
Bone Marrow-Derived IL-1Ra Increases TNF Levels Poststroke

 ,
,  ,
,
Abstract
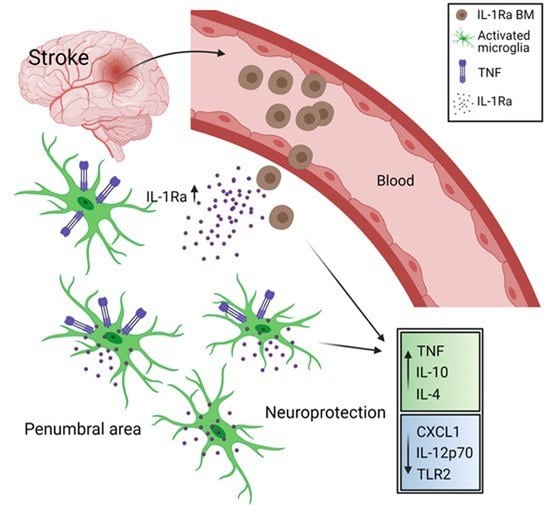
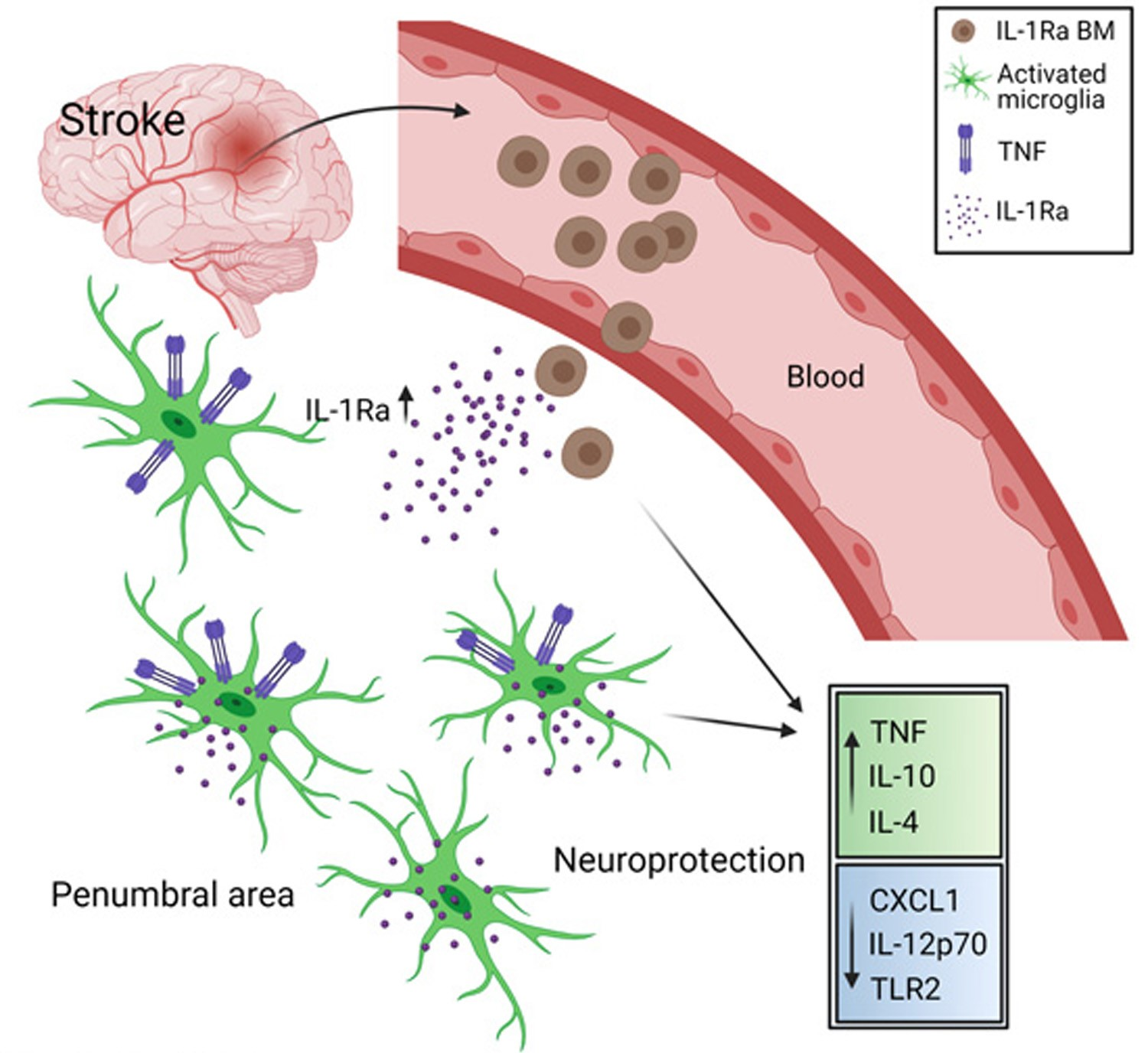
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Study Design and Animal Ethics
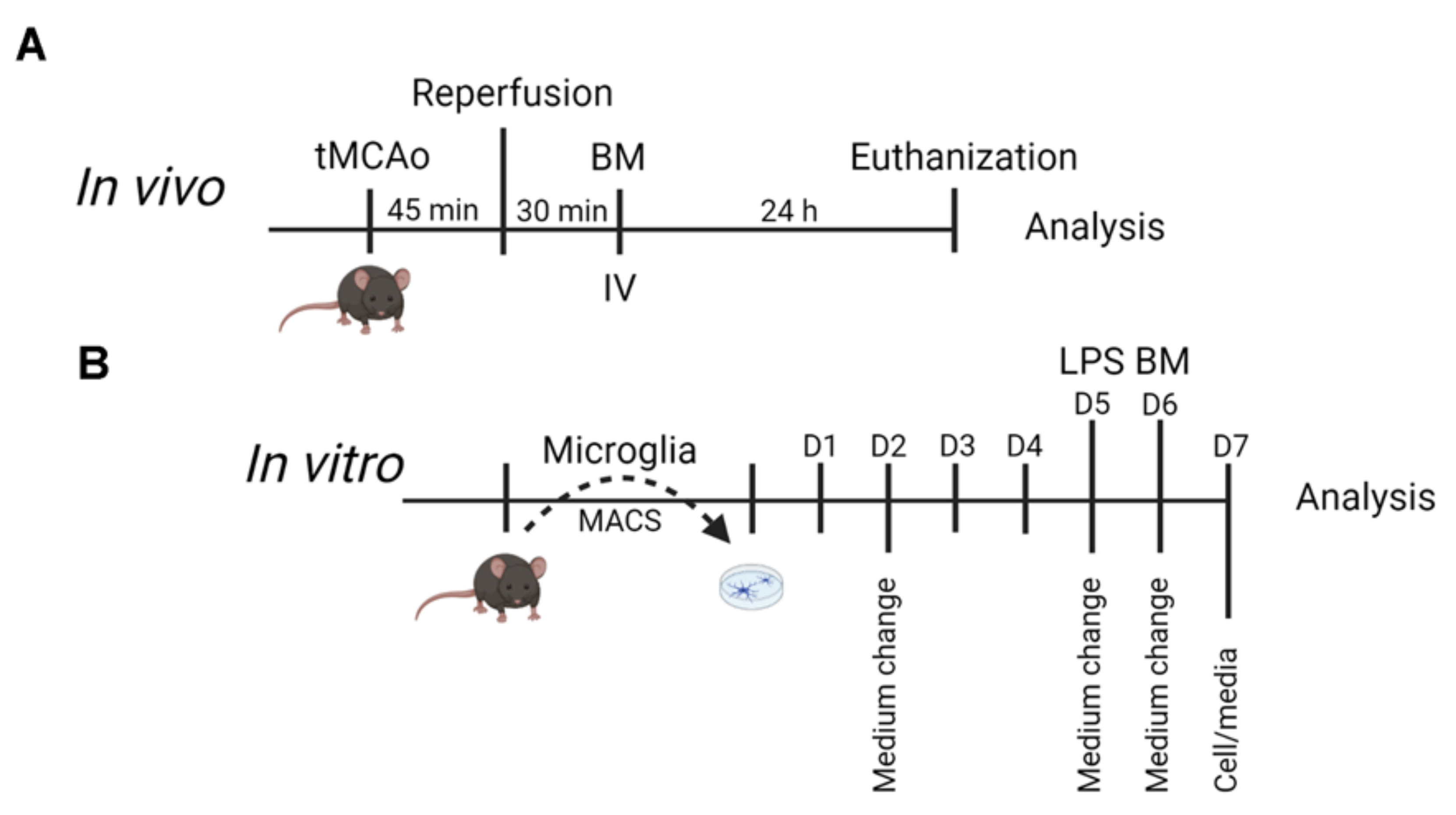
2.2. Mice Used for tMCAo and BM Treatment
2.3. Mice Used for Adult Microglial Cultures
2.4. Tissue Processing
2.5. Electrochemiluminescence and ELISA Analyses
2.6. Quantitative PrimePCR
2.7. Adult Microglia Cultures
2.8. Immunofluorescence
2.9. Statistics
3. Results
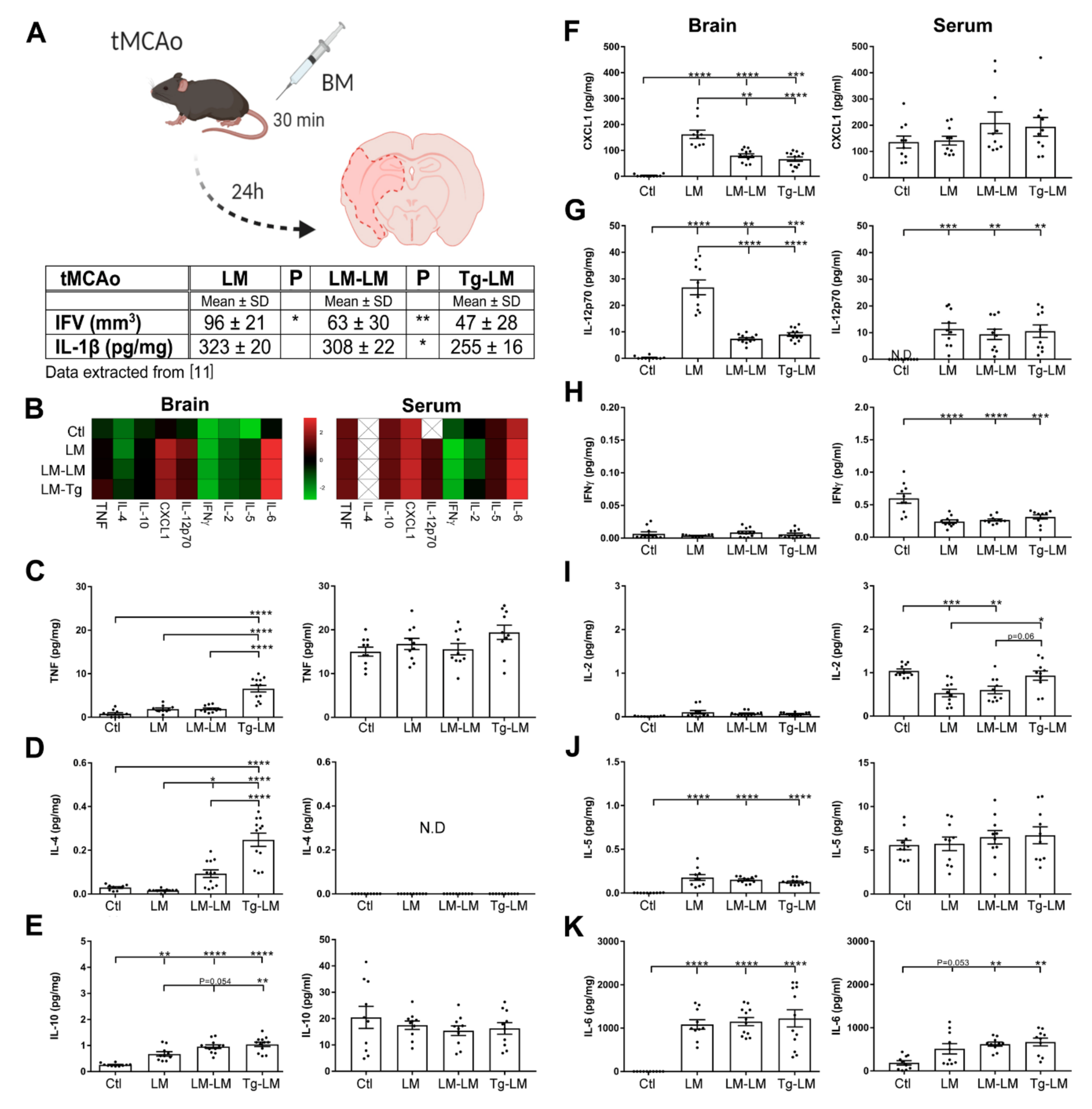
3.1. TNF and IL-1Ra Transcript Levels Are Affected by tMCAo in Mice
3.2. IL-1Ra BM Treatment Increases TNF Protein Levels after tMCAo
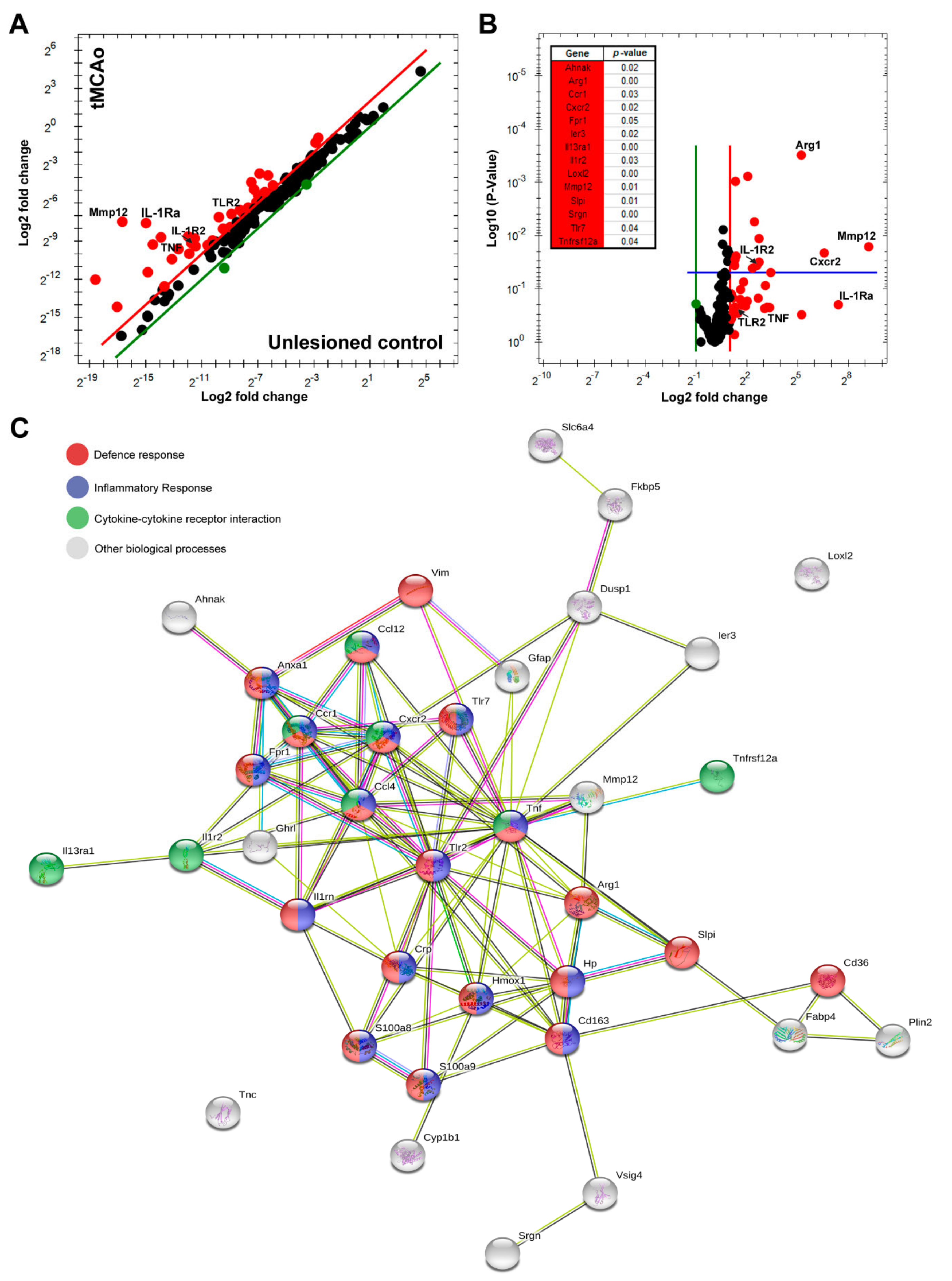
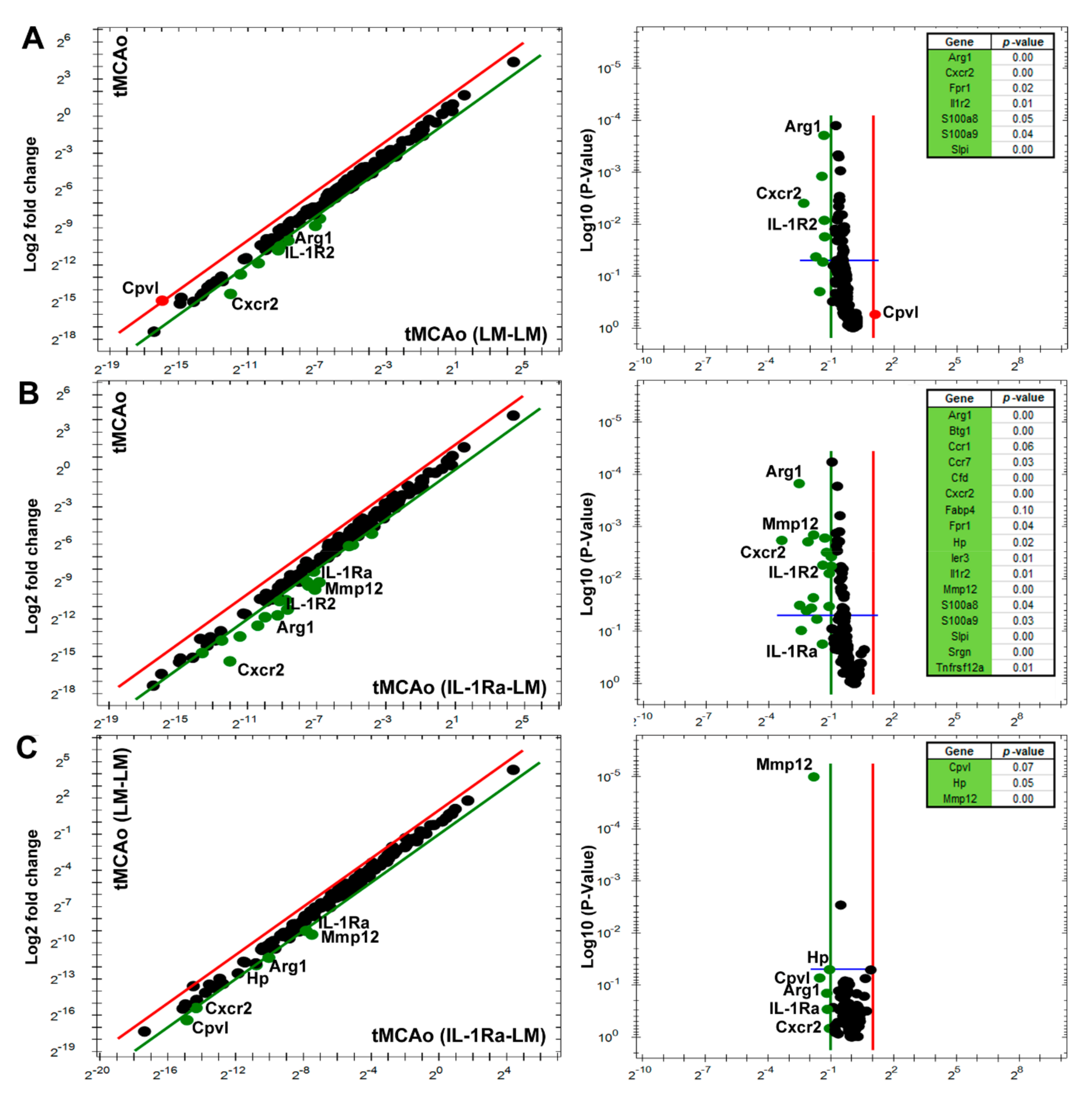
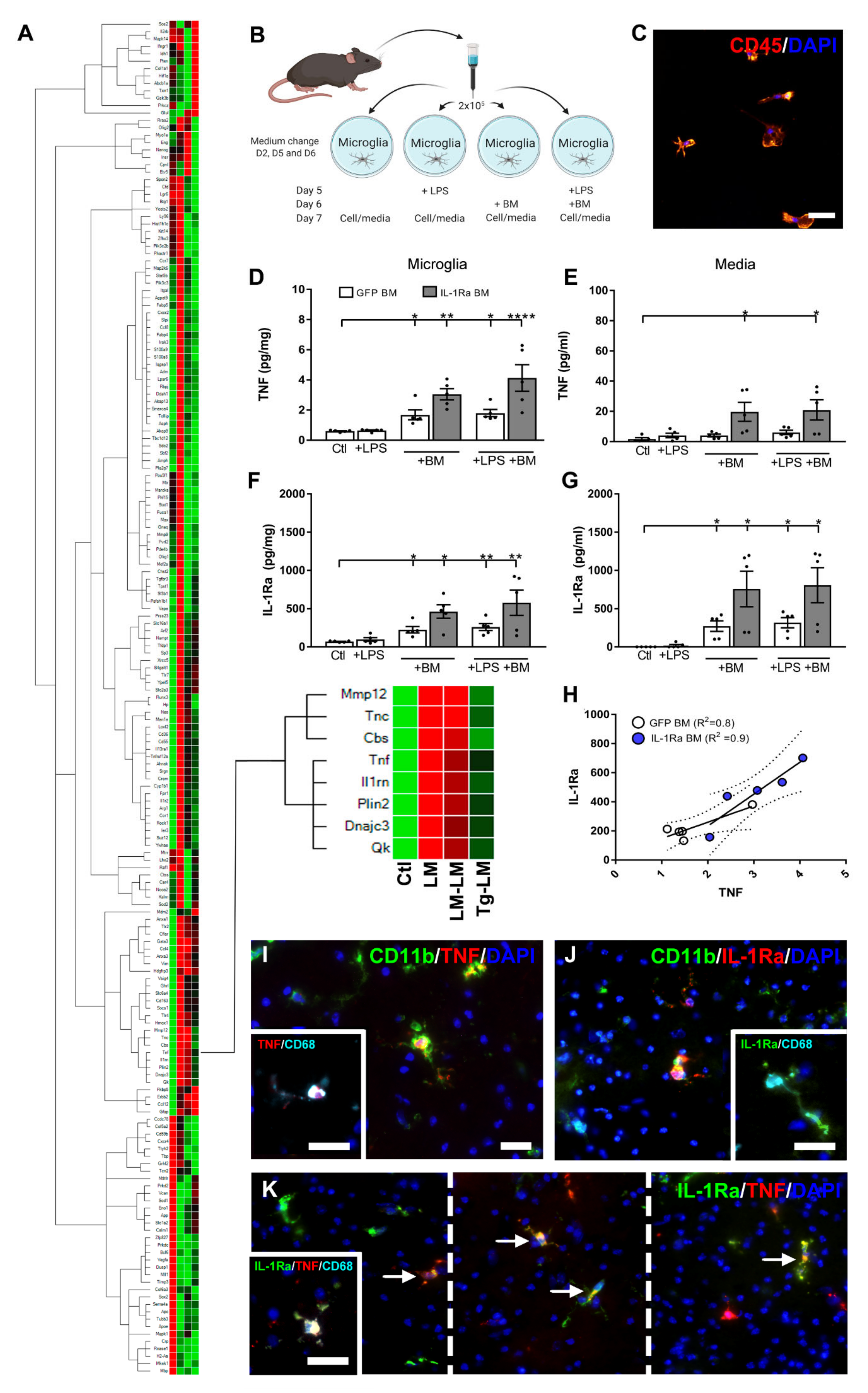
3.3. IL-1Ra BM Treatment Is Associated with a Unique Set of Transcripts after tMCAo
3.4. BM Cells Increase Microglial TNF Expression and Secretion
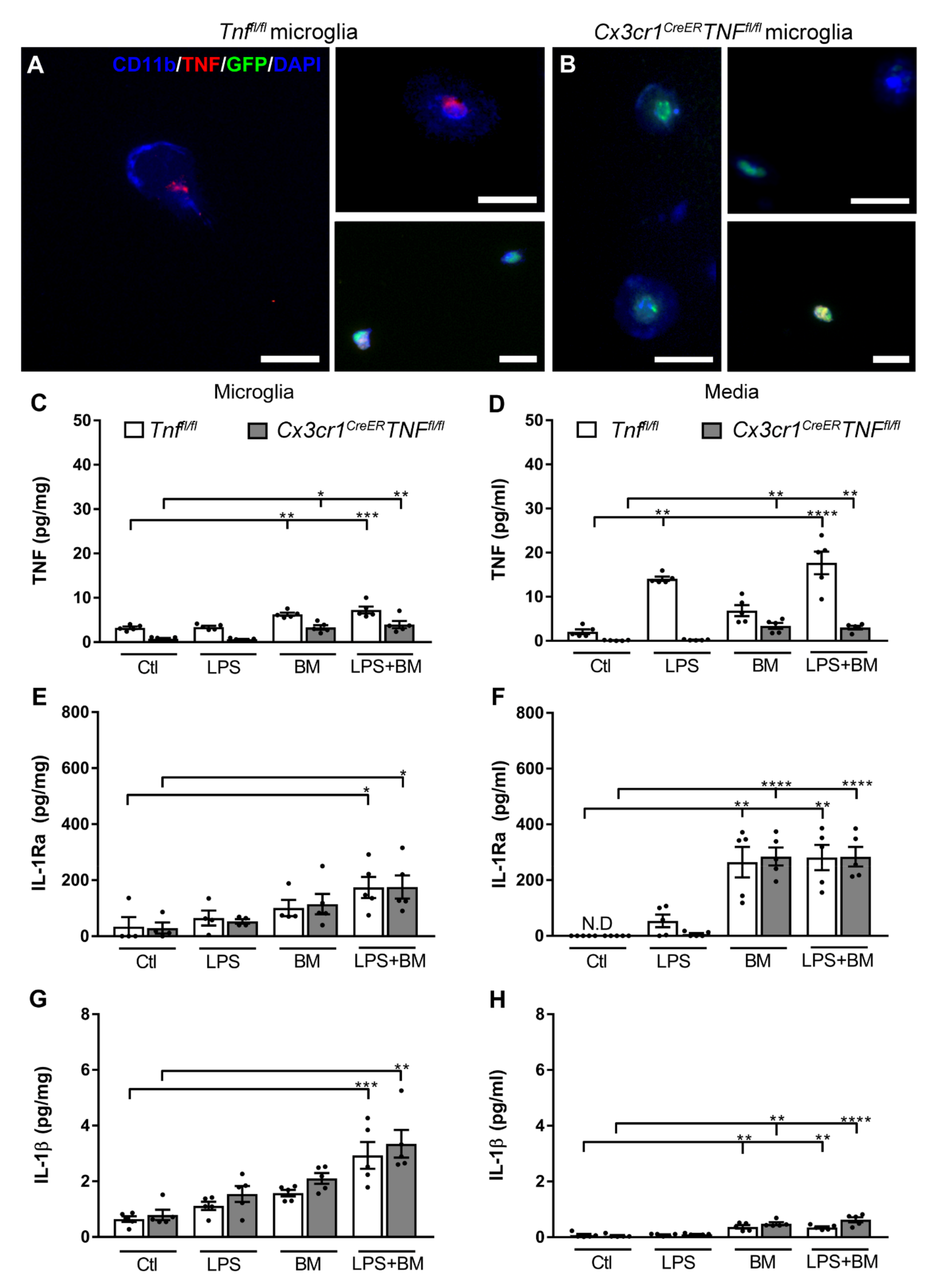
3.5. TNF Does Not Affect IL-1Ra Production in Microglia
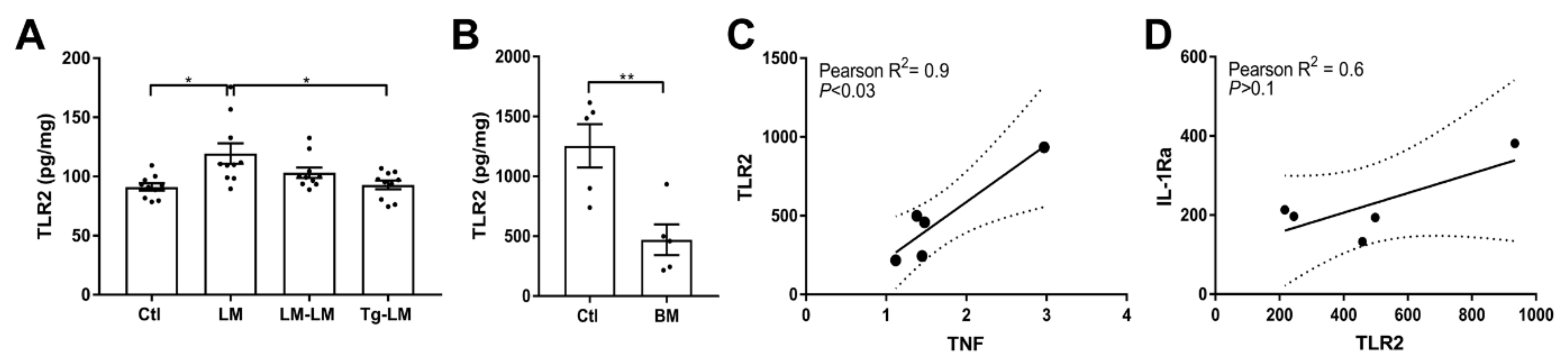
3.6. Toll-Like Receptor 2 Signaling Is Not the Link between TNF and IL-1Ra
