
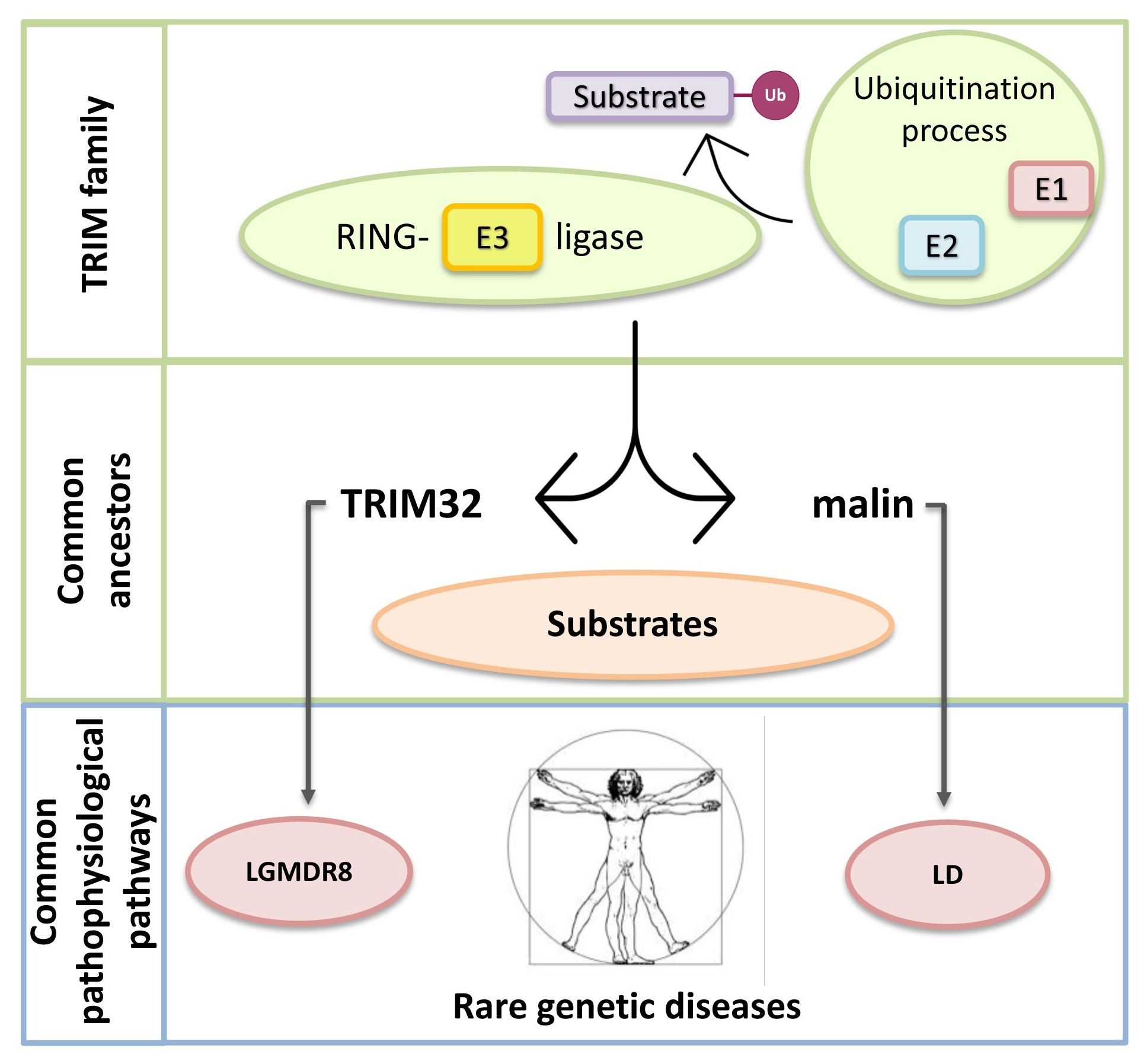
TRIM32 and Malin in Neurological and Neuromuscular Rare Diseases

