmonary smooth muscle cells by sera from patients with scleroderma-associated pulmonary hypertension. Orphanet. J. Rare. Dis. 2014, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Giordo, R.; Thuan, D.T.B.; Posadino, A.M.; Cossu, A.; Zinellu, A.; Erre, G.L.; Pintus, G. Iloprost Attenuates Oxidative Stress-Dependent Activation of Collagen Synthesis Induced by Sera from Scleroderma Patients in Human Pulmonary Microvascular Endothelial Cells. Molecules 2021, 26, 4729. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Makul, A.; Jimenez, S.A. Increased expression of NAPDH oxidase 4 in systemic sclerosis dermal fibroblasts: Regulation by transforming growth factor beta. Arthritis Rheumatol. 2015, 67, 2749–2758. [Google Scholar] [CrossRef] [Green Version]
- Spadoni, T.; Svegliati Baroni, S.; Amico, D.; Albani, L.; Moroncini, G.; Avvedimento, E.V.; Gabrielli, A. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol. 2015, 67, 1611–1622. [Google Scholar] [CrossRef]
- Amico, D.; Spadoni, T.; Rovinelli, M.; Serafini, M.; D’Amico, G.; Campelli, N.; Svegliati Baroni, S.; Gabrielli, A. Intracellular free radical production by peripheral blood T lymphocytes from patients with systemic sclerosis: Role of NADPH oxidase and ERK1/2. Arthritis Res. Ther. 2015, 17, 68. [Google Scholar] [CrossRef] [Green Version]
- Doridot, L.; Jeljeli, M.; Chene, C.; Batteux, F. Implication of oxidative stress in the pathogenesis of systemic sclerosis via inflammation, autoimmunity and fibrosis. Redox. Biol. 2019, 25, 101122. [Google Scholar] [CrossRef]
- Mancini, O.K.; Acevedo, M.; Fazez, N.; Cuillerier, A.; Ruiz, A.F.; Huynh, D.N.; Burelle, Y.; Ferbeyre, G.; Baron, M.; Servant, M.J. Oxidative stress-induced senescence mediates inflammatory and fibrotic phenotypes in fibroblasts from systemic sclerosis patients. Rheumatology 2021, keab477. [Google Scholar] [CrossRef]
- Bueno, M.; Papazoglou, A.; Valenzi, E.; Rojas, M.; Lafyatis, R.; Mora, A.L. Mitochondria, Aging, and Cellular Senescence: Implications for Scleroderma. Curr. Rheumatol. Rep. 2020, 22, 37. [Google Scholar] [CrossRef]
- Movassaghi, S.; Jafari, S.; Falahati, K.; Ataei, M.; Sanati, M.H.; Jadali, Z. Quantification of mitochondrial DNA damage and copy number in circulating blood of patients with systemic sclerosis by a qPCR-based assay. An. Bras. Dermatol. 2020, 95, 314–319. [Google Scholar] [CrossRef]
- Wyman, A.E.; Atamas, S.P. Sirtuins and Accelerated Aging in Scleroderma. Curr. Rheumatol. Rep. 2018, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Bruunsgaard, H.; Pedersen, M.; Pedersen, B.K. Aging and proinflammatory cytokines. Curr. Opin. Hematol. 2001, 8, 131–136. [Google Scholar] [CrossRef]
- Giunta, S. Exploring the complex relations between inflammation and aging (inflamm-aging): Anti-inflamm-aging remodelling of inflamm- aging, from robustness to frailty. Inflamm. Res. 2008, 57, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Verschoor, C.P.; Loukov, D.; Naidoo, A.; Puchta, A.; Johnstone, J.; Millar, J.; Lelic, A.; Novakowski, K.E.; Dorrington, M.G.; Loeb, M.; et al. Circulating TNF and mitochondrial DNA are major determinants of neutrophil phenotype in the advanced-age, frail elderly. Mol. Immunol. 2015, 65, 148–156. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujimoto, M.; Kikuchi, K.; Takehara, K. Elevated serum tumor necrosis factor-alpha levels in patients with systemic sclerosis: Association with pulmonary fibrosis. J. Rheumatol. 1997, 24, 663–665. [Google Scholar]
