
Ionizing Radiation-Induced Brain Cell Aging and the Potential Underlying Molecular Mechanisms



Abstract
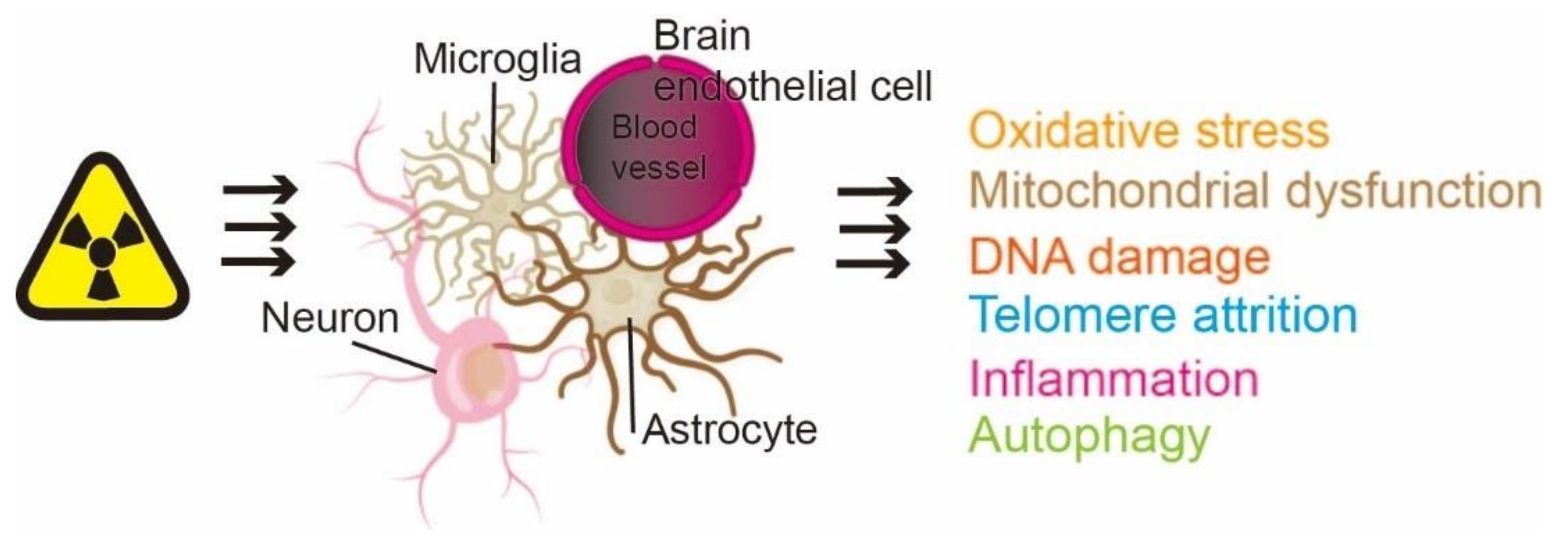
:1. Introduction
2. Radiation-Induced Senescence of Different Types of Brain Cells
2.1. Microglia
2.2. Astrocytes
2.3. Brain Endothelial Cells
2.4. Neurons
3. Effect of Radiation-Induced Brain Aging
3.1. Oxidative Stress
3.2. Mitochondrial Dysfunction
3.3. Telomere Attrition
3.4. DNA Damage
3.5. Inflammation
3.6. Autophagy
4. Conclusions and Future Research Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| BBB | Blood-brain barrier |
| CNS | Central nervous system |
| COX2 | Cyclooxygenases 2 |
| DDR | DNA damage response |
| DSBs | DNA double-strand breaks |
| DAMP | Damage-associated molecular pattern |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| GFAP | Glial fibrillary acidic protein |
| H2O | Water molecules |
| HMGB1 | High-mobility group box 1 |
| IKB | Inhibitory protein |
| iNOS | Inducible nitric oxide synthase |
| IR | Ionizing radiation |
| KLF6 | Kruppel like factor 6 |
| 5-LPO | 5-lypoxygenase |
| MMPs | Matrix metalloproteinases |
| mtDNA | Mitochondrial DNA |
| NADH | Nicotinamide adenine dinucleotide |
| NO | Nitric oxide |
| NF-κB | Nuclear factor kappa B |
| Nrf2 | Nuclear factor E2-related factor 2 |
| O2− | Superoxide anion |
| PGH2 | Prostaglandin H 2 |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SASP | Senescence associated secretion phenotype |
| SA-β-Gal | Senescence-associated-β-galactosidase |
| SIPS | Stress-induced premature senescence |
| TLR4 | Toll-like receptor 4 |
| TLRs | Toll-like receptors |
| TNF-α | Tumor necrosis factor-α |
References
- Martin, L.G. Population aging policies in East Asia and the United States. Science 1991, 251, 527–531. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.J.; Le Couteur, D.G. Aging biology and geriatric clinical pharmacology. Pharmacol. Rev. 2004, 56, 163–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Pluvinage, J.V.; Wyss-Coray, T. Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat. Rev. Neurosci. 2020, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E.; Ryan, L.; Bowers, D.; Foster, T.C.; Bizon, J.L.; Geldmacher, D.S.; Glisky, E.L. Characterizing cognitive aging in humans with links to animal models. Front. Aging Neurosci. 2012, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, G.V.; Pezarat-Correia, P.; Vaz, J.R.; Silva, L.; Heffernan, K.S. Impact of Aging on Endurance and Neuromuscular Physical Performance: The Role of Vascular Senescence. Sports Med. 2017, 47, 583–598. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Bueler, H. Mitochondrial and Autophagic Regulation of Adult Neurogenesis in the Healthy and Diseased Brain. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.; Kim, B.C.; Cho, K.A.; Song, J. The Cerebral Effect of Ammonia in Brain Aging: Blood-Brain Barrier Breakdown, Mitochondrial Dysfunction, and Neuroinflammation. J. Clin. Med. 2021, 10. [Google Scholar] [CrossRef]
- Aliper, A.M.; Bozdaganyan, M.E.; Orekhov, P.S.; Zhavoronkov, A.; Osipov, A.N. Replicative and radiation-induced aging: A comparison of gene expression profiles. Aging 2019, 11, 2378–2387. [Google Scholar] [CrossRef]
- Kuzmic, M.; Galas, S.; Lecomte-Pradines, C.; Dubois, C.; Dubourg, N.; Frelon, S. Interplay between ionizing radiation effects and aging in C. elegans. Free Radic. Biol. Med. 2019, 134, 657–665. [Google Scholar] [CrossRef]
- Kim, H.K.; Song, I.S.; Lee, S.Y.; Jeong, S.H.; Lee, S.R.; Heo, H.J.; Thu, V.T.; Kim, N.; Ko, K.S.; Rhee, B.D.; et al. B7-H4 downregulation induces mitochondrial dysfunction and enhances doxorubicin sensitivity via the cAMP/CREB/PGC1-alpha signaling pathway in HeLa cells. Pflügers Arch.-Eur. J. Physiol. 2014, 466, 2323–2338. [Google Scholar] [CrossRef]
- Kim, S.B.; Heo, J.I.; Kim, H.; Kim, K.S. Acetylation of PGC1alpha by Histone Deacetylase 1 Downregulation Is Implicated in Radiation-Induced Senescence of Brain Endothelial Cells. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 787–793. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Tang, F.R.; Loganovsky, K. Low dose or low dose rate ionizing radiation-induced health effect in the human. J. Environ. Radioact. 2018, 192, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Andreassi, M.G.; Piccaluga, E.; Gargani, L.; Sabatino, L.; Borghini, A.; Faita, F.; Bruno, R.M.; Padovani, R.; Guagliumi, G.; Picano, E. Subclinical carotid atherosclerosis and early vascular aging from long-term low-dose ionizing radiation exposure: A genetic, telomere, and vascular ultrasound study in cardiac catheterization laboratory staff. JACC Cardiovasc. Interv. 2015, 8, 616–627. [Google Scholar] [CrossRef] [PubMed]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers 2019, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Mahamud, O.; So, J.; Chua, M.L.K.; Bristow, R.G. Targeting DNA repair for precision radiotherapy: Balancing the therapeutic ratio. Curr. Probl. Cancer 2017, 41, 265–272. [Google Scholar] [CrossRef]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [Green Version]
