
Chromatin-Independent Interplay of NFATc1 and EZH2 in Pancreatic Cancer

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Mouse Strains and In Vivo Experiments
2.2. Acinar Cell Extraction
2.3. Cell Culture, Transfection, and Treatment
2.4. RNA Extraction and Quantitative Realtime-PCR
2.5. Human PDAC Material and Microdissection of Epithelial Tumor Tissue
2.6. Protein Harvesting, Western Blot, and Immunoprecipitation
2.7. Immunofluorescence and Immunohistochemistry (IHC)
2.8. Chromatin Immunoprecipitation (ChIP)- and RNA-Sequencing Data Analysis
2.9. Statistical Analysis
3. Results
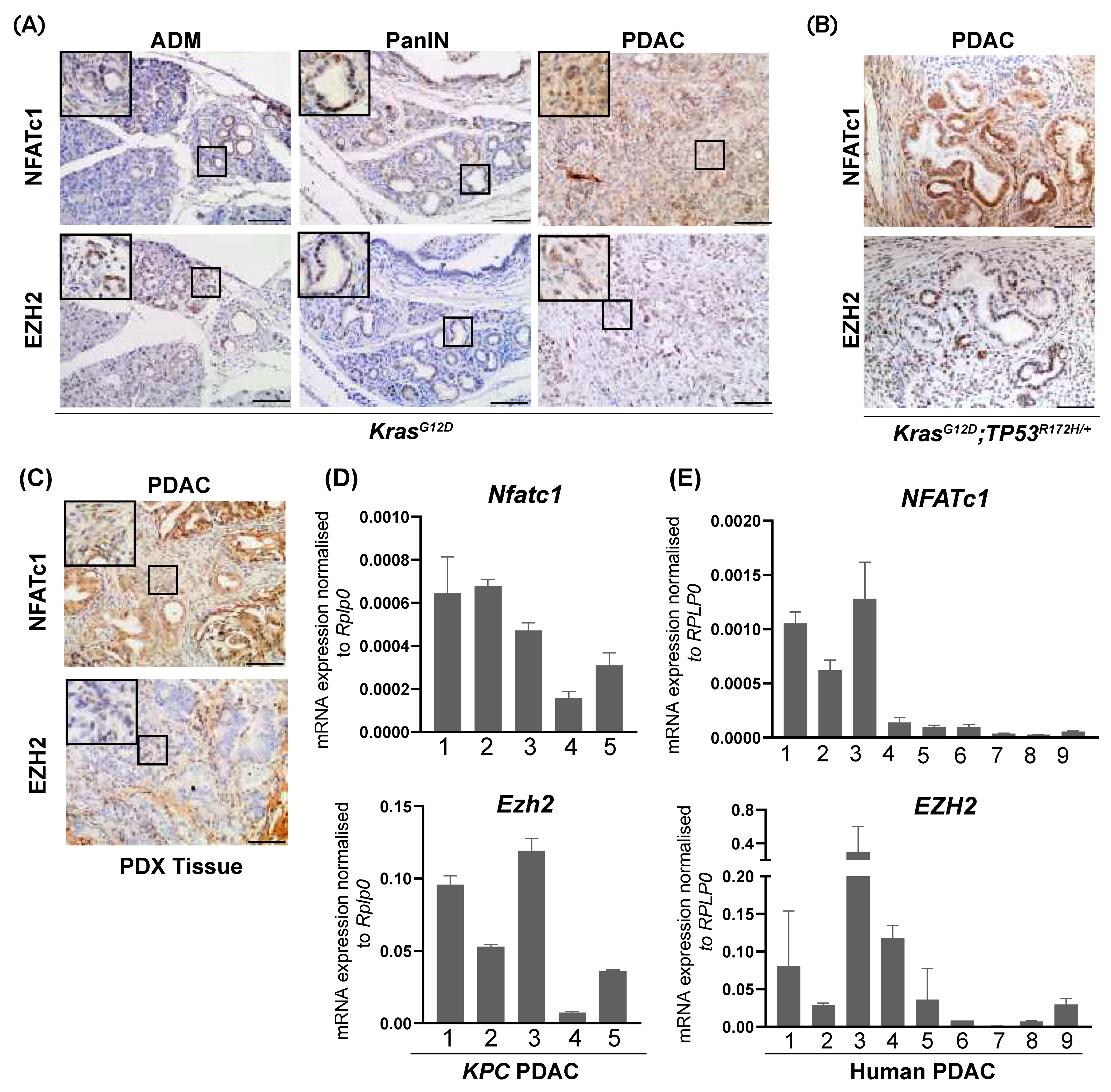
3.1. NFATc1 and EZH2 Are Co-Expressed in a Subset of Murine and Human PDAC Samples
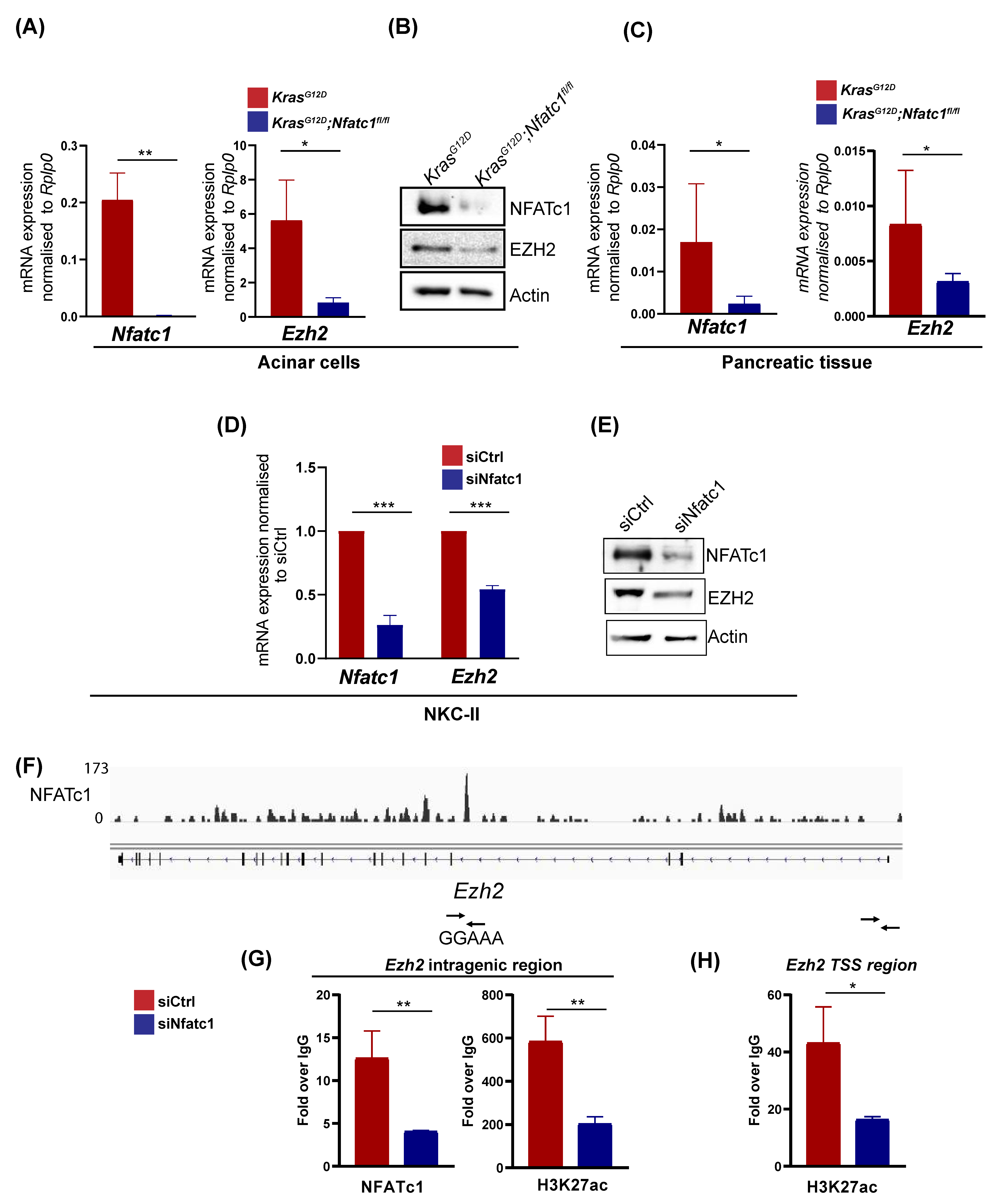
3.2. NFATc1 Induces EZH2 Expression at the Level of Gene Transcription
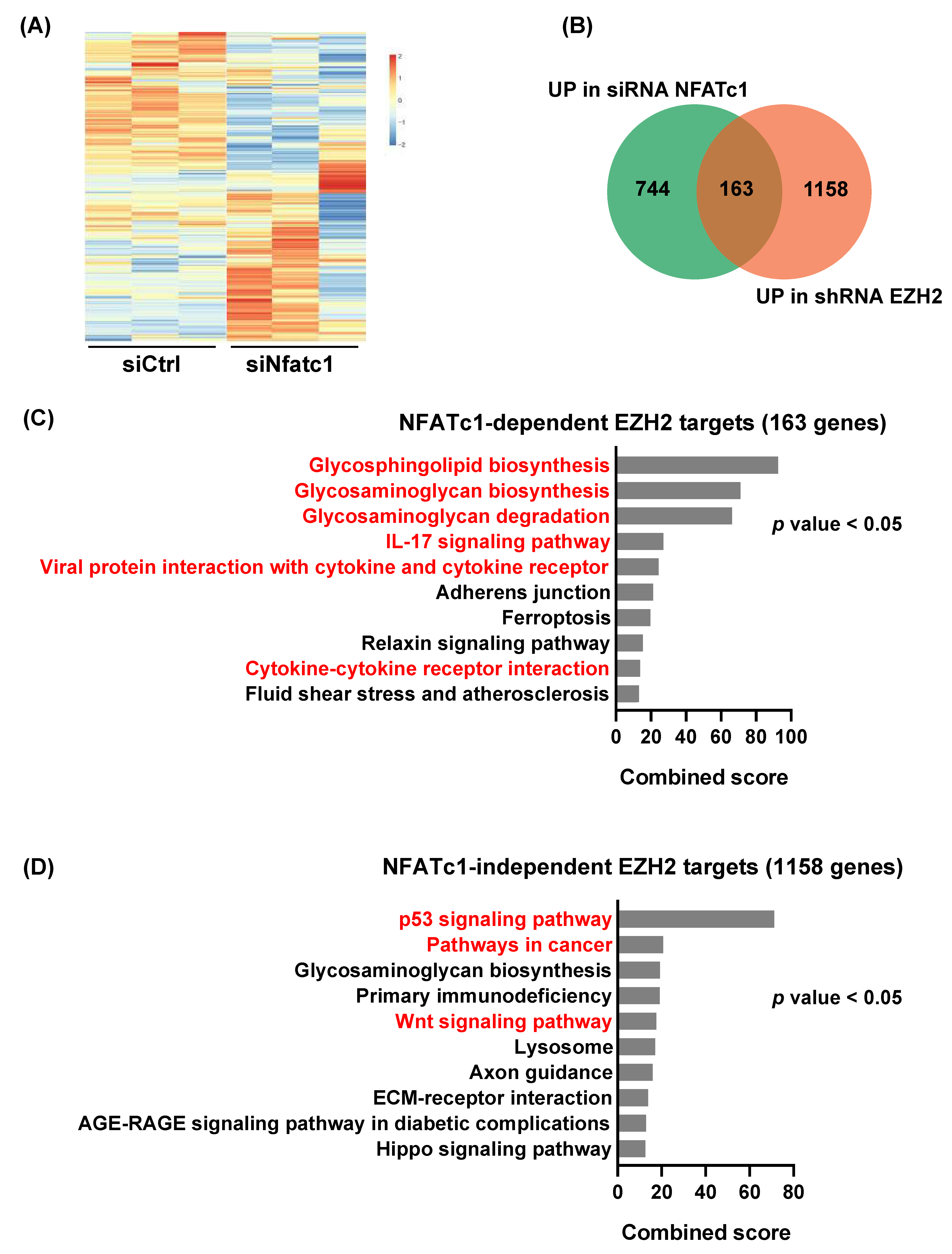
3.3. NFATc1 Is Involved in the Regulation of a Subset of EZH2-Dependent Gene Signatures
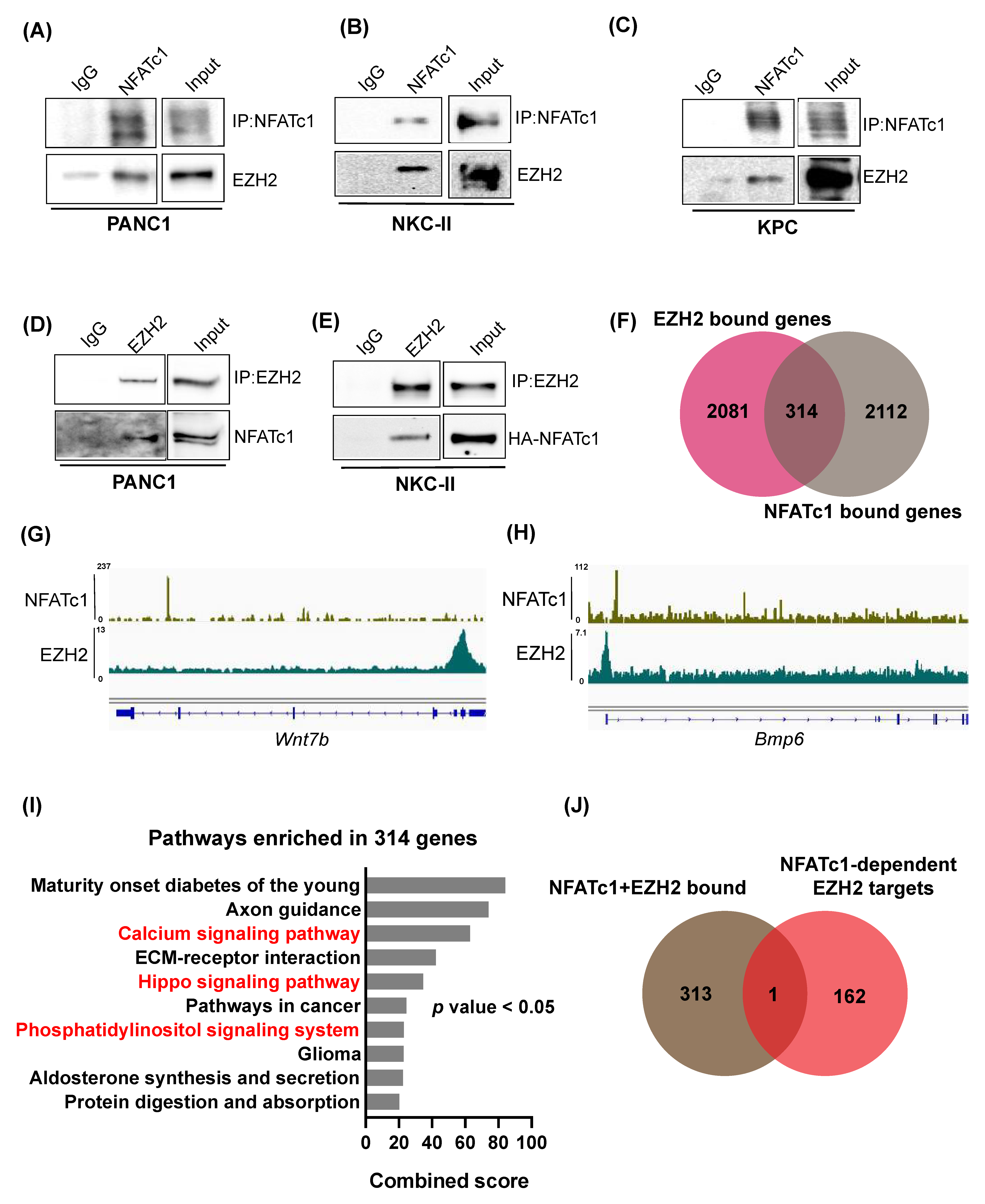
3.4. NFATc1 and EZH2 Biochemically Interact with Each Other but Are Not Involved in Joint Regulation of Direct Target Gene Transcription
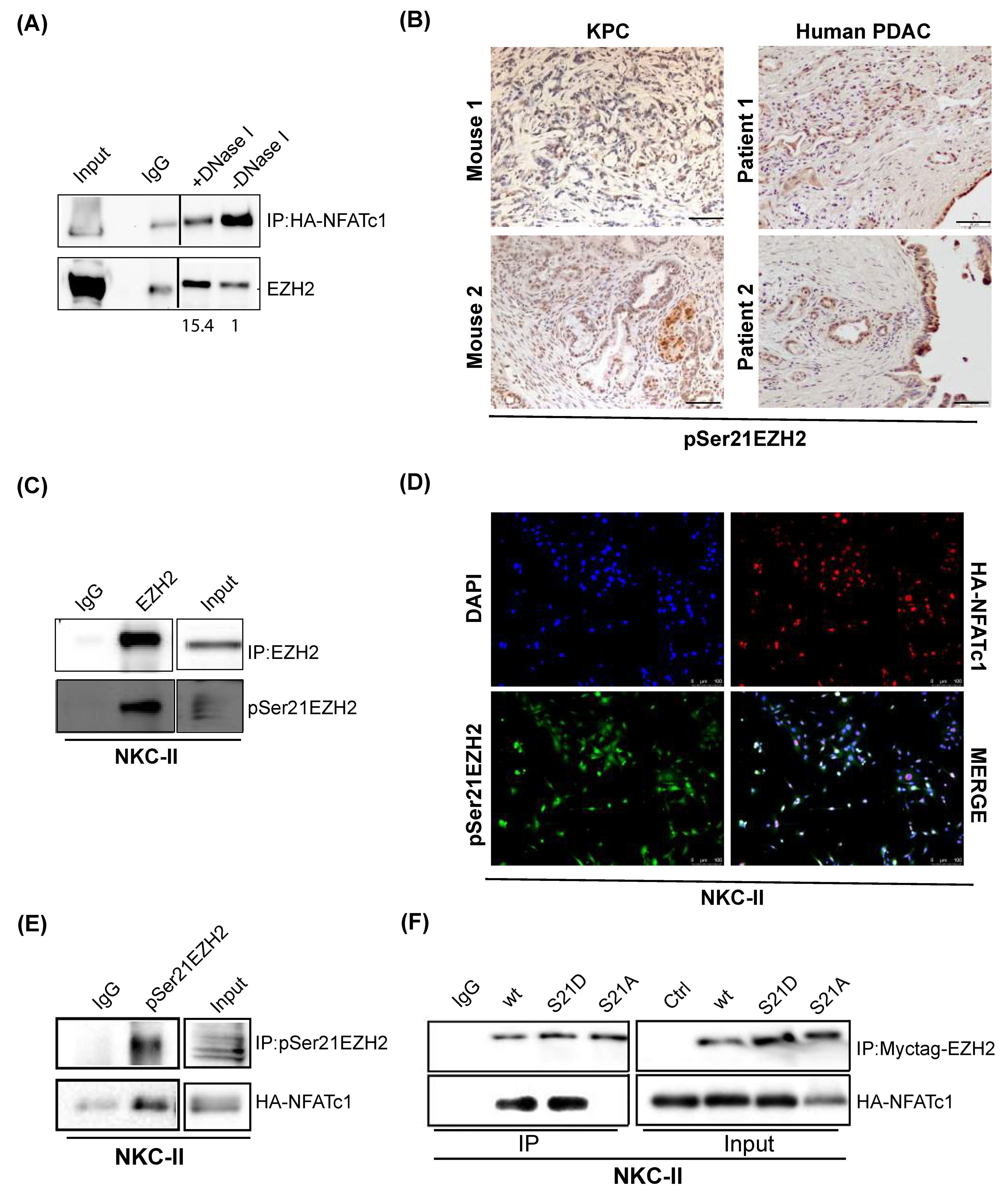
3.5. NFATc1:EZH2 Complex Formation Occurs in a Chromatin-Independent Manner and Requires Posttranslational EZH2 Phosphorylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Waddell, N.; Pajic, M.; Oatch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.K.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, E.R.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Tajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–550. