
Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression

 , , ,
, , ,
Abstract
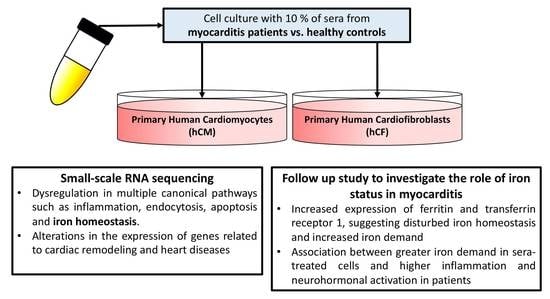
:
1. Introduction
2. Materials and Methods
2.1. Experimental Schedule
2.2. Patients
2.3. Cell Viability Tetrazolium Reduction Assay (MTS)
2.4. RNA Extraction and Library Preparation for mRNA Sequencing
2.5. RNA Sequencing and Transcriptome Analysis
2.6. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
2.7. Western Blotting (WB)
2.8. Statistical Analysis
3. Results
3.1. Cell Viability in hCM and hCF Treated with Patients’ Sera
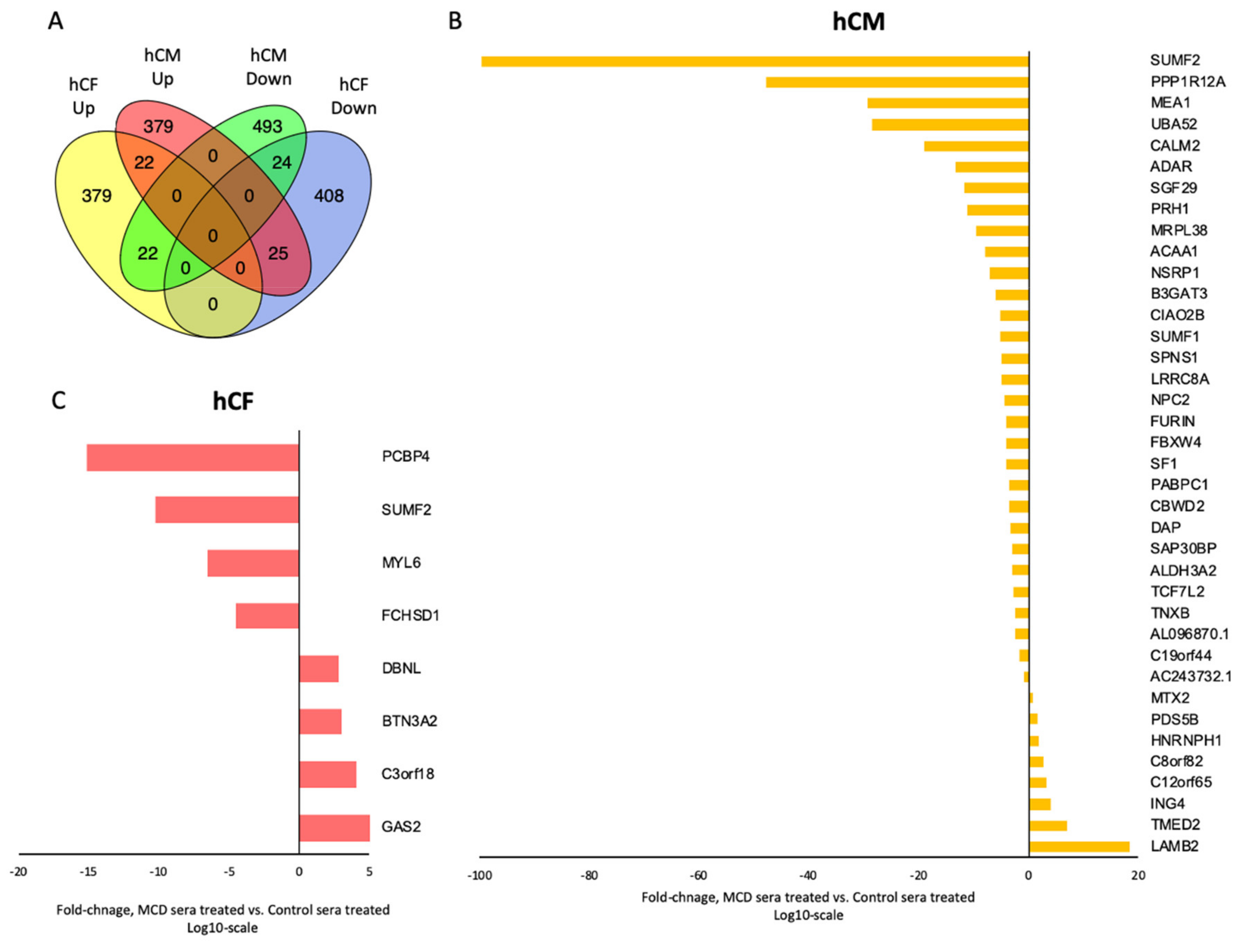
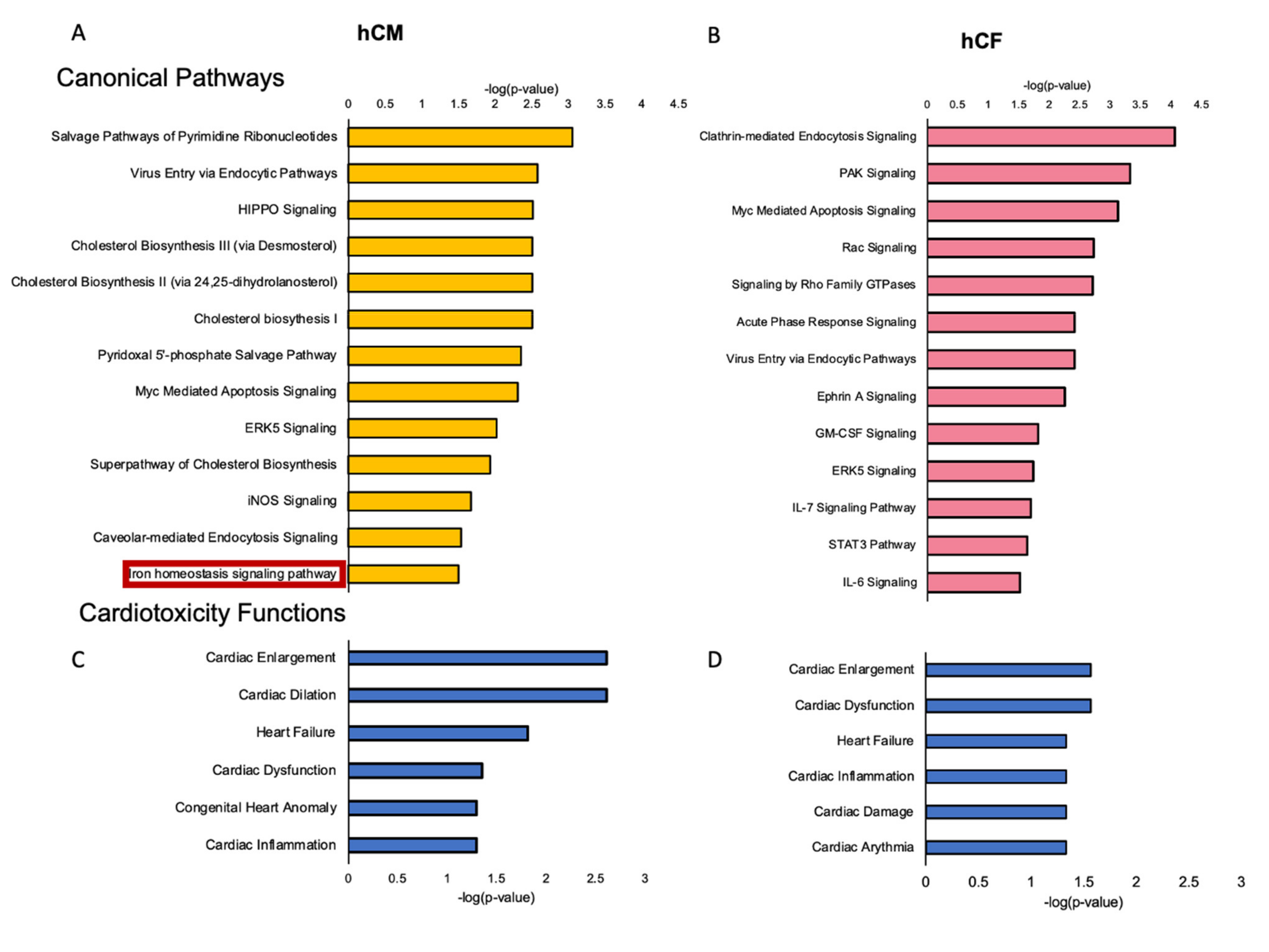
3.2. Transcriptome Profiling Indicates Alterations in the Gene Expression in hCM and hCF Treated with Acute Myocarditis Serum
3.3. Increased Expression of TFR1 Indicates an Iron Depletion in Cells Treated with Sera from Myocarditis Patients
3.4. Increased Expression of Ferritin in the Course of Myocarditis
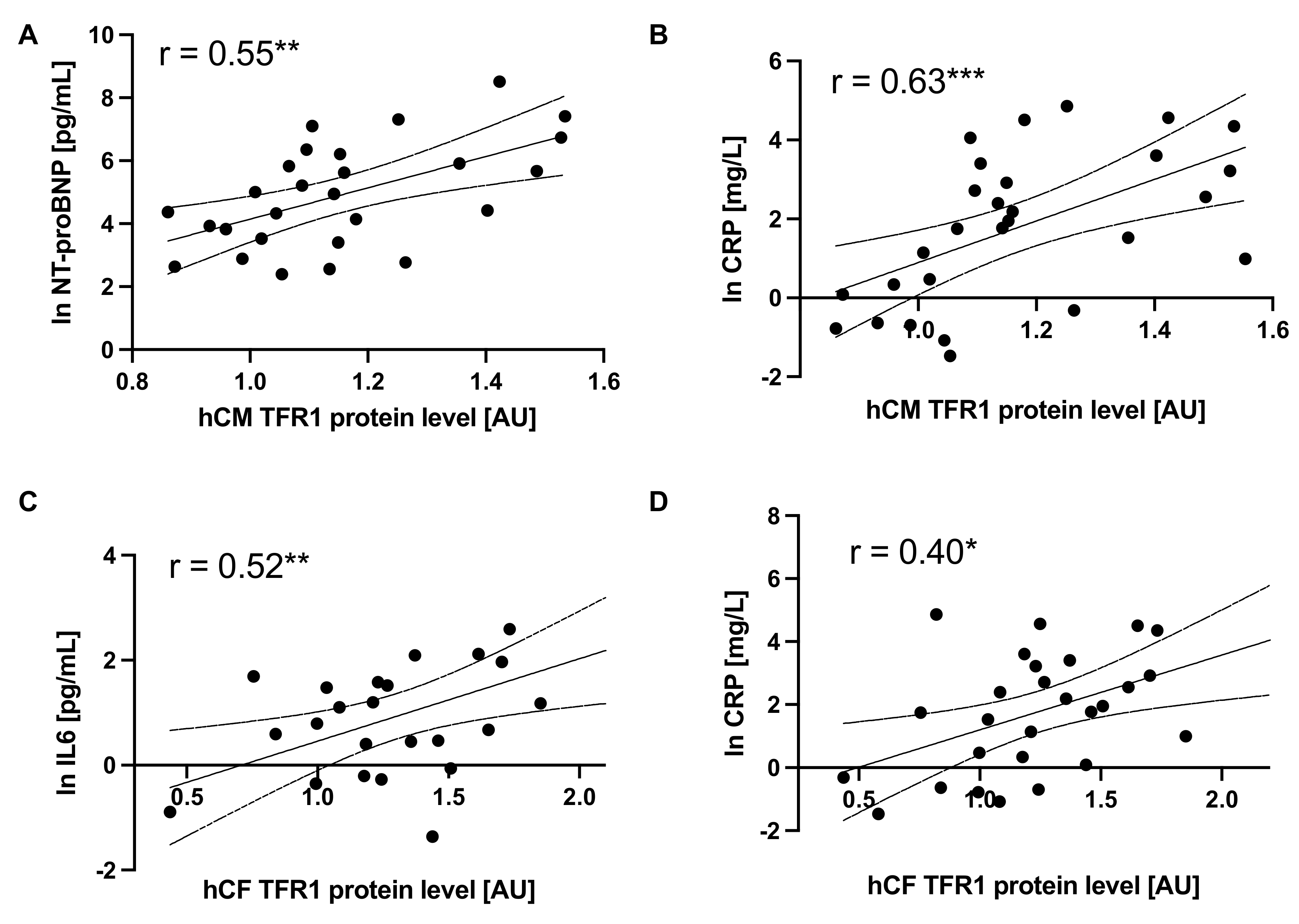
3.5. Disturbed Iron Metabolism in Cells Is Related to Inflammation and Outcome Parameters of Patients
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Błyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Magnani, J.W.; Suk Danik, H.J.; Dec, G.W.; DiSalvo, T.G. Survival in biopsy-proven myocarditis: A long-term retrospective analysis of the histopathologic, clinical, and hemodynamic predictors. Am. Heart J. 2006, 151, 463–470. [Google Scholar] [CrossRef]
- Kindermann, I.; Kindermann, M.; Kandolf, R.; Klingel, K.; Bültmann, B.; Müller, T.; Lindinger, A.; Böhm, M. Predictors of outcome in patients with suspected myocarditis. Circulation 2008, 118, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Grn, S.; Schumm, J.; Greulich, S.; Wagner, A.; Schneider, S.; Bruder, O.; Kispert, E.M.; Hill, S.; Ong, P.; Klingel, K.; et al. Long-term follow-up of biopsy-proven viral myocarditis: Predictors of mortality and incomplete recovery. J. Am. Coll. Cardiol. 2012, 59, 1604–1615. [Google Scholar] [CrossRef]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2020, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Ravingerová, T.; Kindernay, L.; Barteková, M.; Ferko, M.; Adameová, A.; Zohdi, V.; Bernátová, I.; Ferenczyová, K.; Lazou, A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889. [Google Scholar] [CrossRef] [PubMed]