 , , and
, , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
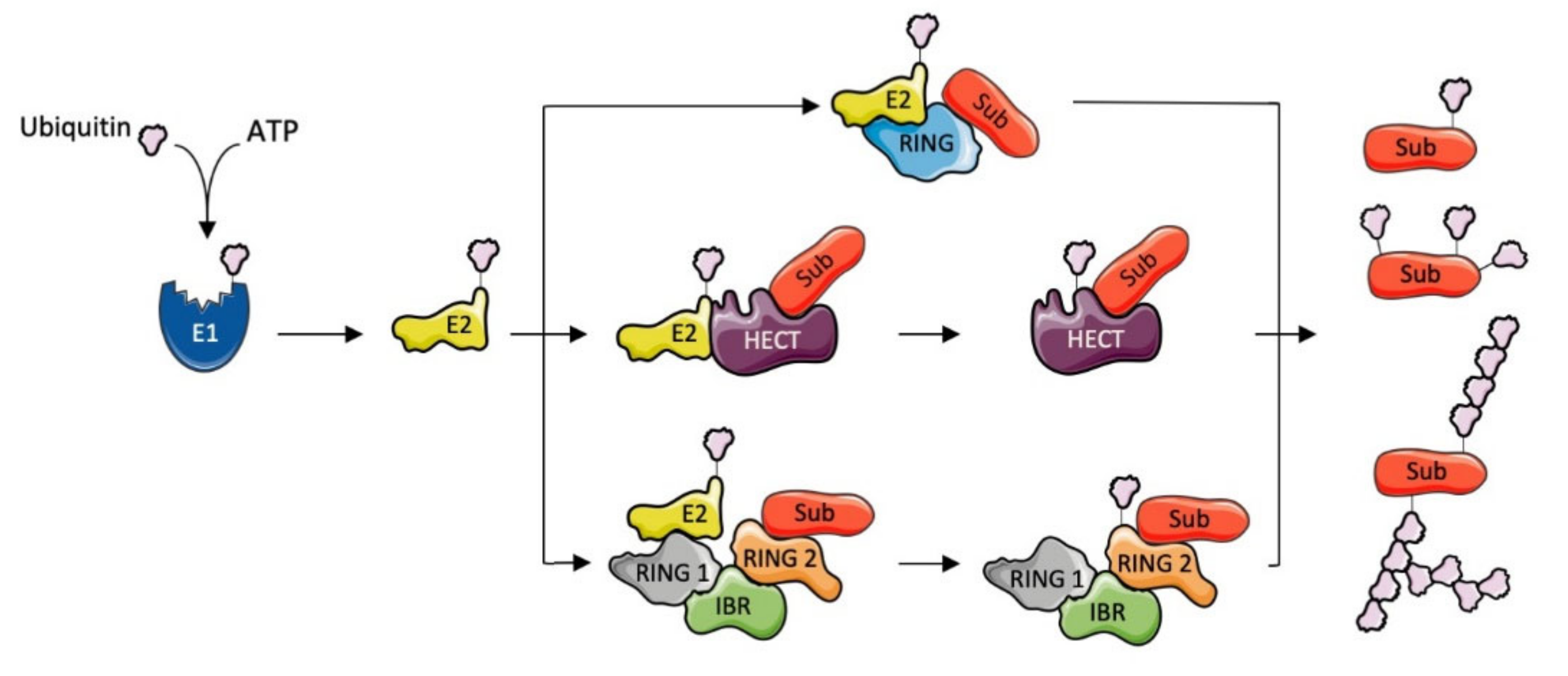
1.1. Ubiquitination
1.2. E3 Ubiquitin Ligases
1.3. TRIpartite Motif (TRIM) Ubiquitin E3 Ligases
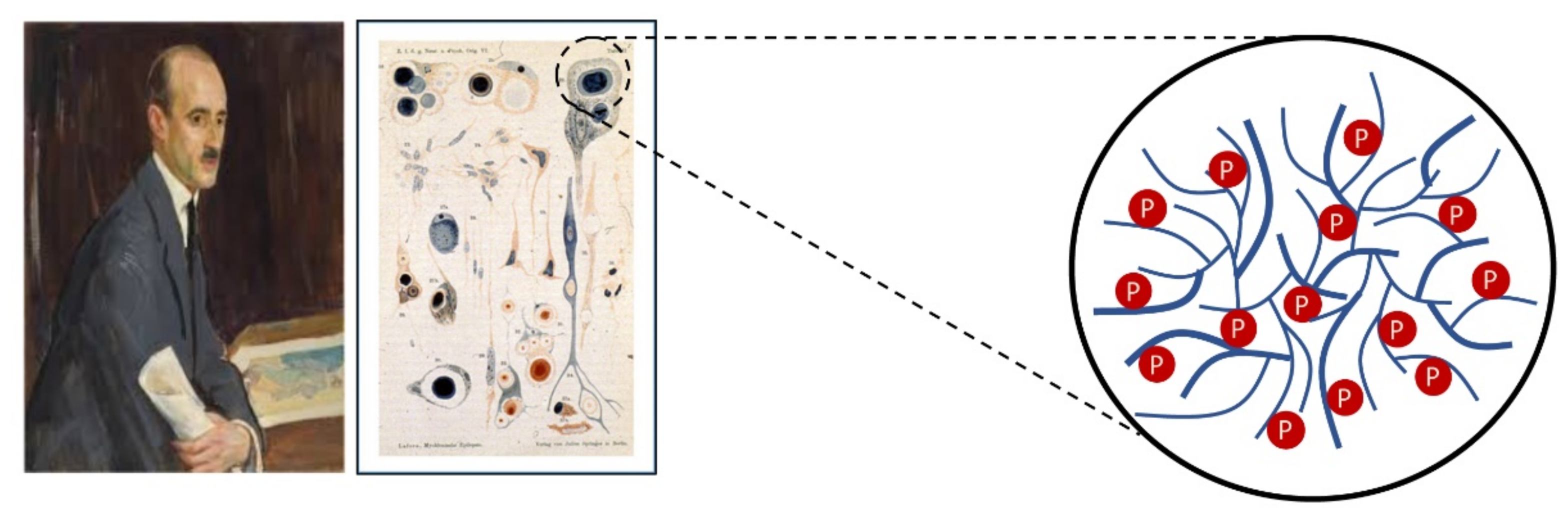
1.4. Limb-Girdle Muscular Dystrophy Type R8 and Lafora Disease
2. Structural Data and Biochemical Function of TRIM32 and Malin
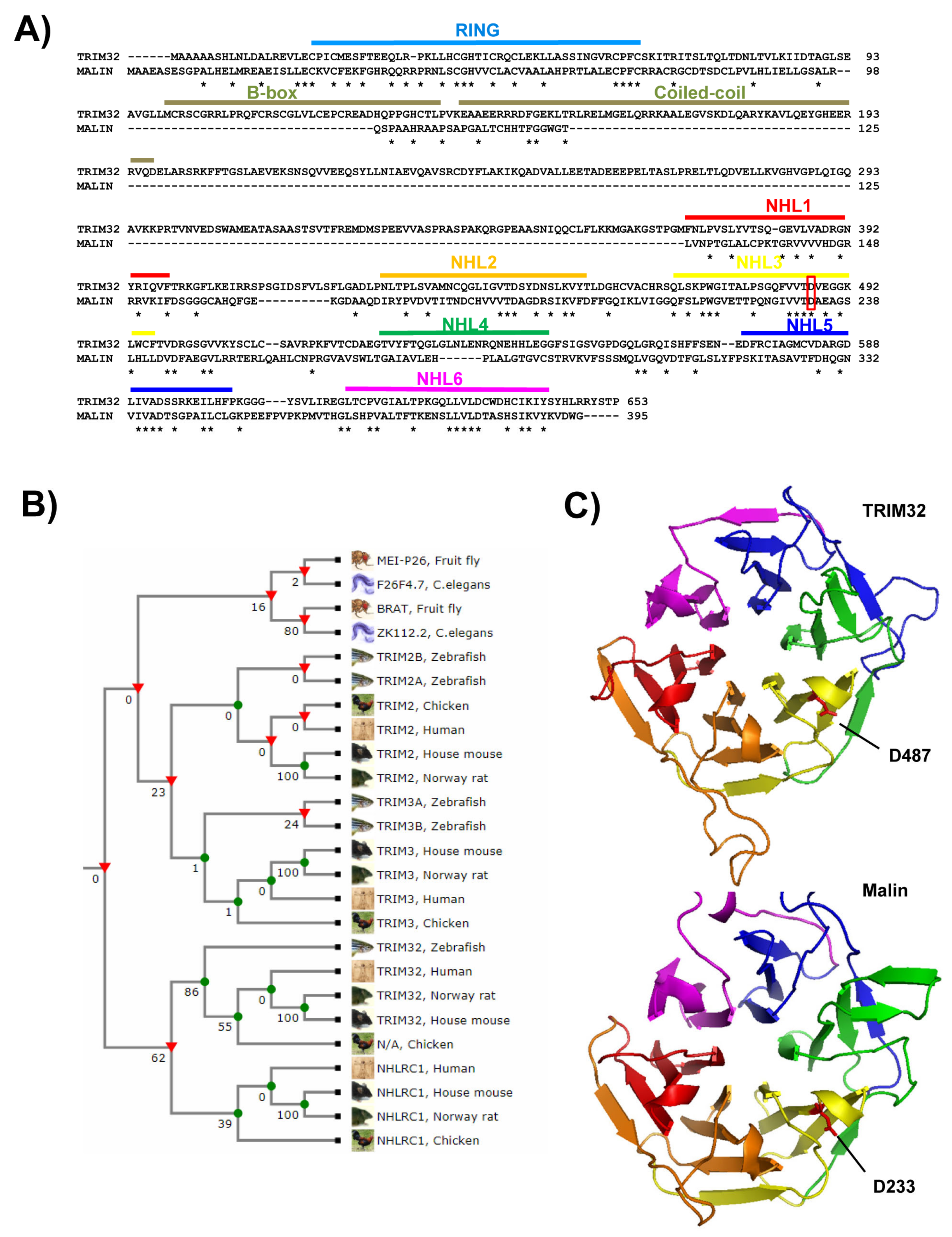
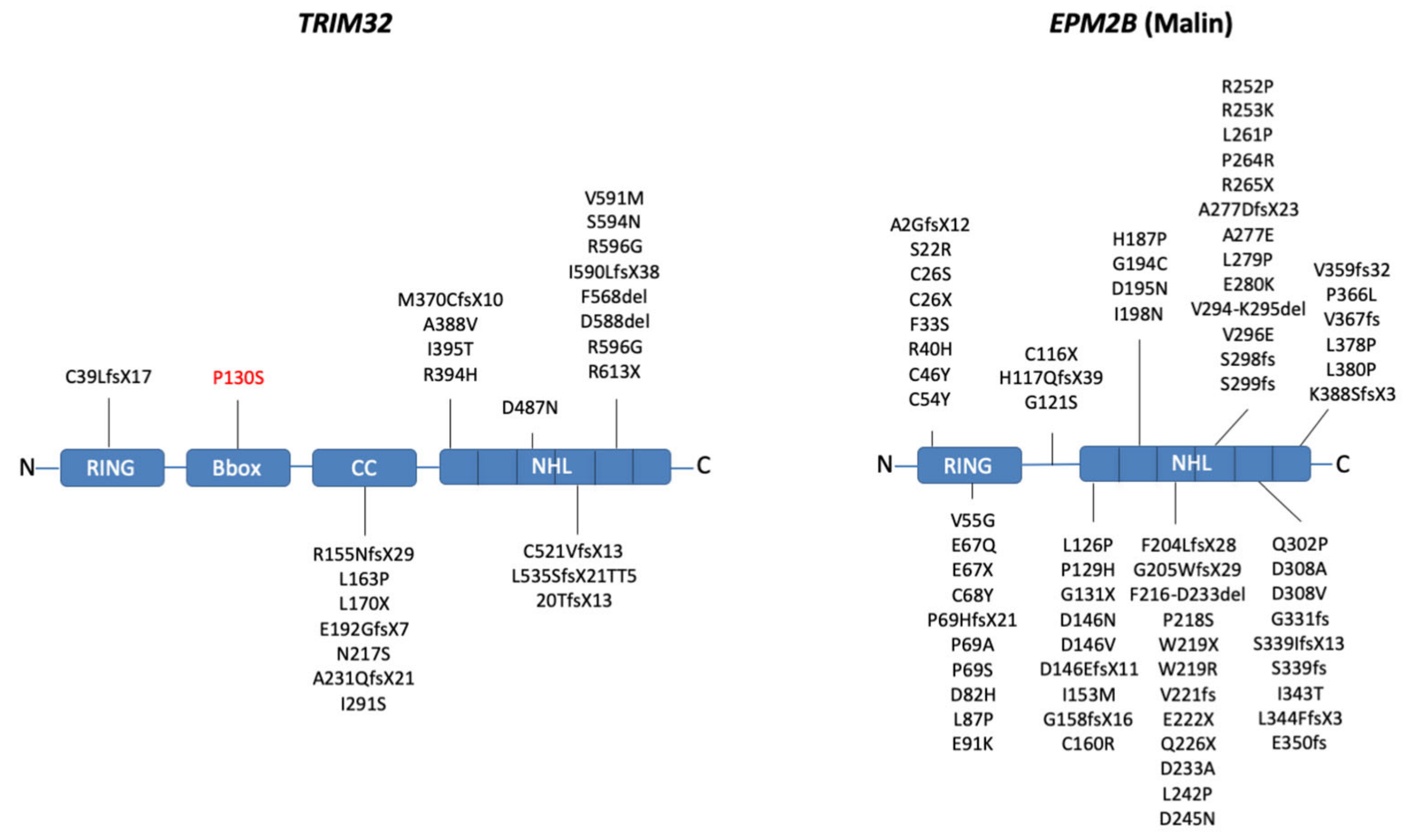
2.1. Domain Composition
2.2. Biochemical Activity
2.3. Mutations Affecting TRIM32 and Malin Activity
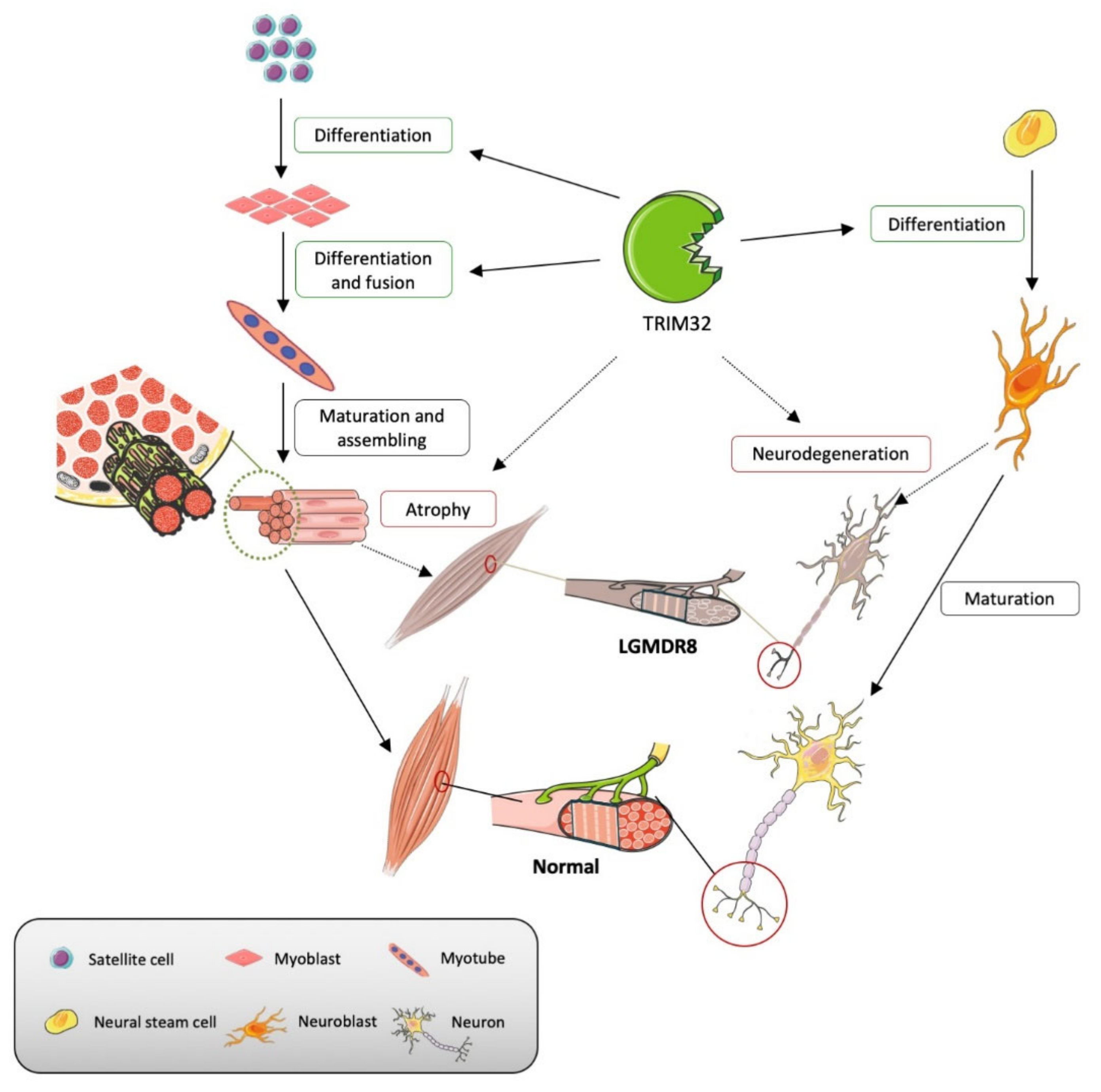
3. TRIM32 and Malin in Normal Physiological Processes
3.1. TRIM32
3.1.1. Maintenance of Sarcomeric Structures
3.1.2. Differentiation and Homeostasis of Satellite Cells
3.1.3. Differentiation and Homeostasis of Motor Neurons
3.2. Malin
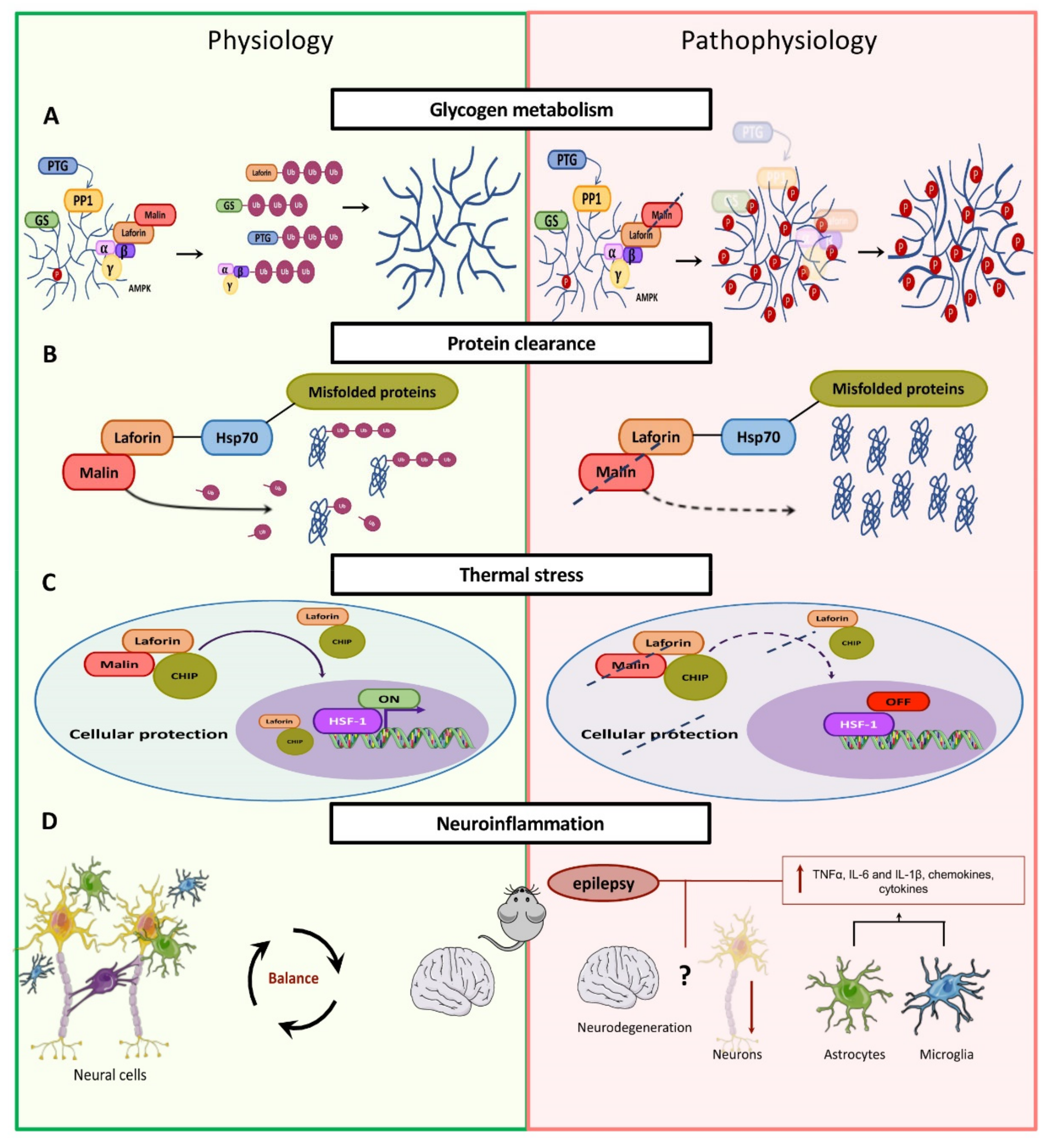
Glycogen Metabolism
4. Malin- and Trim32-Related Pathophysiological Mechanisms
4.1. TRIM32
4.1.1. Muscular Atrophy
4.1.2. Alteration of Satellite Cells
4.1.3. Alteration of Motor Neurons
4.2. Malin
4.2.1. Alteration of Glycogen Metabolism
4.2.2. Clearance of Protein Aggregates
4.2.3. Heat Shock Response
4.2.4. Neuroinflammation
5. Malin and TRIM32 Common Pathways?
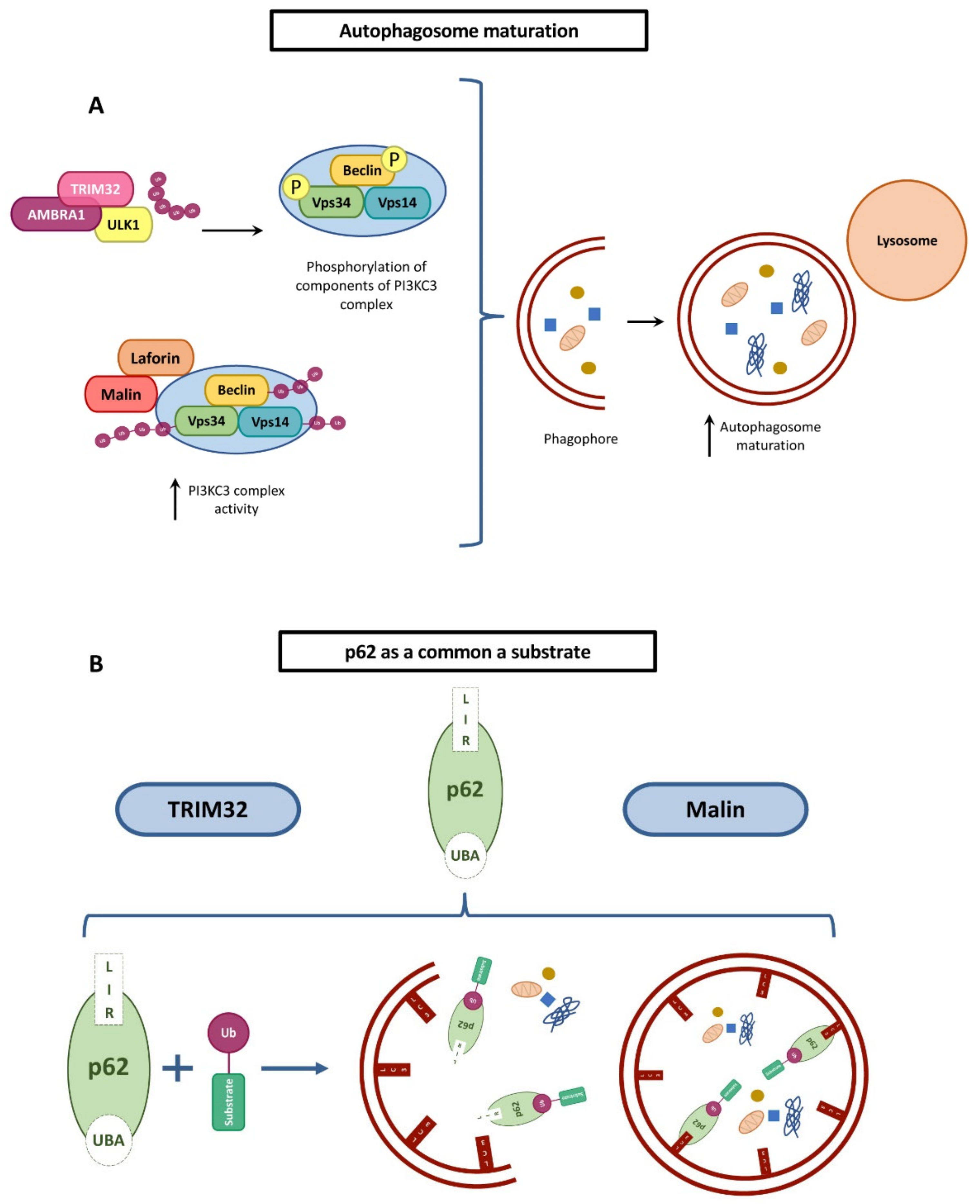
5.1. Autophagy Regulation
5.2. Regulation of WNT Pathway
5.3. Regulation of Glucose Metabolism
6. Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed consent Statement
Data availability Statement
Conflicts of Interest
References
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin e3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallette, F.A.; Richard, S. K48-linked ubiquitination and protein degradation regulate 53bp1 recruitment at DNA damage sites. Cell Res. 2012, 22, 1221–1223. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S. Trim family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Panier, S.; Durocher, D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair. 2009, 8, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Budhidarmo, R.; Nakatani, Y.; Day, C.L. Rings hold the key to ubiquitin transfer. Trends Biochem. Sci. 2012, 37, 58–65. [Google Scholar] [CrossRef]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. Ring-type e3 ligases: Master manipulators of e2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vittal, V.; Stewart, M.D.; Brzovic, P.S.; Klevit, R.E. Regulating the regulators: Recent revelations in the control of e3 ubiquitin ligases. J. Biol. Chem. 2015, 290, 21244–21251. [Google Scholar] [CrossRef] [Green Version]
- Rotin, D.; Kumar, S. Physiological functions of the hect family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Spratt, D.E.; Walden, H.; Shaw, G.S. Rbr e3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef] [Green Version]
- Meroni, G. Trim e3 ubiquitin ligases in rare genetic disorders. Adv. Exp. Med. Biol. 2020, 1233, 311–325. [Google Scholar] [PubMed]
- Slack, F.J.; Ruvkun, G. A novel repeat domain that is often associated with ring finger and b-box motifs. Trends Biochem. Sci. 1998, 23, 474–475. [Google Scholar] [CrossRef]