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stonesifer, C.; Corey, S.; Ghanekar, S.; Diamandis, Z.; Acosta, S.A.; Borlongan, C.V. Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog. Neurobiol. 2017, 158, 94–131. [Google Scholar] [CrossRef]
- Li, L.Z.; Huang, Y.Y.; Yang, Z.H.; Zhang, S.J.; Han, Z.P.; Luo, Y.M. Potential microglia-based interventions for stroke. CNS Neurosci. Ther. 2020, 26, 288–296. [Google Scholar] [CrossRef]
- Eyo, U.B.; Wu, L.J. Microglia: Lifelong patrolling immune cells of the brain. Prog. Neurobiol. 2019, 179, 101614. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonell, W.S.; Murase, S.; Horwitz, A.F.; Mandell, J.W. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: An in situ time-lapse confocal imaging study. J. Neurosci. 2005, 25, 7040–7047. [Google Scholar] [CrossRef] [Green Version]
- Jian, Z.; Liu, R.; Zhu, X.; Smerin, D.; Zhong, Y.; Gu, L.; Fang, W.; Xiong, X. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front. Immunol. 2019, 10, 2167. [Google Scholar] [CrossRef] [Green Version]
- Clausen, B.H.; Lambertsen, K.L.; Babcock, A.A.; Holm, T.H.; Dagnaes-Hansen, F.; Finsen, B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J. Neuroinflammation 2008, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.X.; Kim, H.A.; Lee, S.; Moore, J.P.; Chan, C.T.; Vinh, A.; Gelderblom, M.; Arumugam, T.V.; Broughton, B.R.; Drummond, G.R.; et al. Immune