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, S.; Glesel, E.; Singh, G.; Chen, N.-M.; Reutlinger, K.; Zhang, J.; Billadeau, D.D.; Fernandez-Zapico, M.E.; Gress, T.M.; Singh, S.K.; et al. Restricted heterochromatin formation links NFATc2 repressor activity with growth promotion in pancreatic cancer. Gastroenterology 2012, 142, 388–398.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, S.; Steuber, B.; Kopp, W.; Kari, V.; Urbach, L.; Wang, X.; Küffer, S.; Bohnenberger, H.; Spyropoulou, D.; Zhang, Z.; et al. EZH2 Regulates Pancreatic Cancer Subtype Identity and Tumor Progression via Transcriptional Repression of GATA6. Cancer Res. 2020, 80, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Mazur, K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sánchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.J.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.-U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkel, P.; Dupret, B.; Bourhis, X.J.; Angrand, P.-O. Diverse involvement of EZH2 in cancer epigenetics. Am. J. Transl. Res. 2015, 7, 175–193. [Google Scholar]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [Green Version]
- Mallen-St. Clair, J.; Soydaner-Azeloglu, R.; Lee, K.E.; Taylor, L.; Livanos, A.; Pylaceva-Gupta, Y.; Miller, G.; Margueron, R.; Reinberg, D.; Bar-Sagi, D. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev. 2012, 26, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.