- Frosk, P.; Weiler, T.; Nylen, E.; Sudha, T.; Greenberg, C.R.; Morgan, K.; Fujiwara, T.M.; Wrogemann, K. Limb-girdle muscular dystrophy type 2h associated with mutation in trim32, a putative e3-ubiquitin-ligase gene. Am. J. Hum. Genet. 2002, 70, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.G.; Frosk, P.; Engel, A.G.; Klutzny, U.; Lochmuller, H.; Wrogemann, K. Commonality of trim32 mutation in causing sarcotubular myopathy and lgmd2h. Ann. Neurol. 2005, 57, 591–595. [Google Scholar] [CrossRef]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the e3 ubiquitin ligase trim32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servian-Morilla, E.; Cabrera-Serrano, M.; Rivas-Infante, E.; Carvajal, A.; Lamont, P.J.; Pelayo-Negro, A.L.; Ravenscroft, G.; Junckerstorff, R.; Dyke, J.M.; Fletcher, S.; et al. Altered myogenesis and premature senescence underlie human trim32-related myopathy. Acta Neuropathol. Commun. 2019, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Nectoux, J.; de Cid, R.; Baulande, S.; Leturcq, F.; Urtizberea, J.A.; Penisson-Besnier, I.; Nadaj-Pakleza, A.; Roudaut, C.; Criqui, A.; Orhant, L.; et al. Detection of trim32 deletions in lgmd patients analyzed by a combined strategy of cgh array and massively parallel sequencing. Eur. J. Hum. Genet. 2015, 23, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccone, V.; Palmieri, M.; Passamano, L.; Piluso, G.; Meroni, G.; Politano, L.; Nigro, V. Mutations that impair interaction properties of trim32 associated with limb-girdle muscular dystrophy 2h. Hum. Mutat. 2008, 29, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Shieh, P.B.; Kudryashova, E.; Spencer, M.J. Limb-girdle muscular dystrophy 2h and the role of trim32. Handb. Clin. Neurol. 2011, 101, 125–133. [Google Scholar]
- Kalviainen, R. Progressive myoclonus epilepsies. Semin. Neurol. 2015, 35, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan, T.S.; Delanty, N. Lafora disease: Epidemiology, pathophysiology and management. CNS Drugs 2010, 24, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Tiberia, E.; Striano, P.; Genton, P.; Carpenter, S.; Ackerley, C.A.; Minassian, B.A. Lafora disease. Epileptic Disord. 2016, 18, 38–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gimeno, M.A.; Knecht, E.; Sanz, P. Lafora disease: A ubiquitination-related pathology. Cells 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Abad, C.; Gómez-Garre, P.; Gutiérrez-Delicado, E.; Saygi, S.; Michelucci, R.; Tassinari, C.A.; Rodriguez de Cordoba, S.; Serratosa, J.M. Lafora disease due to epm2b mutations. A clinical and genetic study. Neurology 2005, 64, 982–986. [Google Scholar] [CrossRef]
- Lafora, G.R.; Glueck, B. Beitrag zur histogpathologie der myoklonischen epilepsie. Gesamte Neurol. Psychiatr. 1911, 6, 1–14. [Google Scholar] [CrossRef]
- Singh, S.; Ganesh, S. Phenotype variations in lafora progressive myoclonus epilepsy: Possible involvement of genetic modifiers? J. Hum. Genet. 2012, 57, 283–285. [Google Scholar] [CrossRef]