- Kobak, K.A.; Radwańska, M.; Dzięgała, M.; Kasztura, M.; Josiak, K.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Structural and functional abnormalities in iron-depleted heart. Heart Fail. Rev. 2018, 24, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziegala, M.; Kobak, K.; Kasztura, M.; Bania, J.; Josiak, K.; Banasiak, W.; Ponikowski, P.; Jankowska, E.; Dziegala, M.; Kobak, K.A.; et al. Iron Depletion Affects Genes Encoding Mitochondrial Electron Transport Chain and Genes of Non Oxidative Metabolism, Pyruvate Kinase and Lactate Dehydrogenase, in Primary Human Cardiac Myocytes Cultured upon Mechanical Stretch. Cells 2018, 7, 175. [Google Scholar] [CrossRef] [Green Version]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [Green Version]
- Gordan, R.; Wongjaikam, S.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.C.; Xie, L.H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: An update. Heart Fail. Rev. 2018, 23, 801–816. [Google Scholar] [CrossRef]
- Ponikowski, P.; Kirwan, B.-A.; Anker, S.D.; McDonagh, T.; Dorobantu, M.; Drozdz, J.; Fabien, V.; Filippatos, G.; Göhring, U.M.; Keren, A.; et al. Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: A multicentre, double-blind, randomised, controlled trial. Lancet 2020, 396, 1895–1904. [Google Scholar] [CrossRef]
- Zhang, H.; Zhabyeyev, P.; Wang, S.; Oudit, G.Y. Role of iron metabolism in heart failure: From iron deficiency to iron overload. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1925–1937. [Google Scholar] [CrossRef]
- Murphy, C.J.; Oudit, G.Y. Iron-overload cardiomyopathy: Pathophysiology, diagnosis, and treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Beard, J.L. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J. Nutr. 2001, 131, 568S–580S. [Google Scholar] [CrossRef]
- Huang, M.L.-H.; Lane, D.J.R.; Richardson, D.R. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 2011, 15, 3003–3019. [Google Scholar] [CrossRef] [PubMed]