-M.; Neesse, A.; Dyck, M.L.; Steuber, B.; Koenig, A.O.; Lubeseder-Martellato, C.; Winter, T.; Forster, T.; Bohnenberger, H.; Kitz, J.; et al. Context-Dependent Epigenetic Regulation of Nuclear Factor of Activated T Cells 1 in Pancreatic Plasticity. Gastroenterology 2017, 152, 1507–1520.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, S.; Chen, N.-M.; Siveke, J.T.; König, A.; Zhang, J.-S.; Singh, S.K.; Wolf, E.; Bartkuhn, M.; Esposito, I.; Heßmann, E.; et al. Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov. 2014, 4, 688–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.M.; Singh, G.; Koenig, A.; Liou, G.-Y.; Storz, P.; Zhang, J.-S.; Regul, L.; Nagarajan, S.; Kühnemuth, B.; Johnsen, S.A.; et al. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar-Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015, 148, 1024–1034.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, A.; Linhart, T.; Schlengemann, K.; Reutlinger, K.; Wegele, J.; Adler, G.; Singh, G.; Hofmann, L.; Kunsch, S.; Büch, T.; et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology 2010, 138, 1189–1199.e2. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Chen, N.-M.; Hessmann, E.; Siveke, J.; Lahmann, M.; Singh, G.; Volker, N.; Vogt, S.; Esposito, I.; Schmidt, A.; et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J. 2015, 34, 517–530. [Google Scholar] [CrossRef]
- Hasselluhn, M.C.; Schmidt, G.E.; Ellenrieder, V.; Johnsen, S.A.; Hessmann, E. Aberrant NFATc1 signaling counteracts TGFβ-mediated growth arrest and apoptosis induction in pancreatic cancer progression. Cell Death Dis. 2019, 10, 446. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Daramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Hunt, J.L.; Finkelstein, S.D. Microdissection techniques for molecular testing in surgical pathology. Arch. Pathol. Lab. Med. 2004, 128, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Jo, P.; König, A.; Schirmer, M.; Kitz, J.; Conradi, L.-C.; Azizian, A.; Bernhardt, M.; Wolff, H.A.; Grade, M.; Ghadimi, M.; et al. Heterogeneity of KRAS Mutation Status in Rectal Cancer. PLoS ONE 2016, 11, e0153278. [Google Scholar]
- Buchholz, M.; Schatz, A.; Wagner, M.; Michl, P.; Linhart, T.; Adler, A.; Gress, T.M.; Ellenrieder, V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006, 25, 3714–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, V.; Vaissiere, T.; Herceg, Z. Histone acetylation and chromatin signature in stem cell identity and cancer. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2008, 637, 1–15. [Google Scholar] [CrossRef]
- Baumgart, S.; Ellenrieder, V.; Fernandez-Zapico, M.E. Oncogenic transcription factors: Cornerstones of inflammation-linked pancreatic carcinogenesis. Gut 2013, 62, 310–316. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.-H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Raman, J.D.; Mongan, N.P.; Tickoo, S.K.; Boorjian, S.A.; Scherr, D.S.; Gudas, L.J. Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder. Clin. Cancer Res. 2005, 11, 8570–8576. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Shen, L.; Suzuki, S.; Kurokawa, T.; Masuko, K.; Tanaka, Y.; Kato, H.; Mizuno, Y.; Yokoe, M.; Sugauchi, F.; et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res. 2007, 37, 974–983. [Google Scholar] [CrossRef]