cell infiltration in malignant middle cerebral artery infarction: Comparison with transient cerebral ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 450–459. [Google Scholar] [CrossRef] [Green Version]
- Lambertsen, K.L.; Finsen, B.; Clausen, B.H. Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 2019, 137, 693–714. [Google Scholar] [CrossRef] [Green Version]
- Clausen, B.H.; Lambertsen, K.L.; Dagnaes-Hansen, F.; Babcock, A.A.; von Linstow, C.U.; Meldgaard, M.; Kristensen, B.W.; Deierborg, T.; Finsen, B. Cell therapy centered on IL-1Ra is neuroprotective in experimental stroke. Acta Neuropathol. 2016, 131, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Madsen, P.M.; Clausen, B.H.; Degn, M.; Thyssen, S.; Kristensen, L.K.; Svensson, M.; Ditzel, N.; Finsen, B.; Deierborg, T.; Brambilla, R.; et al. Genetic ablation of soluble tumor necrosis factor with preservation of membrane tumor necrosis factor is associated with neuroprotection after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2016, 36, 1553–1569. [Google Scholar] [CrossRef] [Green Version]
- Simard, A.R.; Rivest, S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004, 18, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Eglitis, M.A.; Dawson, D.; Park, K.W.; Mouradian, M.M. Targeting of marrow-derived astrocytes