- Roma-Mateo, C.; Moreno, D.; Vernia, S.; Rubio, T.; Bridges, T.M.; Gentry, M.S.; Sanz, P. Lafora disease e3-ubiquitin ligase malin is related to trim32 at both the phylogenetic and functional level. BMC Evol. Biol. 2011, 11, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koliopoulos, M.G.; Esposito, D.; Christodoulou, E.; Taylor, I.A.; Rittinger, K. Functional role of trim e3 ligase oligomerization and regulation of catalytic activity. EMBO J. 2016, 35, 1204–1218. [Google Scholar] [CrossRef]
- Bawa, S.; Brooks, D.S.; Neville, K.E.; Tipping, M.; Sagar, M.A.; Kollhoff, J.A.; Chawla, G.; Geisbrecht, B.V.; Tennessen, J.M.; Eliceiri, K.W.; et al. Drosophila trim32 cooperates with glycolytic enzymes to promote cell growth. Elife 2020, 9, e52358. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Gameros, S.; Baumann, K.; Brooks, D.S.; Kollhoff, J.A.; Zolkiewski, M.; Re Cecconi, A.D.; Panini, N.; Russo, M.; Piccirillo, R.; et al. Costameric integrin and sarcoglycan protein levels are altered in a drosophila model for limb-girdle muscular dystrophy type 2h. Mol. Biol. Cell 2021, 32, 260–273. [Google Scholar] [CrossRef]
- Gentry, M.S.; Worby, C.A.; Dixon, J.E. Insights into lafora disease: Malin is an e3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA 2005, 102, 8501–8506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohi, H.; Ianzano, L.; Zhao, X.C.; Chan, E.M.; Turnbull, J.; Scherer, S.W.; Ackerley, C.A.; Minassian, B.A. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 2005, 14, 2727–2736. [Google Scholar] [CrossRef]
- Lazzari, E.; El-Halawany, M.S.; De March, M.; Valentino, F.; Cantatore, F.; Migliore, C.; Onesti, S.; Meroni, G. Analysis of the zn-binding domains of trim32, the e3 ubiquitin ligase mutated in limb girdle muscular dystrophy 2h. Cells 2019, 8, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Kudryashov, D.; Kramerova, I.; Spencer, M.J. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2h that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005, 354, 413–424. [Google Scholar] [CrossRef]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. Trim32 senses and restricts influenza a virus by ubiquitination of pb1 polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overa, K.S.; Garcia-Garcia, J.; Bhujabal, Z.; Jain, A.; Overvatn, A.; Larsen, K.B.; Deretic, V.; Johansen, T.; Lamark, T.; Sjottem, E. Trim32, but not its muscular dystrophy-associated mutant, positively regulates and is targeted to autophagic degradation by p62/sqstm1. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. Trim32 protein modulates type i interferon induction and cellular antiviral response by targeting mita/sting protein for k63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [Green Version]
- Solaz-Fuster, M.C.; Gimeno-Alcaniz, J.V.; Ros, S.; Fernandez-Sanchez, M.E.; Garcia-Fojeda, B.; Criado Garcia, O.; Vilchez, D.; Dominguez, J.; Garcia-Rocha, M.; Sanchez-Piris, M.; et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the amp-activated protein kinase pathway. Hum. Mol. Genet. 