- Pehlivan, Y.; Onat, A.M.; Ceylan, N.; Turkbeyler, I.H.; Buyukhatipoglu, H.; Comez, G.; Babacan, T.; Tarakcioglu, M. Serum leptin, resistin and TNF-alpha levels in patients with systemic sclerosis: The role of adipokines in scleroderma. Int. J. Rheum. Dis. 2012, 15, 374–379. [Google Scholar] [CrossRef]
- Murdaca, G.; Spano, F.; Contatore, M.; Guastalla, A.; Puppo, F. Potential use of TNF-alpha inhibitors in systemic sclerosis. Immunotherapy 2014, 6, 283–289. [Google Scholar] [CrossRef]
- Pandey, J.P.; Takeuchi, F. TNF-alpha and TNF-beta gene polymorphisms in systemic sclerosis. Hum. Immunol. 1999, 60, 1128–1130. [Google Scholar] [CrossRef]
- Lomeli-Nieto, J.A.; Munoz-Valle, J.F.; Banos-Hernandez, C.J.; Navarro-Zarza, J.E.; Ramirez-Duenas, M.G.; Sanchez-Hernandez, P.E.; Machado-Sulbaran, A.C.; Parra-Rojas, I.; Garcia-Chagollan, M.; Hernandez-Bello, J. TNFA -308G>A and -238G>A polymorphisms and risk to systemic sclerosis: Impact on TNF-alpha serum levels, TNFA mRNA expression, and autoantibodies. Clin. Exp. Med. 2019, 19, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujimoto, M.; Kikuchi, K.; Takehara, K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J. Rheumatol. 1997, 24, 328–332. [Google Scholar]
- Gourh, P.; Arnett, F.C.; Assassi, S.; Tan, F.K.; Huang, M.; Diekman, L.; Mayes, M.D.; Reveille, J.D.; Agarwal, S.K. Plasma cytokine profiles in systemic sclerosis: Associations with autoantibody subsets and clinical manifestations. Arthritis. Res. Ther. 2009, 11, R147. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.; Arend, W.; Sims, J.; Smith, D.; Blumberg, H.; O’Neill, L.; Goldbach-Mansky, R.; Pizarro, T.; Hoffman, H.; Bufler, P.; et al. IL-1 family nomenclature. Nat. Immunol. 2010, 11, 973. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Mu, R.; Wei, X. The Roles of IL-1 Family Cytokines in the Pathogenesis of Systemic Sclerosis. Front. Immunol. 2019, 10, 2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, G.; Cavalli, G.; Campochiaro, C.; Tresoldi, M.; Dagna, L. Myocarditis: An Interleukin-1-Mediated Disease? Front. Immunol. 2018, 9, 1335. [Google Scholar] [CrossRef] [Green Version]
- De Luca, G.; Cavalli, G.; Campochiaro, C.; Bruni, C.; Tomelleri, A.; Dagna, L.; Matucci-Cerinic, M. Interleukin-1 and Systemic Sclerosis: Getting to the Heart of Cardiac Involvement. Front. Immunol. 2021, 12, 653950. [Google Scholar] [CrossRef]
- Hasegawa, M.; Sato, S.; Fujimoto, M.; Ihn, H.; Kikuchi, K.; Takehara, K. Serum levels of interleukin 6 (IL-6), oncostatin M, soluble IL-6 receptor, and soluble gp130 in patients with systemic sclerosis. J. Rheumatol. 1998, 25, 308–313. [Google Scholar]
- Ohtsuka, T. Serum interleukin-6 level is reflected in elevated high-sensitivity C-reactive protein level in patients with systemic sclerosis. J. Dermatol. 2010, 37, 801–806. [Google Scholar] [CrossRef]
- Denton, C.P.; Ong, V.H.; Xu, S.; Chen-Harris, H.; Modrusan, Z.; Lafyatis, R.; Khanna, D.; Jahreis, A.; Siegel, J.; Sornasse, T. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: Insights from the faSScinate clinical trial in systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1362–1371. [Google Scholar] [CrossRef]