- Kempf, S.J.; Azimzadeh, O.; Atkinson, M.J.; Tapio, S. Long-term effects of ionising radiation on the brain: Cause for concern? Radiat. Environ. Biophys 2013, 52, 5–16. [Google Scholar] [CrossRef]
- Hendry, J.H.; Simon, S.L.; Wojcik, A.; Sohrabi, M.; Burkart, W.; Cardis, E.; Laurier, D.; Tirmarche, M.; Hayata, I. Human exposure to high natural background radiation: What can it teach us about radiation risks? J. Radiol. Prot. 2009, 29, A29–A42. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Sharma, R.; Mathur, D.; Sharad, S.; Minhas, G.; Bhatia, K.; Anand, A.; Ghosh, S.P. Role of Ionizing Radiation in Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, M.; Tokonami, S.; Omori, Y.; Sahoo, S.K.; Akiba, S.; Sorimachi, A.; Ishikawa, T.; Nair, R.R.; Jayalekshmi, P.A.; Sebastian, P.; et al. Estimation of external dose by car-borne survey in Kerala, India. PLoS ONE 2015, 10, e0124433. [Google Scholar] [CrossRef] [Green Version]
- Kudo, H.; Tokonami, S.; Omori, Y.; Ishikawa, T.; Iwaoka, K.; Sahoo, S.K.; Akata, N.; Hosoda, M.; Wanabongse, P.; Pornnumpa, C.; et al. Comparative dosimetry for radon and thoron in high background radiation areas in China. Radiat. Prot. Dosim. 2015, 167, 155–159. [Google Scholar] [CrossRef]
- Omori, Y.; Tokonami, S.; Sahoo, S.K.; Ishikawa, T.; Sorimachi, A.; Hosoda, M.; Kudo, H.; Pornnumpa, C.; Nair, R.R.; Jayalekshmi, P.A.; et al. Radiation dose due to radon and thoron progeny inhalation in high-level natural radiation areas of Kerala, India. J. Radiol. Prot. 2017, 37, 111–126. [Google Scholar] [CrossRef]
- Nugraha, E.D.; Hosoda, M.; Mellawati, J.; Tamakuma, Y.; Ikram, A.; Syaifudin, M.; Yamada, R.; Akata, N.; Sasaki, M.; Furukawa, M.; et al. Comprehensive exposure assessments from the viewpoint of health in a unique high natural background radiation area, Mamuju, Indonesia. Sci. Rep. 2021, 11, 14578. [Google Scholar] [CrossRef] [PubMed]
- Yablokov, A.V.; Nesterenko, V.B.; Nesterenko, A.V. Consequences of the Chernobyl catastrophe for public health and the environment 23 years later. Ann. N. Y. Acad. Sci. 2009, 1181, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Eglite, M.E.; Zvagule, T.J.; Rainsford, K.D.; Reste, J.D.; Curbakova, E.V.; Kurjane, N.N. Clinical aspects of the health disturbances in Chernobyl Nuclear Power Plant accident clean-up workers (liquidators) from Latvia. Inflammopharmacology 2009, 17, 163–169. [Google Scholar] [CrossRef]
- Sasaki, H.; Kodama, K.; Yamada, M. A review of forty-five years study of Hiroshima and Nagasaki atomic bomb survivors. Aging. J. Radiat. Res. 1991, 32, 310–326. [Google Scholar] [CrossRef] [Green Version]
- Yablokov, A.V. Accelerated aging as a consequence of the Chernobyl catastrophe. Ann. N. Y. Acad. Sci. 2009, 1181, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Holodova, N.B.; Zhavoronkova, L.A.; Ryzhov, B.N. [Complex pathogenetic treatment schemes of vascular dyscirculatory disorders in the remote period after exposure to low dose radiation]. Radiat. Biol. Radioecol. 2013, 53, 525–535. [Google Scholar]
- Polyukhov, A.M.; Kobsar, I.V.; Grebelnik, V.I.; Voitenko, V.P. The accelerated occurrence of age-related changes of organism in Chernobyl workers: A radiation-induced progeroid syndrome? Exp. Gerontol. 2000, 35, 105–115. [Google Scholar] [CrossRef]
- Brizzee, K.R. Quantitative histological studies on aging changes in cerebral cortex of Rhesus monkey and albino rat with notes on effects of prolonged low-dose ionizing irradiation in the rat. Prog. Brain Res. 1973, 40, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Lowe, X.R.; Bhattacharya, S.; Marchetti, F.; Wyrobek, A.J. Early brain response to low-dose radiation exposure involves molecular networks and pathways associated with cognitive functions, advanced aging and Alzheimer’s disease. Radiat. Res. 2009, 171, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Lowe, X.; Wyrobek, A. Characterization of the early CNS stress biomarkers and profiles associated with neuropsychiatric diseases. Curr. Genomics 2012, 13, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Meng, Z.; Zhang, G.; Xing, Y.; Feng, L.; Fan, S.; Fan, F.; Buren, B.; Liu, Q. N-acetylcysteine relieves oxidative stress and protects hippocampus of rat from radiation-induced apoptosis by inhibiting caspase-3. Biomed. Pharmacother. 2015, 70, 1–6. [Google Scholar] [CrossRef]