to the ischemic brain. Neuroreport 1999, 10, 1289–1292. [Google Scholar] [CrossRef]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Janowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflammation 2019, 16, 178. [Google Scholar] [CrossRef] [Green Version]
- He, J.Q.; Sussman, E.S.; Steinberg, G.K. Revisiting Stem Cell-Based Clinical Trials for Ischemic Stroke. Front. Aging Neurosci. 2020, 12, 575990. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J.; Oh, J.Y. Mesenchymal stem/stromal cells (MSCs): Role as guardians of inflammation. Mol. Ther. 2012, 20, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyurkchiev, D.; Bochev, I.; Ivanova-Todorova, E.; Mourdjeva, M.; Oreshkova, T.; Belemezova, K.; Kyurkchiev, S. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J. Stem Cells 2014, 6, 552–570. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Correa, D. The MSC: An injury drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, B.H.; Wirenfeldt, M.; Hogedal, S.S.; Frich, L.H.; Nielsen, H.H.; Schroder, H.D.; Ostergaard, K.; Finsen, B.; Kristensen, B.W.; Lambertsen, K.L. Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol. Commun. 2020, 8, 81. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Taoufik, E.; Petit, E.; Divoux, D.; Tseveleki, V.; Mengozzi, M.; Roberts, M.L.; Valable, S.; Ghezzi, P.; Quackenbush, J.; Brines, M.; et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc. Natl. Acad. Sci. USA 2008, 105, 6185–6190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Meldgaard, M.; Ladeby, R.; Finsen, B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2005, 25, 119–135. [Google Scholar] [CrossRef] [Green Version]
- Marie, C.; Pitton, C.; Fitting, C.; Cavaillon, J.M. IL-10 and IL-4 synergize with TNF-alpha to induce IL-1ra production by human neutrophils. Cytokine 1996, 8, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Koba, S.; Kobayashi, Y. Roles of bone-marrow-derived cells and inflammatory cytokines in neointimal hyperplasia after vascular injury. Biomed. Res. Int. 2014, 2014, 945127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.; Irikura, V.M.; Paul, S.M.; Hirsh, D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc. Natl. Acad. Sci. USA 1996, 93, 11008–11013. [Google Scholar] [CrossRef] [Green Version]
- Nygren, J.; Wieloch, T. Enriched environment enhances recovery of motor function after focal ischemia in mice, and downregulates the transcription factor NGFI-A. J. Cereb. Blood Flow Metab. 2005, 25, 1625–1633. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Tumanov, A.V.; Liepinsh, D.J.; Kruglov, A.A.; Marakusha, B.I.; Shakhov, A.N.; Murakami, T.; Drutskaya, L.N.; Forster, I.; Clausen, B.E.; et al. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: Protective and deleterious effects. Immunity 2005, 22, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Dalby-Hansen, C.; Ellman, D.G.; Madsen, P.M.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing Functions of Microglial and Macrophagic TNFR2 in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Clausen, B.H.; Lambertsen, K.L.; Meldgaard, M.; Finsen, B. A quantitative in situ hybridization and polymerase chain reaction study of microglial-macrophage expression of interleukin-1beta mRNA following permanent middle cerebral artery occlusion in mice. Neuroscience 2005, 132, 879–892. [Google Scholar] [CrossRef]
- Yli-Karjanmaa, M.; Larsen, K.S.; Fenger, C.D.; Kristensen, L.K.; Martin, N.A.; Jensen, P.T.; Breton, A.; Nathanson, L.; Nielsen, P.V.; Lund, M.C.; et al. TNF deficiency causes alterations in the spatial organization of neurogenic zones and alters the number of microglia and neurons in the cerebral cortex. Brain Behav. Immun. 2019, 82, 279–297. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Luo, H.; Huang, C.; Li, S.; Liu, A.; Jiang, Y. Systematic Identification and Analysis of Expression Profiles of mRNAs and Incrnas in Macrophage Inflammatory Response. Shock 2019, 51, 770–779. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucl. Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Clausen, B.H.; Degn, M.; Sivasaravanaparan, M.; Fogtmann, T.; Andersen, M.G.; Trojanowsky, M.D.; Gao, H.; Hvidsten, S.; Baun, C.; Deierborg, T.; et al. Conditional ablation of myeloid TNF increases lesion volume after experimental stroke in mice, possibly via altered ERK1/2 signaling. Sci. Rep. 2016, 6, 29291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColl, S.R.; Paquin, R.; Menard, C.; Beaulieu, A.D. Human neutrophils produce high levels of the interleukin 1 receptor antagonist in response to granulocyte/macrophage colony-stimulating factor and tumor necrosis factor alpha. J. Exp. Med. 1992, 176, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Re, F.; Mengozzi, M.; Muzio, M.; Dinarello, C.A.; Mantovani, A.; Colotta, F. Expression of interleukin-1 receptor antagonist (IL-1ra) by human circulating polymorphonuclear cells. Eur. J. Immunol. 1993, 23, 570–573. [Google Scholar] [CrossRef]
- Grilli, M.; Barbieri, I.; Basudev, H.; Brusa, R.; Casati, C.; Lozza, G.; Ongini, E. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur. J. Neurosci. 2000, 12, 2265–2272. [Google Scholar] [CrossRef]