- Rao, Z.Y.; Cai, M.-Y.; Yang, G.-F.; He, L.-R.; Mai, S.-J.; Hua, W.-F.; Lioa, Y.-J.; Deng, H.-X.; Chen, Y.-C.; Guan, X.-Y.; et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-β1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.; Solis, L.M.; Lin, H.; Yuan, P.; Tang, X.; Kadara, H.; Riquelme, E.; Galindo, H.; Moran, C.A.; Kalhor, N.; et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin. Cancer Res. 2013, 19, 6556–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Cardoso, C.; Mignon, C.; Hetet, G.; Grandchamps, B.; Fontes, M.; Colleaux, L. The human EZH2 gene: Genomic organisation and revised mapping in 7q35 within the critical region for malignant myeloid disorders. Eur. J. Hum. Genet. 2000, 8, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Graves, A.P.; Ganji, G.; Diaz, E.; Halsey, W.S.; Jiang, Y.; Smitheman, K.N.; Ott, H.M.; Pappalardi, M.B.; Allen, K.E.; et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. USA 2012, 109, 2989–2994. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.-W.G.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [Green Version]
- Coe, B.P.; Thu, K.L.; Aviel-Ronen, S.; Vucic, E.A.; Gazdar, A.F.; Lam, S.; Tsao, M.-S.; Lam, W.L. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS ONE 2013, 8, e71670. [Google Scholar]