2008, 17, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Eddins, M.J.; Carlile, C.M.; Gomez, K.M.; Pickart, C.M.; Wolberger, C. Mms2-ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat. Struct. Mol. Biol. 2006, 13, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, P.; Roma-Mateo, C.; Viana, R.; Sanz, P. Ubiquitin conjugating enzyme e2-n and sequestosome-1 (p62) are components of the ubiquitination process mediated by the malin-laforin e3-ubiquitin ligase complex. Int. J. Biochem. Cell Biol. 2015, 69, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional interactions between ubiquitin e2 enzymes and trim proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plechanovova, A.; Jaffray, E.G.; McMahon, S.A.; Johnson, K.A.; Navratilova, I.; Naismith, J.H.; Hay, R.T. Mechanism of ubiquitylation by dimeric ring ligase rnf4. Nat. Struct. Mol. Biol. 2011, 18, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.G.; Chiang, J.J.; Sparrer, K.M.J.; Alam, S.L.; Chi, M.; Roganowicz, M.D.; Sankaran, B.; Gack, M.U.; Pornillos, O. Mechanism of trim25 catalytic activation in the antiviral rig-i pathway. Cell Rep. 2016, 16, 1315–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. Ring dimerization links higher-order assembly of trim5alpha to synthesis of k63-linked polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Bushby, K.M. Making sense of the limb-girdle muscular dystrophies. Brain 1999, 122 Pt 8, 1403–1420. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Ganesh, S. Lafora progressive myoclonus epilepsy: A meta-analysis of reported mutations in the first decade following the discovery of the epm2a and nhlrc1 genes. Hum. Mutat. 2009, 30, 715–723. [Google Scholar] [CrossRef]
- Couarch, P.; Vernia, S.; Gourfinkel-An, I.; Lesca, G.; Gataullina, S.; Fedirko, E.; Trouillard, O.; Depienne, C.; Dulac, O.; Steschenko, D.; et al. Lafora progressive myoclonus epilepsy: Nhlrc1 mutations affect glycogen metabolism. J. Mol. Med. 2011, 89, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Nallamilli, B.R.R.; Chakravorty, S.; Kesari, A.; Tanner, A.; Ankala, A.; Schneider, T.; da Silva, C.; Beadling, R.; Alexander, J.J.; Askree, S.H.; et al. Genetic landscape and novel disease mechanisms from a large lgmd cohort of 4656 patients. Ann. Clin. Transl. Neurol. 2018, 5, 1574–1587. [Google Scholar] [CrossRef]
- Chiang, A.P.; Beck, J.S.; Yen, H.J.; Tayeh, M.K.; Scheetz, T.E.; Swiderski, R.E.; Nishimura, D.Y.; Braun, T.A.; Kim, K.Y.; Huang, J.; et al. Homozygosity mapping with snp arrays identifies trim32, an e3 ubiquitin ligase, as a bardet-biedl syndrome gene (bbs11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Struyk, A.; Mokhonova, E.; Cannon, S.C.; Spencer, M.J. The common missense mutation d489n in trim32 causing limb girdle muscular dystrophy 2h leads to loss of the mutated protein in knock-in mice resulting in a trim32-null phenotype. Hum. Mol. Genet. 2011, 20, 3925–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, K.; Stucka, R.; Locke, M.; Melin, E.; Ahlberg, G.; Klutzny, U.; Hagen, M.; Huebner, A.; Lochmuller, H.; Wrogemann, K.; et al. Intragenic deletion of trim32 in compound heterozygotes with sarcotubular myopathy/lgmd2h. Hum. Mutat. 2009, 30, E831–E844. [Google Scholar] [CrossRef] [PubMed]