- Shima, Y. The benefits and prospects of interleukin-6 inhibitor on systemic sclerosis. Mod. Rheumatol. 2019, 29, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Iorio, G.C.; Ammendolia, A.; Marotta, N.; Ricardi, U.; de Sire, A. A bond between rheumatic diseases and cancer in the elderly: The interleukin-6 pathway. Int. J. Rheum. Dis. 2021, 24, 1317–1320. [Google Scholar] [CrossRef]
- Brembilla, N.C.; Chizzolini, C. T cell abnormalities in systemic sclerosis with a focus on Th17 cells. Eur. Cytokine. Netw. 2012, 23, 128–139. [Google Scholar] [CrossRef]
- Lei, L.; Zhao, C.; Qin, F.; He, Z.Y.; Wang, X.; Zhong, X.N. Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin. Exp. Rheumatol. 2016, 34 (Suppl. 100), 14–22. [Google Scholar]
- Robak, E.; Gerlicz-Kowalczuk, Z.; Dziankowska-Bartkowiak, B.; Wozniacka, A.; Bogaczewicz, J. Serum concentrations of IL-17A, IL-17B, IL-17E and IL-17F in patients with systemic sclerosis. Arch. Med. Sci. 2019, 15, 706–712. [Google Scholar] [CrossRef]
- Ahmed, S.; Misra, D.P.; Agarwal, V. Interleukin-17 pathways in systemic sclerosis-associated fibrosis. Rheumatol. Int. 2019, 39, 1135–1143. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Y.; Wu, B.; Qi, Q.; Li, H.; Lu, H.; Fan, C.; Feng, C.; Zuo, J.; Niu, L.; et al. DZ2002 ameliorates fibrosis, inflammation, and vasculopathy in experimental systemic sclerosis models. Arthritis Res. Ther. 2019, 21, 290. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhao, C.; Lei, L.; Tao, Z.; Zheng, L.; Wen, J.; Li, X. Effects of thalidomide on Th17, Treg cells and TGF-beta1/Smad3 pathway in a mouse model of systemic sclerosis. Int. J. Rheum. Dis. 2020, 23, 406–419. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A. Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Acharya, P.; Talahalli, R.R. Aging and Hyperglycemia Intensify Dyslipidemia-Induced Oxidative Stress and Inflammation in Rats: Assessment of Restorative Potentials of ALA and EPA + DHA. Inflammation 2019, 42, 946–952. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Graves, C.L.; Gonzalez, O.A.; Dawson, D., III; Morford, L.A.; Huja, P.E.; Hartsfield, J.K., Jr.; Huja, S.S.; Pandruvada, S.; Wallet, S.M. Aging, inflammation, immunity and periodontal disease. Periodontol 2000 2016, 72, 54–75. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of Bone Resorption in Periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef] [Green Version]
- Ohlrich, E.J.; Cullinan, M.P.; Seymour, G.J. The immunopathogenesis of periodontal disease. Aust. Dent. J. 2009, 54 (Suppl. 1), S2–S10. [Google Scholar] [CrossRef]
- Tan, F.K.; Zhou, X.; Mayes, M.D.; Gourh, P.; Guo, X.; Marcum, C.; Jin, L.; Arnett, F.C., Jr. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology 2006, 45, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Skaug, B.; Bi, X.; Mills, T.; Salazar, G.; Zhou, X.; Reveille, J.; Agarwal, S.K.; Blackburn, M.R.; Mayes, M.D.; et al. Interferon regulatory factor 7 (IRF7) represents a link between inflammation and fibrosis in the pathogenesis of systemic sclerosis. Ann. Rheum. Dis. 2019, 78, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Dai, A.; Pan, L.; Zhang, L.; Wang, Z.; Ke, T.; Sun, W.; Wu, Y.; Ding, P.H.; Chen, L. Inflamm-Aging-Related Cytokines of IL-17 and IFN-gamma Accelerate Osteoclastogenesis and Periodontal Destruction. J. Immunol. Res. 2021, 2021, 9919024. [Google Scholar] [CrossRef]