- Fujii, S.; Tokita, K.; Wada, N.; Ito, K.; Yamacchi, C.; Ito, Y.; Ochiai, A. MEK-ERK pathway regulates EZH2 overexpression in association with aggressive breast cancer subtypes. Oncogene 2011, 30, 4118–4128. [Google Scholar] [CrossRef] [Green Version]
- Feber, A.; Clark, J.; Goodwin, G.; Dodson, A.R.; Smith, P.H.; Fletcher, A.; Edwards, S.; Flohr, P.; Falconer, A.; Roe, T.; et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene 2004, 23, 1627–1630. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Iwata, T.; Zheng, Q.; Bethel, C.; Yegnasubramanian, S.; De Marzo, A.M. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget 2011, 2, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Garipov, A.; Li, H.; Bitler, B.G.; Thapa, R.J.; Balachandran, S.; Zhang, R. NF-YA underlies EZH2 upregulation and is essential for proliferation of human epithelial ovarian cancer cells. Mol. Cancer Res. 2013, 11, 360–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.W.; Ren, L.-L.; Xiong, H.; Du, W.; Yu, Y.-N.; Sun, T.-T.; Wenig, Y.-R.; Wang, Z.-H.; Wang, J.-L.; Wang, Y.-C.; et al. Role of STAT3 and vitamin D receptor in EZH2-mediated invasion of human colorectal cancer. J. Pathol. 2013, 230, 277–290. [Google Scholar] [CrossRef]
- Kunderfranco, P.; Mello-Grand, M.; Cangemi, R.; Pellini, S.; Mensah, A.; Albertini, V.; Malek, A.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS ONE 2010, 5, e10547. [Google Scholar] [CrossRef] [Green Version]