- Locke, M.; Tinsley, C.L.; Benson, M.A.; Blake, D.J. Trim32 is an e3 ubiquitin ligase for dysbindin. Hum. Mol. Genet. 2009, 18, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, S.; Otto, A.; Wu, X.; Miller, P.; Stelzer, S.; Wen, Y.; Kuang, S.; Wrogemann, K.; Patel, K.; Ding, H.; et al. Trim32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PLoS ONE 2012, 7, e30445. [Google Scholar] [CrossRef] [Green Version]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The trim-nhl protein trim32 activates micrornas and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Lazzari, E.; Meroni, G. Trim32 ubiquitin e3 ligase, one enzyme for several pathologies: From muscular dystrophy to tumours. Int. J. Biochem. Cell Biol. 2016, 79, 469–477. [Google Scholar] [CrossRef]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by trim32 causes coupled loss of desmin, z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef]
- Hillje, A.L.; Worlitzer, M.M.; Palm, T.; Schwamborn, J.C. Neural stem cells maintain their stemness through protein kinase c zeta-mediated inhibition of trim32. Stem Cells 2011, 29, 1437–1447. [Google Scholar]
- Mokhonova, E.I.; Avliyakulov, N.K.; Kramerova, I.; Kudryashova, E.; Haykinson, M.J.; Spencer, M.J. The e3 ubiquitin ligase trim32 regulates myoblast proliferation by controlling turnover of ndrg2. Hum. Mol. Genet. 2015, 24, 2873–2883. [Google Scholar] [CrossRef] [Green Version]
- Albor, A.; El-Hizawi, S.; Horn, E.J.; Laederich, M.; Frosk, P.; Wrogemann, K.; Kulesz-Martin, M. The interaction of piasy with trim32, an e3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2h, promotes piasy degradation and regulates uvb-induced keratinocyte apoptosis through nfkappab. J. Biol. Chem. 2006, 281, 25850–25866. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.S.; Guinovart, J.J.; Minassian, B.A.; Roach, P.J.; Serratosa, J.M. Lafora disease offers a unique window into neuronal glycogen metabolism. J. Biol. Chem. 2018, 293, 7117–7125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C., Jr.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase a. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilchez, D.; Ros, S.; Cifuentes, D.; Pujadas, L.; Valles, J.; Garcia-Fojeda, B.; Criado-Garcia, O.; Fernandez-Sanchez, E.; Medrano-Fernandez, I.; Dominguez, J.; et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 2007, 10, 1407–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, S.; Cuthbertson, D.J.; Cunningham, J.; Sales, M.; Cohen, P.T. Human skeletal muscle expresses a glycogen-targeting subunit of pp1 that is identical to the insulin-sensitive glycogen-targeting subunit g(l) of liver. Diabetes 2002, 51, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (ptg). J. Biol. Chem. 2008, 283, 4069–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, D.; Towler, M.C.; Hardie, D.G.; Knecht, E.; Sanz, P. The laforin-malin complex, involved in lafora disease, promotes the incorporation of k63-linked ubiquitin chains into amp-activated protein kinase beta subunits. Mol. Biol. Cell 2010, 21, 2578–2588. [Google Scholar] [CrossRef] [Green Version]