- Hower, V.; Mendes, P.; Torti, F.M.; Laubenbacher, R.; Akman, S.; Shulaev, V.; Torti, S.V. A general map of iron metabolism and tissue-specific subnetworks. Mol. Biosyst. 2009, 5, 422–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, L.L.; Suryo Rahmanto, Y.; Richardson, D.R. Iron uptake and metabolism in the new millennium. Trends Cell Biol. 2007, 17, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Tong, W.-H. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. Iron behaving badly: Inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med. Genom. 2009, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.A.; Sauve, A.A.A.; Andrews, N.C.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasztura, M.; Dzięgała, M.; Kobak, K.; Bania, J.; Mazur, G.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A.E.A.; Dziegała, M.; Kobak, K.; et al. Both iron excess and iron depletion impair viability of rat H9C2 cardiomyocytes and L6G8C5 myocytes. Kardiol. Pol. 2017, 75, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Dziegala, M.; Kasztura, M.; Kobak, K.; Bania, J.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Influence of the availability of iron during hypoxia on the genes associated with apoptotic activity and local iron metabolism in rat H9C2 cardiomyocytes and L6G8C5 skeletal myocytes. Mol. Med. Rep. 2016, 14, 3969–33977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Walker, J.M. The Bicinchoninic Acid (BCA) Assay for protein quantitation. In Basic Protein and Peptide Protocols; Humana Press: Totowa, NJ, USA, 1994; Volume 32, pp. 5–8. [Google Scholar]
- Garcia, A.M.; Nakano, S.J.; Karimpour-Fard, A.; Nunley, K.; Blain-Nelson, P.; Stafford, N.M.; Stauffer, B.L.; Sucharov, C.C.; Miyamoto, S.D. Phosphodiesterase-5 Is Elevated in Failing Single Ventricle Myocardium and Affects Cardiomyocyte Remodeling In Vitro. Circ. Heart Fail. 2018, 11, e004571. [Google Scholar] [CrossRef]