- Ungvari, Z.; Podlutsky, A.; Sosnowska, D.; Tucsek, Z.; Toth, P.; Deak, F.; Gautam, T.; Csiszar, A.; Sonntag, W.E. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: Role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1443–1457. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Dong, Y.; Wang, Y.; Wang, Y.; Guan, B.; Lu, Y.; Wu, J.; Wang, X.; Li, D.; Meng, A.; et al. Potential role of senescent macrophages in radiation-induced pulmonary fibrosis. Cell Death Dis. 2021, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.B. Ionizing radiation and aging: Rejuvenating an old idea. Aging 2009, 1, 887–902. [Google Scholar] [CrossRef] [Green Version]
- Yarilin, A.A.; Belyakov, I.M.; Kusmenok, O.I.; Arshinov, V.Y.; Simonova, A.V.; Nadezhina, N.M.; Gnezditskaya, E.V. Late T cell deficiency in victims of the Chernobyl radiation accident: Possible mechanisms of induction. Int. J. Radiat. Biol. 1993, 63, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, W.; Jia, Y.; Liu, J.; Tan, G.; Zou, J.; Li, X.; Su, Y.; Lei, S.; Sun, Q. Long-term immune effects of high-level natural radiation on Yangjiang inhabitants in China. Int. J. Radiat. Biol. 2019, 95, 764–770. [Google Scholar] [CrossRef]
- Little, M.P. The proportion of thyroid cancers in the Japanese atomic bomb survivors associated with natural background radiation. J. Radiol. Prot. 2002, 22, 279–291. [Google Scholar] [CrossRef]
- Little, M.P.; Wakeford, R.; Lubin, J.H.; Kendall, G.M. The statistical power of epidemiological studies analyzing the relationship between exposure to ionizing radiation and cancer, with special reference to childhood leukemia and natural background radiation. Radiat. Res. 2010, 174, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Rasband, M.N. Glial Contributions to Neural Function and Disease. Mol. Cell. Proteom. 2016, 15, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, F. Endothelial cells of the brain and other organ systems: Some similarities and differences. Prog. Neurobiol. 1996, 48, 255–273. [Google Scholar] [CrossRef]
- Xu, A.; Li, R.; Ren, A.; Jian, H.; Huang, Z.; Zeng, Q.; Wang, B.; Zheng, J.; Chen, X.; Zheng, N.; et al. Regulatory coupling between long noncoding RNAs and senescence in irradiated microglia. J. Neuroinflamm. 2020, 17, 321. [Google Scholar] [CrossRef]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-induced astrocyte senescence is rescued by Delta133p53. Neuro. Oncol. 2019, 21, 474–485. [Google Scholar] [CrossRef] [Green Version]
- McRobb, L.S.; McKay, M.J.; Gamble, J.R.; Grace, M.; Moutrie, V.; Santos, E.D.; Lee, V.S.; Zhao, Z.; Molloy, M.P.; Stoodley, M.A. Ionizing radiation reduces ADAM10 expression in brain microvascular endothelial cells undergoing stress-induced senescence. Aging 2017, 9, 1248–1268. [Google Scholar] [CrossRef] [Green Version]
- Schindler, M.K.; Forbes, M.E.; Robbins, M.E.; Riddle, D.R. Aging-dependent changes in the radiation response of the adult rat brain. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 826–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Harry, G.J.; Kraft, A.D. Neuroinflammation and microglia: Considerations and approaches for neurotoxicity assessment. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ullrich, R.K. Molecular targets in radiation oncology. Oncogene 2003, 22, 5730–5733. [Google Scholar] [CrossRef] [Green Version]
- Lumniczky, K.; Szatmari, T.; Safrany, G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Xu, P.; Xu, Y.; Hu, B.; Wang, J.; Pan, R.; Murugan, M.; Wu, L.J.; Tang, Y. Extracellular ATP enhances radiation-induced brain injury through microglial activation and paracrine signaling via P2X7 receptor. Brain Behav. Immun. 2015, 50, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, M.; Nguyen, T.; Silva, K.K.; Westerhout, R.; Antoszewski, J.; Keating, A.; Savvides, N.; Musca, C.; Dell, J.; Faraone, L. Short-wavelength infrared tuneable filters on HgCdTe photoconductors. Opt. Express 2005, 13, 9683–9694. [Google Scholar] [CrossRef] [PubMed]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tha, K.K.; Okuma, Y.; Miyazaki, H.; Murayama, T.; Uehara, T.; Hatakeyama, R.; Hayashi, Y.; Nomura, Y. Changes in expressions of proinflammatory cytokines IL-1beta, TNF-alpha and IL-6 in the brain of senescence accelerated mouse (SAM) P8. Brain Res. 2000, 885, 25–31. [Google Scholar] [CrossRef]