- Kudryashova, E.; Kramerova, I.; Spencer, M.J. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2h. J. Clin. Investig. 2012, 122, 1764–1776. [Google Scholar] [CrossRef] [Green Version]
- Brewer, M.K.; Putaux, J.L.; Rondon, A.; Uittenbogaard, A.; Sullivan, M.A.; Gentry, M.S. Polyglucosan body structure in lafora disease. Carbohydr. Polym. 2020, 240, 116260. [Google Scholar] [CrossRef]
- Cavanagh, J.B. Corpora-amylacea and the family of polyglucosan diseases. Brain Res. Rev. 1999, 29, 265–295. [Google Scholar] [CrossRef]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 2006, 281, 30412–30418. [Google Scholar] [CrossRef] [Green Version]
- Nitschke, F.; Wang, P.; Schmieder, P.; Girard, J.M.; Awrey, D.E.; Wang, T.; Israelian, J.; Zhao, X.; Turnbull, J.; Heydenreich, M.; et al. Hyperphosphorylation of glucosyl c6 carbons and altered structure of glycogen in the neurodegenerative epilepsy lafora disease. Cell Metab. 2013, 17, 756–767. [Google Scholar] [CrossRef] [Green Version]
- Roma-Mateo, C.; Sanz, P.; Gentry, M.S. Deciphering the role of malin in the lafora progressive myoclonus epilepsy. IUBMB Life 2012, 64, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garyali, P.; Siwach, P.; Singh, P.K.; Puri, R.; Mittal, S.; Sengupta, S.; Parihar, R.; Ganesh, S. The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum. Mol. Genet. 2009, 18, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Badhwar, I.; Upadhyay, M.; Singh, S.; Ganesh, S. Malin and laforin are essential components of a protein complex that protects cells from thermal stress. J. Cell Sci. 2011, 124, 2277–2286. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.N.; Sharma, J.; Maity, R.; Jana, N.R. Co-chaperone chip stabilizes aggregate-prone malin, a ubiquitin ligase mutated in lafora disease. J. Biol. Chem. 2010, 285, 1404–1413. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gonzalez, I.; Viana, R.; Sanz, P.; Ferrer, I. Inflammation in lafora disease: Evolution with disease progression in laforin and malin knock-out mouse models. Mol. Neurobiol. 2017, 54, 3119–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahuerta, M.; Gonzalez, D.; Aguado, C.; Fathinajafabadi, A.; Garcia-Gimenez, J.L.; Moreno-Estelles, M.; Roma-Mateo, C.; Knecht, E.; Pallardo, F.V.; Sanz, P. Reactive glia-derived neuroinflammation: A novel hallmark in lafora progressive myoclonus epilepsy that progresses with age. Mol. Neurobiol. 2020, 57, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Bauer, S.; Bozzi, Y.; Caleo, M.; Dingledine, R.; Gorter, J.A.; Henshall, D.C.; Kaufer, D.; Koh, S.; Loscher, W.; et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017, 58 (Suppl. S3), 27–38. [Google Scholar] [CrossRef]
- Auge, E.; Pelegri, C.; Manich, G.; Cabezon, I.; Guinovart, J.J.; Duran, J.; Vilaplana, J. Astrocytes and neurons produce distinct types of polyglucosan bodies in lafora disease. Glia 2018, 66, 2094–2107. [Google Scholar] [CrossRef]