- Agnoletti, L.; Curello, S.; Bachetti, T.; Malacarne, F.; Gaia, G.; Comini, L.; Volterrani, M.; Bonetti, P.; Parrinello, G.; Cadei, M.; et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic. Role of tumor necrosis factor-α. Circulation 1999, 100, 1983–1991. [Google Scholar] [CrossRef] [Green Version]
- Valgimigli, M.; Agnoletti, L.; Curello, S.; Comini, L.; Francolini, G.; Mastrorilli, F.; Merli, E.; Pirani, R.; Guardigli, G.; Grigolato, P.G.; et al. Serum from patients with acute coronary syndromes displays a proapoptotic effect on human endothelial cells: A possible link to pan-coronary syndromes. Circulation 2003, 107, 264–270. [Google Scholar] [CrossRef]
- Glück, B.; Merkle, I.; Dornberger, G.; Stelzner, A. Expression of inducible nitric oxide synthase in experimental viral myocarditis. Herz 2000, 25, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, J.A.; Lesnefsky, E.J.; Hoit, B.D.; Smith, M.A.; Lee, H.G. Therapeutic potential of c-Myc inhibition in the treatment of hypertrophic cardiomyopathy. Ther. Adv. Chronic Dis. 2011, 2, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Lai, Y.; Sun, L.; Zhang, H. GW26-e4567 The role of Hippo signal transduction pathway in the development of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 66, C58. [Google Scholar] [CrossRef] [Green Version]
- Lindner, D.; Li, J.; Savvatis, K.; Klingel, K.; Blankenberg, S.; Tschöpe, C.; Westermann, D. Cardiac Fibroblasts Aggravate Viral Myocarditis: Cell Specific Coxsackievirus B3 Replication. Mediat. Inflamm. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Yang, W.H.; Ding, C.K.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501.e4–2508.e4. [Google Scholar] [CrossRef]
- Weiss, G.; Werner-Felmayer, G.; Werner, E.R.; Grünewald, K.; Wachter, H.; Hentze, M.W. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J. Exp. Med. 1994, 180, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Iyer, J.; Zhu, S.; Frick, K.K.; Wada, R.K.; Eskenazi, A.E.; Berg, P.E.; Ikegaki, N.; Kennett, R.H.; Frantz, C.N. Inhibition of N-myc Expression and Induction of Apoptosis by Iron Chelation in Human Neuroblastoma. Cancer Res. 2001, 61, 1073–1079. [Google Scholar]