- Osman, A.M.; Sun, Y.; Burns, T.C.; He, L.; Kee, N.; Oliva-Vilarnau, N.; Alevyzaki, A.; Zhou, K.; Louhivuori, L.; Uhlen, P.; et al. Radiation Triggers a Dynamic Sequence of Transient Microglial Alterations in Juvenile Brain. Cell Rep. 2020, 31, 107699. [Google Scholar] [CrossRef]
- Betlazar, C.; Middleton, R.J.; Banati, R.B.; Liu, G.J. The impact of high and low dose ionising radiation on the central nervous system. Redox Biol. 2016, 9, 144–156. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.M.; Zhao, Y.M.; Luo, X.G.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.Y.; Luo, X.M.; Chen, S.D.; Wang, X.Y. Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Le, O.; Palacio, L.; Bernier, G.; Batinic-Haberle, I.; Hickson, G.; Beausejour, C. INK4a/ARF Expression Impairs Neurogenesis in the Brain of Irradiated Mice. Stem Cell Rep. 2018, 10, 1721–1733. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.; Pellegatta, S.; Favaro, R.; Pisati, F.; Roncaglia, P.; Testa, G.; Nicolis, S.K.; Finocchiaro, G.; d’Adda di Fagagna, F. DNA damage in mammalian neural stem cells leads to astrocytic differentiation mediated by BMP2 signaling through JAK-STAT. Stem Cell Rep. 2013, 1, 123–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta). J. Neuroinflamm. 2011, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Priego, N.; Valiente, M. The Potential of Astrocytes as Immune Modulators in Brain Tumors. Front. Immunol. 2019, 10, 1314. [Google Scholar] [CrossRef]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Newington, J.T.; Harris, R.A.; Cumming, R.C. Reevaluating Metabolism in Alzheimer’s Disease from the Perspective of the Astrocyte-Neuron Lactate Shuttle Model. J. Neurodegener. Dis. 2013, 2013, 234572. [Google Scholar] [CrossRef]
- Lecuyer, M.A.; Kebir, H.; Prat, A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochim. Biophys. Acta 2016, 1862, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, N.; Ellerby, L.M.; Davalos, A.R.; Zeng, X.; Campisi, J.; Desprez, P.Y. Responses of human embryonic stem cells and their differentiated progeny to ionizing radiation. Biochem. Biophys. Res. Commun. 2012, 426, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef]
- Crowe, E.P.; Tuzer, F.; Gregory, B.D.; Donahue, G.; Gosai, S.J.; Cohen, J.; Leung, Y.Y.; Yetkin, E.; Nativio, R.; Wang, L.S.; et al. Changes in the Transcriptome of Human Astrocytes Accompanying Oxidative Stress-Induced Senescence. Front. Aging Neurosci. 2016, 8, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hladik, D.; Dalke, C.; von Toerne, C.; Hauck, S.M.; Azimzadeh, O.; Philipp, J.; Ung, M.C.; Schlattl, H.; Rossler, U.; Graw, J.; et al. CREB Signaling Mediates Dose-Dependent Radiation Response in the Murine Hippocampus Two Years after Total Body Exposure. J. Proteome Res. 2020, 19, 337–345. [Google Scholar] [CrossRef]
- Seol, M.A.; Jung, U.; Eom, H.S.; Kim, S.H.; Park, H.R.; Jo, S.K. Prolonged expression of senescence markers in mice exposed to gamma-irradiation. J. Vet. Sci. 2012, 13, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimzadeh, O.; Sievert, W.; Sarioglu, H.; Merl-Pham, J.; Yentrapalli, R.; Bakshi, M.V.; Janik, D.; Ueffing, M.; Atkinson, M.J.; Multhoff, G.; et al. Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long-term radiation-induced vascular dysfunction. J. Proteome Res. 2015, 14, 1203–1219. [Google Scholar] [CrossRef]
- Le, O.N.; Rodier, F.; Fontaine, F.; Coppe, J.P.; Campisi, J.; DeGregori, J.; Laverdiere, C.; Kokta, V.; Haddad, E.; Beausejour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Tarantini, S.; Hertelendy, P.; Valcarcel-Ares, M.N.; Fulop, G.A.; Logan, S.; Kiss, T.; Farkas, E.; Csiszar, A.; Yabluchanskiy, A. Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation-induced accelerated brain senescence. Geroscience 2017, 39, 33–42. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Palmer, T.D.; Willhoite, A.R.; Gage, F.H. Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 2000, 425, 479–494. [Google Scholar] [CrossRef]