- Yang, C.; Tang, S.; Zhu, D.; Ding, Y.; Qiao, J. Classical Disease-Specific Autoantibodies in Systemic Sclerosis: Clinical Features, Gene Susceptibility, and Disease Stratification. Front. Med. 2020, 7, 587773. [Google Scholar] [CrossRef]
- Shen, C.Y.; Li, K.J.; Lai, P.H.; Yu, C.L.; Hsieh, S.C. Anti-CENP-B and anti-TOPO-1-containing sera from systemic sclerosis-related diseases with Raynaud’s phenomenon induce vascular endothelial cell senescence not via classical p53-p21 pathway. Clin. Rheumatol. 2018, 37, 749–756. [Google Scholar] [CrossRef]
- Raschi, E.; Privitera, D.; Bodio, C.; Lonati, P.A.; Borghi, M.O.; Ingegnoli, F.; Meroni, P.L.; Chighizola, C.B. Scleroderma-specific autoantibodies embedded in immune complexes mediate endothelial damage: An early event in the pathogenesis of systemic sclerosis. Arthritis Res. Ther. 2020, 22, 265. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, F.; Li, Y.; Zhao, X.Z.; Xu, D.; Li, Y.N.; Li, Y.H.; Sun, X.L. Significance of anti-tubulin-alpha-1C autoantibody in systemic sclerosis. Beijing Da Xue Xue Bao Yi Xue Ban 2020, 52, 1009–1013. [Google Scholar] [PubMed]
- Adler, B.L.; Boin, F.; Wolters, P.J.; Bingham, C.C.O.; Shah, A.A.; Greider, C.; Casciola-Rosen, L.; Rosen, A. Autoantibodies targeting telomere-associated proteins in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 912–919. [Google Scholar] [CrossRef]
- Saito, E.; Fujimoto, M.; Hasegawa, M.; Komura, K.; Hamaguchi, Y.; Kaburagi, Y.; Nagaoka, T.; Takehara, K.; Tedder, T.F.; Sato, S. CD19-dependent B lymphocyte signaling thresholds influence skin fibrosis and autoimmunity in the tight-skin mouse. J. Clin. Invest. 2002, 109, 1453–1462. [Google Scholar] [CrossRef]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K. Altered blood B lymphocyte homeostasis in systemic sclerosis: Expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004, 50, 1918–1927. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Kuroki, K.; Fujimoto, M.; Murakami, Y.; Tedder, T.F.; Tokunaga, K.; Takehara, K.; Sato, S. Association of a functional CD19 polymorphism with susceptibility to systemic sclerosis. Arthritis Rheum. 2004, 50, 4002–4007. [Google Scholar] [CrossRef]
- Simon, D.; Balogh, P.; Erdo-Bonyar, S.; Borocz, K.; Minier, T.; Czirjak, L.; Berki, T. Increased Frequency of Activated Switched Memory B Cells and Its Association With the Presence of Pulmonary Fibrosis in Diffuse Cutaneous Systemic Sclerosis Patients. Front. Immunol. 2021, 12, 686483. [Google Scholar] [CrossRef]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K.; Tedder, T.F. Altered B lymphocyte function induces systemic autoimmunity in systemic sclerosis. Mol. Immunol. 2004, 41, 1123–1133. [Google Scholar] [CrossRef]
- Fuschiotti, P. T cells and cytokines in systemic sclerosis. Curr. Opin. Rheumatol. 2018, 30, 594–599. [Google Scholar] [CrossRef]