- Chorghade, S.; Seimetz, J.; Emmons, R.; Yang, J.; Bresson, S.M.; de Lisio, M.; Parise, G.; Conrad, N.K.; Kalsotra, A. Poly(A) tail length regulates PABPC1 expression to tune translation in the heart. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Dupays, L.; Towers, N.; Wood, S.; David, A.; Stuckey, D.J.; Mohun, T. Furin, a transcriptional target of NKX2-5, has an essential role in heart development and function. PLoS ONE 2019, 14, e0212992. [Google Scholar] [CrossRef]
- Altaf, F.; Vesely, C.; Sheikh, A.M.; Munir, R.; Shah, S.T.A.; Tariq, A. Modulation of ADAR mRNA expression in patients with congenital heart defects. PLoS ONE 2019, 14, e0200968. [Google Scholar] [CrossRef]
- Sun, M.S.; Zhang, J.; Jiang, L.Q.; Pan, Y.X.; Tan, J.Y.; Yu, F.; Guo, L.; Yin, L.; Shen, C.; Shu, H.B.; et al. TMED2 Potentiates Cellular IFN Responses to DNA Viruses by Reinforcing MITA Dimerization and Facilitating Its Trafficking. Cell Rep. 2018, 25, 3086.e3–3098.e3. [Google Scholar] [CrossRef] [Green Version]
- Ponka, P.; Lok, C.N. The transferrin receptor: Role in health and disease. Int. J. Biochem. Cell Biol. 1999, 31, 1111–1137. [Google Scholar] [CrossRef]
- Maeder, M.T.; Khammy, O.; dos Remedios, C.; Kaye, D.M. Myocardial and Systemic Iron Depletion in Heart Failure. J. Am. Coll. Cardiol. 2011, 58, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure. Circulation 2018, 138, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Suchdev, P.S.; Williams, A.M.; Mei, Z.; Flores-Ayala, R.; Pasricha, S.R.; Rogers, L.M.; Namaste, S.M.L. Assessment of iron status in settings of inflammation: Challenges and potential approaches. Am. J. Clin. Nutr. 2017, 106, 1626S–1633S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northrop-Clewes, C.A. Interpreting indicators of iron status during an acute phase response—Lessons from malaria and human immunodeficiency virus. Ann. Clin. Biochem. 2008, 45, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Catharine Ross, A. Impact of chronic and acute inflammation on extra- and intracellular iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1581S–1587S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnahan, K.A.; Tanumihardjo, S.A. Undernutrition, the Acute Phase Response to Infection, and Its Effects on Micronutrient Status Indicators. Adv. Nutr. 2014, 5, 702–711. [Google Scholar] [CrossRef] [Green Version]