- Remes, T.M.; Suo-Palosaari, M.H.; Koskenkorva, P.K.T.; Sutela, A.K.; Toiviainen-Salo, S.M.; Arikoski, P.M.; Arola, M.O.; Heikkila, V.P.; Kapanen, M.; Lahteenmaki, P.M.; et al. Radiation-induced accelerated aging of the brain vasculature in young adult survivors of childhood brain tumors. Neurooncol. Pract. 2020, 7, 415–427. [Google Scholar] [CrossRef]
- Flanary, B. The role of microglial cellular senescence in the aging and Alzheimer diseased brain. Rejuvenation Res. 2005, 8, 82–85. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Mata-Garrido, J.; Tapia, O.; Casafont, I.; Berciano, M.T.; Cuadrado, A.; Lafarga, M. Persistent accumulation of unrepaired DNA damage in rat cortical neurons: Nuclear organization and ChIP-seq analysis of damaged DNA. Acta Neuropathol. Commun. 2018, 6, 68. [Google Scholar] [CrossRef]
- De, A.K.; Chipalkatti, S.; Aiyar, A.S. Effects of chronic irradiation on age-related biochemical changes in mice. Radiat. Res. 1983, 95, 637–645. [Google Scholar] [CrossRef]
- Tang, F.R.; Liu, L.; Wang, H.; Ho, K.J.N.; Sethi, G. Spatiotemporal dynamics of gammaH2AX in the mouse brain after acute irradiation at different postnatal days with special reference to the dentate gyrus of the hippocampus. Aging 2021, 13, 15815–15832. [Google Scholar] [CrossRef]
- Fike, J.R.; Rosi, S.; Limoli, C.L. Neural precursor cells and central nervous system radiation sensitivity. Semin Radiat. Oncol. 2009, 19, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazzaglia, S.; Briganti, G.; Mancuso, M.; Saran, A. Neurocognitive Decline Following Radiotherapy: Mechanisms and Therapeutic Implications. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Li, Y.Q.; Wong, C.S. Effects of Aging on Hippocampal Neurogenesis after Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 1181–1189. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, J.M.; Gatti, R.A. Clinical radiation sensitivity with DNA repair disorders: An overview. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahas, S.A.; Gatti, R.A. DNA double strand break repair defects, primary immunodeficiency disorders, and ‘radiosensitivity’. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 510–516. [Google Scholar] [CrossRef]
- Andreassen, C.N.; Schack, L.M.; Laursen, L.V.; Alsner, J. Radiogenomics—Current status, challenges and future directions. Cancer Lett. 2016, 382, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Foray, N.; Bourguignon, M.; Hamada, N. Individual response to ionizing radiation. Mutat. Res. Rev. Mutat. Res. 2016, 770, 369–386. [Google Scholar] [CrossRef]
- Borgmann, K.; Kocher, S.; Kriegs, M.; Mansour, W.Y.; Parplys, A.C.; Rieckmann, T.; Rothkamm, K. DNA Repair. Recent Results Cancer Res. 2016, 198, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.L. Modulating Both Tumor Cell Death and Innate Immunity Is Essential for Improving Radiation Therapy Effectiveness. Front. Immunol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Livingston, K.; Schlaak, R.A.; Puckett, L.L.; Bergom, C. The Role of Mitochondrial Dysfunction in Radiation-Induced Heart Disease: From Bench to Bedside. Front. Cardiovasc. Med. 2020, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Cabuy, E.; Newton, C.; Joksic, G.; Woodbine, L.; Koller, B.; Jeggo, P.A.; Slijepcevic, P. Accelerated telomere shortening and telomere abnormalities in radiosensitive cell lines. Radiat. Res. 2005, 164, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Castella, M.; Puerto, S.; Creus, A.; Marcos, R.; Surralles, J. Telomere length modulates human radiation sensitivity in vitro. Toxicol. Lett. 2007, 172, 29–36. [Google Scholar] [CrossRef]
- Slijepcevic, P. Is there a link between telomere maintenance and radiosensitivity? Radiat. Res. 2004, 161, 82–86. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Schaue, D.; Kachikwu, E.L.; McBride, W.H. Cytokines in radiobiological responses: A review. Radiat. Res. 2012, 178, 505–523. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.M.; Kang, C.M.; Cho, E.S.; Kang, S.M.; Lee, S.B.; Um, H.D. Ionizing radiation-induced micronucleus formation is mediated by reactive oxygen species that are produced in a manner dependent on mitochondria, Nox1, and JNK. Oncol. Rep. 2007, 17, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, Y.; Sasabe, E.; Ueta, E.; Yamamoto, T. Ionizing irradiation induces apoptotic damage of salivary gland acinar cells via NADPH oxidase 1-dependent superoxide generation. Biochem. Biophys. Res. Commun. 2008, 366, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Todd, D.G.; Mikkelsen, R.B. Ionizing radiation induces a transient increase in cytosolic free [Ca2+] in human epithelial tumor cells. Cancer Res. 1994, 54, 5224–5230. [Google Scholar]