- Saramaki, O.R.; Tammela, T.L.J.; Martikainen, P.M.; Vessela, R.L.; Visakorpi, T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer 2006, 45, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Liang, J.; Yang, X.; Wang, Y.; Zhao, Y.; Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Li, R.; et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 2007, 27, 5105–5119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.M.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.Y.; Jun, S.; Lee, M.; Kim, H.-C.; Wang, X.; Ji, H.; McCrea, P.D.; Park, J.-I. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol. Cell 2013, 52, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Li, M.; Wang, D.; Hou, P.; Chen, X.; Chu, S.; Chai, D.; Zheng, J.; Bai, J. Post-translational modifications of EZH2 in cancer. Cell Biosci. 2020, 10, 143. [Google Scholar] [CrossRef]
- Ko, H.W.; Lee, H.-H.; Huo, L.; Xia, W.; Yang, C.-C.; Hsu, J.L.; Li, L.-Y.; Lai, C.-C.; Chan, L.-C.; Cheng, C.-C.; et al. GSK3β inactivation promotes the oncogenic functions of EZH2 and enhances methylation of H3K27 in human breast cancers. Oncotarget 2016, 7, 57131–57144. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, S.; Chen, N.-M.; Zhang, J.-S.; Billadeau, D.D.; Gaisina, I.N.; Kozikowski, A.P.; Singh, S.K.; Fink, D.; Ströbel, P.; Klindt, C.; et al. GSK-3β Governs Inflammation-Induced NFATc2 Signaling Hubs to Promote Pancreatic Cancer Progression. Mol. Cancer Ther. 2016, 15, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugolkov, A.; Gaisina, I.; Zhang, J.-S.; Billadeau, D.D.; White, K.; Kozikowski, A.; Jain, S.; Cristofenilli, M.; Giles, F.; O’Halloran, T.; et al. GSK-3 inhibition overcomes chemoresistance in human breast cancer. Cancer Lett. 2016, 380, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Herreros-Villanueva, M.; Koenig, A.; Deng, Z.; de Narvajas, A.A.