- Fontes, J.D.; Yamamoto, J.F.; Larson, M.G.; Wang, N.; Dallmeier, D.; Rienstra, M.; Schnabel, R.B.; Vasan, R.S.; Keaney, J.F.; Benjamin, E.J. Clinical correlates of change in inflammatory biomarkers: The Framingham Heart Study. Atherosclerosis 2013, 228, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Kyto, V.; Sipila, J.; Rautava, P. Gender differences in myocarditis: A nationwide study in Finland. Eur. Heart J. 2013, 34, 3505. [Google Scholar] [CrossRef] [Green Version]
- Fairweather, D.L.; Cooper, L.T.; Blauwet, L.A. Sex and Gender Differences in Myocarditis and Dilated Cardiomyopathy. Curr. Probl. Cardiol. 2013, 38, 7–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Reference | |
|---|---|---|
| TfR1 | qHsaCID0022106 (Bio-Rad PrimePCR™) | |
| UBC | ge-SY-12 (PrimerDesign) | |
| ATP5B | ge-SY-12; PrimerDesign | |
| RQ1/RQ2 | qHsaCtlD0001002 (Bio-Rad PrimePCR™) | |
| Gene | Sequence forward (5′–3′) | Sequence reverse (5′–3′) |
| FTH | GCTCTACGCCTCCTACGTTT | GAAGATTCGGCCACCTCGTT |
| FTL | ATTTCGACCGCGATGATGTG | CATGGCGTCTGGGGTTTTAC |
| Antigen | Dilution | Manufacturer | Ref. Number |
|---|---|---|---|
| TFR1 | 1:500 | Abcam | ab84036 |
| FTH | 1:500 | Abcam | ab81444 |
| FTL | 1:5000 | Abcam | ab186871 |
| Actin HRP | 1:5000 | Santa Cruz Biotechnology | sc-1616 HRP |
| Rabbit IgG HRP | 1:40,000 | Jackson ImmunoResearch | 111-035-045 |
| Parameter | N | Healthy Controls | N | Myocarditis (Acute Phase) | Acute Phase vs. Healthy p-Value | N | Myocarditis (After 6 Weeks of Clinical Recovery) |
|---|---|---|---|---|---|---|---|
| Age (years) | 10 | 46 ± 13 | 18 | 33 ± 8.8 | 0.005 | 18 | 33 ± 8.8 |
| Sex (% Male) | 10 | 100% | 18 | 100% | - | 18 | 100% |
| LVEF (%) | 10 | 65 ± 4.7 | 18 | 58 ± 9.4 | 0.027 | 17 | 58 ± 7.8 |
| CRP (mg/L) | 10 | 0.63(0.43–1.50) | 18 | 17(6.8–63.0) | <0.0001 | 17 | 1(0.6–3.8) |