- Fiocco, U.; Rosada, M.; Cozzi, L.; Ortolani, C.; De Silvestro, G.; Ruffatti, A.; Cozzi, E.; Gallo, C.; Todesco, S. Early phenotypic activation of circulating helper memory T cells in scleroderma: Correlation with disease activity. Ann. Rheum. Dis. 1993, 52, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Fuschiotti, P.; Medsger, T.A., Jr.; Morel, P.A. Effector CD8+ T cells in systemic sclerosis patients produce abnormally high levels of interleukin-13 associated with increased skin fibrosis. Arthritis Rheum. 2009, 60, 1119–1128. [Google Scholar] [CrossRef]
- Medsger, T.A., Jr.; Ivanco, D.E.; Kardava, L.; Morel, P.A.; Lucas, M.R.; Fuschiotti, P. GATA-3 up-regulation in CD8+ T cells as a biomarker of immune dysfunction in systemic sclerosis, resulting in excessive interleukin-13 production. Arthritis Rheum. 2011, 63, 1738–1747. [Google Scholar] [CrossRef]
- Frantz, C.; Auffray, C.; Avouac, J.; Allanore, Y. Regulatory T Cells in Systemic Sclerosis. Front. Immunol. 2018, 9, 2356. [Google Scholar] [CrossRef]
- Gumkowska-Sroka, O.; Jagoda, K.; Owczarek, A.; Helbig, G.; Giemza-Stoklosa, J.; Kotyla, P.J. Cytometric Characterization of Main Immunocompetent Cells in Patients with Systemic Sclerosis: Relationship with Disease Activity and Type of Immunosuppressive Treatment. J. Clin. Med. 2019, 8, 625. [Google Scholar] [CrossRef] [Green Version]
- Fox, D.A.; Lundy, S.K.; Whitfield, M.L.; Berrocal, V.; Campbell, P.; Rasmussen, S.; Ohara, R.; Stinson, A.; Gurrea-Rubio, M.; Wiewiora, E.; et al. Correction to: Lymphocyte subset abnormalities in early diffuse cutaneous systemic sclerosis. Arthritis Res. Ther. 2021, 23, 73. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Lee, S.Y.; Choi, J.W.; Lee, A.R.; Yoo, J.J.H.; Moon, S.J.; Park, S.H.; Cho, M.L. Correction to: Metformin ameliorates scleroderma via inhibiting Th17 cells and reducing mTOR-STAT3 signaling in skin fibroblasts. J. Transl. Med. 2021, 19, 266. [Google Scholar] [CrossRef]
- Paleja, B.; Low, A.H.L.; Kumar, P.; Saidin, S.; Lajam, A.; Nur Hazirah, S.; Chua, C.; Li Yun, L.; Albani, S. Systemic Sclerosis Perturbs the Architecture of the Immunome. Front. Immunol. 2020, 11, 1602. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, C.-Y.; Lu, C.-H.; Wu, C.-H.; Li, K.-J.; Kuo, Y.-M.; Hsieh, S.-C.; Yu, C.-L. Molecular Basis of Accelerated Aging with Immune Dysfunction-Mediated Inflammation (Inflamm-Aging) in Patients with Systemic Sclerosis. Cells 2021, 10, 3402. https://doi.org/10.3390/cells10123402
Shen C-Y, Lu C-H, Wu C-H, Li K-J, Kuo Y-M, Hsieh S-C, Yu C-L. Molecular Basis of Accelerated Aging with Immune Dysfunction-Mediated Inflammation (Inflamm-Aging) in Patients with Systemic Sclerosis. Cells. 2021; 10(12):3402. https://doi.org/10.3390/cells10123402
Chicago/Turabian StyleShen, Chieh-Yu, Cheng-Hsun Lu, Cheng-Han Wu, Ko-Jen Li, Yu-Min Kuo, Song-Chou Hsieh, and Chia-Li Yu. 2021. "Molecular Basis of Accelerated Aging with Immune Dysfunction-Mediated Inflammation (Inflamm-Aging) in Patients with Systemic Sclerosis" Cells 10, no. 12: 3402. https://doi.org/10.3390/cells10123402
APA StyleShen, C. -Y., Lu, C. -H., Wu, C. -H., Li, K. -J., Kuo, Y. -M., Hsieh, S. -C., & Yu, C. -L. (2021). Molecular Basis of Accelerated Aging with Immune Dysfunction-Mediated Inflammation (Inflamm-Aging) in Patients with Systemic Sclerosis. Cells, 10(12), 3402. https://doi.org/10.3390/cells10123402