- Rubio-Villena, C.; Viana, R.; Bonet, J.; Garcia-Gimeno, M.A.; Casado, M.; Heredia, M.; Sanz, P. Astrocytes: New players in progressive myoclonus epilepsy of lafora type. Hum. Mol. Genet. 2018, 27, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. Trim-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, P.; Lahuerta, M.; Viana, R.; Knecht, E.; Sanz, P. Regulation of the autophagic pi3kc3 complex by laforin/malin e3-ubiquitin ligase, two proteins involved in lafora disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118613. [Google Scholar] [CrossRef] [PubMed]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. P62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. P62/sqstm1 binds directly to atg8/lc3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Di Rienzo, M.; Antonioli, M.; Fusco, C.; Liu, Y.; Mari, M.; Orhon, I.; Refolo, G.; Germani, F.; Corazzari, M.; Romagnoli, A.; et al. Autophagy induction in atrophic muscle cells requires ulk1 activation by trim32 through unanchored k63-linked polyubiquitin chains. Sci. Adv. 2019, 5, eaau8857. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Mulherkar, S.; Mukherjee, D.; Jana, N.R. Malin regulates wnt signaling pathway through degradation of dishevelled2. J. Biol. Chem. 2012, 287, 6830–6839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.; Li, L.; Mubarokah, S.N.; Meech, R. Wnt/beta-catenin signaling induces the myomirs mir-133b and mir-206 to suppress pax7 and induce the myogenic differentiation program. J. Cell. Biochem. 2019, 120, 12740–12751. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Guo, Q.; Chen, Q.; Han, Z.; Zhou, X.; Wu, L.; Guo, X.; Ni, B.; Yang, J. Trim32 triggers beta-catenin signaling through ubiquitylation of axin1 to promote inflammatory factor-induced apoptosis of rat nucleus pulposus cells. Am. J. Physiol. Cell Physiol. 2020, 318, C695–C703. [Google Scholar] [CrossRef]
- Wang, C.; Xu, J.; Fu, H.; Zhang, Y.; Zhang, X.; Yang, D.; Zhu, Z.; Wei, Z.; Hu, Z.; Yan, R.; et al. Trim32 promotes cell proliferation and invasion by activating beta-catenin signalling in gastric cancer. J. Cell. Mol. Med. 2018, 22, 5020–5028. [Google Scholar] [CrossRef]
- Cohen, S.; Lee, D.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Trim32 reduces pi3k-akt-foxo signaling in muscle atrophy by promoting plakoglobin-pi3k dissociation. J. Cell Biol. 2014, 204, 747–758. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumarasinghe, L.; Xiong, L.; Garcia-Gimeno, M.A.; Lazzari, E.; Sanz, P.; Meroni, G. TRIM32 and Malin in Neurological and Neuromuscular Rare Diseases. Cells 2021, 10, 820. https://doi.org/10.3390/cells10040820
Kumarasinghe L, Xiong L, Garcia-Gimeno MA, Lazzari E, Sanz P, Meroni G. TRIM32 and Malin in Neurological and Neuromuscular Rare Diseases. Cells. 2021; 10(4):820. https://doi.org/10.3390/cells10040820
Chicago/Turabian StyleKumarasinghe, Lorena, Lu Xiong, Maria Adelaida Garcia-Gimeno, Elisa Lazzari, Pascual Sanz, and Germana Meroni. 2021. "TRIM32 and Malin in Neurological and Neuromuscular Rare Diseases" Cells 10, no. 4: 820. https://doi.org/10.3390/cells10040820
APA StyleKumarasinghe, L., Xiong, L., Garcia-Gimeno, M. A., Lazzari, E., Sanz, P., & Meroni, G. (2021). TRIM32 and Malin in Neurological and Neuromuscular Rare Diseases. Cells, 10(4), 820. https://doi.org/10.3390/cells10040820