- Ohshima, Y.; Tsukimoto, M.; Takenouchi, T.; Harada, H.; Suzuki, A.; Sato, M.; Kitani, H.; Kojima, S. gamma-Irradiation induces P2X(7) receptor-dependent ATP release from B16 melanoma cells. Biochim. Biophys. Acta 2010, 1800, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Zeh, H.J., III; Lotze, M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 2011, 14, 1315–1335. [Google Scholar] [CrossRef] [Green Version]
- Veskoukis, A.S.; Tsatsakis, A.M.; Kouretas, D. Dietary oxidative stress and antioxidant defense with an emphasis on plant extract administration. Cell Stress Chaperones 2012, 17, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.R.; Nielsen, J.B.; Nielsen, F.; Grandjean, P. Antioxidative enzyme activities in human erythrocytes. Clin. Chem. 1997, 43, 562–568. [Google Scholar] [CrossRef]
- Inal, M.E.; Kanbak, G.; Sunal, E. Antioxidant enzyme activities and malondialdehyde levels related to aging. Clin. Chim. Acta 2001, 305, 75–80. [Google Scholar] [CrossRef]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc. Soc. Exp. Biol. Med. 1999, 222, 236–245. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kasapovic, J.; Stojiljkovic, V.; Gavrilovic, L.; Popovic, N.; Milicevic, Z. Antioxidant protection against curative and palliative doses of ionizing irradiation in human blood decreases with aging. Sci. World J. 2012, 2012, 982594. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.T.; Wu, J.; Holstein, D.; Upadhyay, G.; Rourk, W.; Muller, E.; Lechleiter, J.D. Ca2+ signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol. Aging 2007, 28, 99–111. [Google Scholar] [CrossRef]
- Ghosh, D.; LeVault, K.R.; Barnett, A.J.; Brewer, G.J. A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg-AD mouse neurons. J. Neurosci. 2012, 32, 5821–5832. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. The Free Radical Theory of Aging: Effect of Age on Serum Copper Levels. J. Gerontol. 1965, 20, 151–153. [Google Scholar] [CrossRef]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a ‘good’ look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; Rizzuto, R. Mitochondrial longevity pathways. Biochim. Biophys. Acta 2011, 1813, 260–268. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Stahon, K.E.; Bastian, C.; Griffith, S.; Kidd, G.J.; Brunet, S.; Baltan, S. Age-Related Changes in Axonal and Mitochondrial Ultrastructure and Function in White Matter. J. Neurosci. 2016, 36, 9990–10001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lores-Arnaiz, S.; Lombardi, P.; Karadayian, A.G.; Orgambide, F.; Cicerchia, D.; Bustamante, J. Brain cortex mitochondrial bioenergetics in synaptosomes and non-synaptic mitochondria during aging. Neurochem. Res. 2016, 41, 353–363. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Larsson, N.G. Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 2011, 193, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnane, A.W.; Marzuki, S.; Ozawa, T.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 1, 642–645. [Google Scholar] [CrossRef]
- Fattal, O.; Budur, K.; Vaughan, A.J.; Franco, K. Review of the literature on major mental disorders in adult patients with mitochondrial diseases. Psychosomatics 2006, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Goto, S.; Kawakatsu, M.; Urata, Y.; Li, T.S. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free Radic. Res. 2012, 46, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Limoli, C.L.; Giedzinski, E.; Morgan, W.F.; Swarts, S.G.; Jones, G.D.; Hyun, W. Persistent oxidative stress in chromosomally unstable cells. Cancer Res. 2003, 63, 3107–3111. [Google Scholar]
- Robbins, M.E.; Zhao, W. Chronic oxidative stress and radiation-induced late normal tissue injury: A review. Int. J. Radiat. Biol. 2004, 80, 251–259. [Google Scholar] [CrossRef]
- Wallace, S.S. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat. Res. 1998, 150, S60–S79. [Google Scholar] [CrossRef]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [Green Version]
- Dekker, C.J.; Geurts van Kessel, W.S.; Klomp, J.P.; Pieters, J.; De Kruijff, B. Synthesis and polymorphic phase behaviour of polyunsaturated phosphatidylcholines and phosphatidylethanolamines. Chem. Phys. Lipids 1983, 33, 93–106. [Google Scholar] [CrossRef]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Blasco, M.A. Role of shelterin in cancer and aging. Aging Cell 2010, 9, 653–666. [Google Scholar] [CrossRef]