| ALT (IU/L) | 10 | 29(21–37) | 18 | 30(25–55) | 0.23 | 17 | 22(17–39) |
| Troponin | 10 | 0.01 (0.01–0.01) | 18 | 0.87(0.26–5.7) | <0.0001 | 17 | 0.01(0.01–0.01) |
| Serum creatinine (mg/dL) | 10 | 0.88(0.86–0.97) | 0.94(0.82–1.00) | 0.99 | 17 | 0.90(0.82–0.98) | |
| NT-proBNP (pg/mL) | 10 | 40(16–77) | 18 | 315(98–939) | 0.0005 | 16 | 32(26–62) |
| IL-6 (pg/mL) | 10 | 0.73(0.2–1.9) | 16 | 4.5(1.7–7.9) | 0.0003 | 17 | 0.69(0.1–0.69) |
| Hematological Parameters and Indices of Iron Status | |||||||
| Hemoglobin concentration (g/dL) | 10 | 15.2 ± 1.2 | 18 | 15 ± 1.1 | 0.29 | 17 | 15 ± 0.94 |
| Serum iron (μg/dL) | 10 | 102 ± 47 | 18 | 69 ± 29 | 0.029 | 16 | 88 ± 20 |
| Serum ferritin (μg/L) | 10 | 154(78–213) | 18 | 234(150–418) | 0.026 | 16 | 160(112–187) |
| Soluble transferrin receptor (mg/L) | 8 | 1.2(1.0–1.6) | 18 | 1.2(1.0–1.4) | 0.81 | 17 | 1.2(1.1–1.3) |
| Transferrin saturation (%) | 10 | 30 ± 13 | 18 | 21 ± 8.4 | 0.052 | 17 | 25 ± 5.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobak, K.A.; Franczuk, P.; Schubert, J.; Dzięgała, M.; Kasztura, M.; Tkaczyszyn, M.; Drozd, M.; Kosiorek, A.; Kiczak, L.; Bania, J.; et al. Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression. Cells 2021, 10, 818. https://doi.org/10.3390/cells10040818
Kobak KA, Franczuk P, Schubert J, Dzięgała M, Kasztura M, Tkaczyszyn M, Drozd M, Kosiorek A, Kiczak L, Bania J, et al. Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression. Cells. 2021; 10(4):818. https://doi.org/10.3390/cells10040818
Chicago/Turabian StyleKobak, Kamil A., Paweł Franczuk, Justyna Schubert, Magdalena Dzięgała, Monika Kasztura, Michał Tkaczyszyn, Marcin Drozd, Aneta Kosiorek, Liliana Kiczak, Jacek Bania, and et al. 2021. "Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression" Cells 10, no. 4: 818. https://doi.org/10.3390/cells10040818
APA StyleKobak, K. A., Franczuk, P., Schubert, J., Dzięgała, M., Kasztura, M., Tkaczyszyn, M., Drozd, M., Kosiorek, A., Kiczak, L., Bania, J., Ponikowski, P., & Jankowska, E. A. (2021). Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression. Cells, 10(4), 818. https://doi.org/10.3390/cells10040818