-M.; Gomez, T.S.; Meng, X.; Bujanda, L.; Ellenrieder, V.; Li, X.K.; et al. Differential activity of GSK-3 isoforms regulates NF-KB and TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell Death Dis. 2014, 5, e1142. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, S.; Forster, T.; Reutlinger, K.; Kopp, W.; Versemann, L.; Spitalieri, J.; Gaedcke, J.; Ströbel, P.; Singh, S.K.; Ellenrieder, V.; et al. Chromatin-Independent Interplay of NFATc1 and EZH2 in Pancreatic Cancer. Cells 2021, 10, 3463. https://doi.org/10.3390/cells10123463
Patil S, Forster T, Reutlinger K, Kopp W, Versemann L, Spitalieri J, Gaedcke J, Ströbel P, Singh SK, Ellenrieder V, et al. Chromatin-Independent Interplay of NFATc1 and EZH2 in Pancreatic Cancer. Cells. 2021; 10(12):3463. https://doi.org/10.3390/cells10123463
Chicago/Turabian StylePatil, Shilpa, Teresa Forster, Kristina Reutlinger, Waltraut Kopp, Lennart Versemann, Jessica Spitalieri, Jochen Gaedcke, Philipp Ströbel, Shiv K. Singh, Volker Ellenrieder, and et al. 2021. "Chromatin-Independent Interplay of NFATc1 and EZH2 in Pancreatic Cancer" Cells 10, no. 12: 3463. https://doi.org/10.3390/cells10123463
APA StylePatil, S., Forster, T., Reutlinger, K., Kopp, W., Versemann, L., Spitalieri, J., Gaedcke, J., Ströbel, P., Singh, S. K., Ellenrieder, V., Neesse, A., & Hessmann, E. (2021). Chromatin-Independent Interplay of NFATc1 and EZH2 in Pancreatic Cancer. Cells, 10(12), 3463. https://doi.org/10.3390/cells10123463