- Ilyenko, I.; Lyaskivska, O.; Bazyka, D. Analysis of relative telomere length and apoptosis in humans exposed to ionising radiation. Exp. Oncol. 2011, 33, 235–238. [Google Scholar] [PubMed]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Ojo, J.O.; Rezaie, P.; Gabbott, P.L.; Stewart, M.G. Impact of age-related neuroglial cell responses on hippocampal deterioration. Front. Aging Neurosci. 2015, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unryn, B.M.; Hao, D.; Gluck, S.; Riabowol, K.T. Acceleration of telomere loss by chemotherapy is greater in older patients with locally advanced head and neck cancer. Clin. Cancer Res. 2006, 12, 6345–6350. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Furukawa, K.; Opresko, P.L.; Xu, X.; Bohr, V.A.; Mattson, M.P. TRF2 dysfunction elicits DNA damage responses associated with senescence in proliferating neural cells and differentiation of neurons. J. Neurochem. 2006, 97, 567–581. [Google Scholar] [CrossRef]
- Crompton, N.E. Telomeres, senescence and cellular radiation response. Cell. Mol. Life Sci. 1997, 53, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Ayouaz, A.; Raynaud, C.; Heride, C.; Revaud, D.; Sabatier, L. Telomeres: Hallmarks of radiosensitivity. Biochimie 2008, 90, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Mikhel’son, V.M.; Gamalei, I.A. [Telomere shortening is the main mechanism of natural and radiation aging]. Radiat. Biol. Radioecol. 2010, 50, 269–275. [Google Scholar] [CrossRef]
- Bucher, N.; Britten, C.D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; You, L.; Xue, J.; Lu, Y. Ionizing Radiation-Induced Cellular Senescence in Normal, Non-transformed Cells and the Involved DNA Damage Response: A Mini Review. Front. Pharmacol. 2018, 9, 522. [Google Scholar] [CrossRef]
- Robello, E.; Bonetto, J.G.; Puntarulo, S. Cellular Oxidative/Antioxidant Balance in gamma-Irradiated Brain: An Update. Mini-Rev. Med. Chem. 2016, 16, 937–946. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Su, Y.; Ming, G.L.; Song, H. DNA damage and repair regulate neuronal gene expression. Cell Res. 2015, 25, 993–994. [Google Scholar] [CrossRef] [Green Version]
- Gudkov, A.V.; Komarova, E.A. The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer 2003, 3, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 2016, 37-38, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [Green Version]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Shtutman, M.; Chang, B.D.; Schools, G.P.; Broude, E.V. Cellular Model of p21-Induced Senescence. Methods Mol. Biol. 2017, 1534, 31–39. [Google Scholar] [CrossRef]
- Zhang, H. Molecular signaling and genetic pathways of senescence: Its role in tumorigenesis and aging. J. Cell Physiol. 2007, 210, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Hansen, G.; Murray, D. Role of p16(INK4A) in Replicative Senescence and DNA Damage-Induced Premature Senescence in p53-Deficient Human Cells. Biochem. Res. Int. 2012, 2012, 951574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef] [Green Version]
- Greene-Schloesser, D.; Robbins, M.E. Radiation-induced cognitive impairment—From bench to bedside. Neuro. Oncol. 2012, 14, iv37–iv44. [Google Scholar] [CrossRef] [Green Version]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015, 6, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K. The emerging role of senescent cells in tissue homeostasis and pathophysiology. Pathobiol. Aging Age Relat. Dis. 2015, 5, 27743. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. TLR-induced NF-kappaB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012, 586, 1022–1026. [Google Scholar] [CrossRef] [Green Version]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [Green Version]
- Meeren, A.V.; Bertho, J.M.; Vandamme, M.; Gaugler, M.H. Ionizing radiation enhances IL-6 and IL-8 production by human endothelial cells. Mediat. Inflamm. 1997, 6, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Morishita, Y.; Kubo, Y.; Kusunoki, Y.; Hayashi, I.; Kasagi, F.; Hakoda, M.; Kyoizumi, S.; Nakachi, K. Long-term effects of radiation dose on inflammatory markers in atomic bomb survivors. Am. J. Med. 2005, 118, 83–86. [Google Scholar] [CrossRef]
- Heissig, B.; Rafii, S.; Akiyama, H.; Ohki, Y.; Sato, Y.; Rafael, T.; Zhu, Z.; Hicklin, D.J.; Okumura, K.; Ogawa, H.; et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J. Exp. Med. 2005, 202, 739–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panes, J.; Granger, D.N. Neutrophils generate oxygen free radicals in rat mesenteric microcirculation after abdominal irradiation. Gastroenterology 1996, 111, 981–989. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, M.; Bhatt, A.N.; Das, A.; Dwarakanath, B.S.; Sharma, K. Radiation-induced autophagy: Mechanisms and consequences. Free Radic. Res. 2016, 50, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Gorbunov, N.V.; Kiang, J.G. Up-regulation of autophagy in small intestine Paneth cells in response to total-body gamma-irradiation. J. Pathol. 2009, 219, 242–252. [Google Scholar] [CrossRef]
- Kiang, J.G.; Smith, J.T.; Agravante, N.G. Geldanamycin analog 17-DMAG inhibits iNOS and caspases in gamma-irradiated human T cells. Radiat. Res. 2009, 172, 321–330. [Google Scholar] [CrossRef]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta 2007, 1776, 86–107. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Lemasters, J.J. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signal. 2011, 14, 1919–1928. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Kumar, A.V.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.D.; Jurk, D.; Khosla, S.; Kirkland, J.L.; LeBrasseur, N.K.; Miller, J.D.; Passos, J.F.; Pignolo, R.J.; Tchkonia, T.; Niedernhofer, L.J. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 779–803. [Google Scholar] [CrossRef]
- Qing, Y.; Li, H.; Zhao, Y.; Hu, P.; Wang, X.; Yu, X.; Zhu, M.; Wang, H.; Wang, Z.; Guo, Q.; et al. One-Two Punch Therapy for the Treatment of T-Cell Malignancies Involving p53-Dependent Cellular Senescence. Oxid. Med. Cell Longev. 2021, 2021, 5529518. [Google Scholar] [CrossRef]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Radiation Source | Effects | References |
|---|---|---|
| Medical radiation (radiographs, computed tomography scans) | Cardiovascular disease, premature aging, inflammation, and neurodegenerative diseases | [16,17,21] |
| Natural background radiation | Inflammation, immunosenescence, thyroid cancer, and childhood leukemia | [41,42] [43,44] |
| Nuclear disasters | “Chernobyl AIDS,” CNS damage, premature aging, atherosclerosis, and senile encephalopathy | [28,29,30] [31,32] |
| Cell Types | Models | Radiation Type & Dose/Dose-Rate | Radiation-Induced Changes | Reference |
|---|---|---|---|---|
| Microglia | Murine microglial cells BV2 and neuronal cells HT22 | 3 Gy/min (Clinac iX) (X-ray) 2 Gy/min (X-ray irradiator) | SA-β-Gal, p16INK4a, MMP3↑ | [47] |
| Primary microglia from adult male C57BI6/J mice | Single dose of 10/20 Gy at a dose rate of 3 Gy/min (Clinac iX) (X-ray) | SA-β-Gal, p16INK4a↑ | [47] | |
| Astrocytes | Non-cancerous tissue from cancer patients having received cranial radiation | IR (X-Rad 320 biologic irradiator) (X-ray) | p16INK4a, Hp1γ↑ | [48] |
| Primary human astrocytes | 0.5–20 Gy (X-ray) | SA-β-Gal, p16INK4a, p21, IL-1, IL-6, IL-8↑IGF-1, GFAP↓ DNA damage | [48] | |
| Brain endothelial cells | ATCC-derived murine brain endothelial cells, bEnd.3 | X-ray (20 Gy) | SA-β-Gal, p21, p16INK4a, ICAM-1, PAI-1↑ | [49] |
| Neurons | Male rats aged 8, 18 or 28 months | Whole-brain radiation with a single dose of 10 Gy (X-ray) | Greater inflammatory response; decrease in newborn neurons | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.-Q.; Yin, G.; Huang, J.-R.; Xi, S.-J.; Qian, F.; Lee, R.-X.; Peng, X.-C.; Tang, F.-R. Ionizing Radiation-Induced Brain Cell Aging and the Potential Underlying Molecular Mechanisms. Cells 2021, 10, 3570. https://doi.org/10.3390/cells10123570
Wang Q-Q, Yin G, Huang J-R, Xi S-J, Qian F, Lee R-X, Peng X-C, Tang F-R. Ionizing Radiation-Induced Brain Cell Aging and the Potential Underlying Molecular Mechanisms. Cells. 2021; 10(12):3570. https://doi.org/10.3390/cells10123570
Chicago/Turabian StyleWang, Qin-Qi, Gang Yin, Jiang-Rong Huang, Shi-Jun Xi, Feng Qian, Rui-Xue Lee, Xiao-Chun Peng, and Feng-Ru Tang. 2021. "Ionizing Radiation-Induced Brain Cell Aging and the Potential Underlying Molecular Mechanisms" Cells 10, no. 12: 3570. https://doi.org/10.3390/cells10123570
APA StyleWang, Q. -Q., Yin, G., Huang, J. -R., Xi, S. -J., Qian, F., Lee, R. -X., Peng, X. -C., & Tang, F. -R. (2021). Ionizing Radiation-Induced Brain Cell Aging and the Potential Underlying Molecular Mechanisms. Cells, 10(12), 3570. https://doi.org/10.3390/cells10123570