- Perez-de Puig, I.; Miro, F.; Salas-Perdomo, A.; Bonfill-Teixidor, E.; Ferrer-Ferrer, M.; Marquez-Kisinousky, L.; Planas, A.M. IL-10 deficiency exacerbates the brain inflammatory response to permanent ischemia without preventing resolution of the lesion. J. Cereb. Blood Flow Metab. 2013, 33, 1955–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, J.; Song, Y.; Yang, P.; Yang, Y.; Huang, Z.; Wang, K. Deficiency of anti-inflammatory cytokine IL-4 leads to neural hyperexcitability and aggravates cerebral ischemia-reperfusion injury. Acta Pharm. Sin. B 2020, 10, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, H.; Sun, G.; Zhang, J.; Edwards, N.J.; Aronowski, J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J. Neurosci. 2015, 35, 11281–11291. [Google Scholar] [CrossRef] [Green Version]
- Sawant, K.V.; Poluri, K.M.; Dutta, A.K.; Sepuru, K.M.; Troshkina, A.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci. Rep. 2016, 6, 33123. [Google Scholar] [CrossRef] [Green Version]
- Cassatella, M.A.; Meda, L.; Gasperini, S.; D’Andrea, A.; Ma, X.; Trinchieri, G. Interleukin-12 production by human polymorphonuclear leukocytes. Eur. J. Immunol. 1995, 25, 1–5. [Google Scholar] [CrossRef]
- Call, D.R.; Nemzek, J.A.; Ebong, S.J.; Bolgos, G.L.; Newcomb, D.E.; Remick, D.G. Ratio of local to systemic chemokine concentrations regulates neutrophil recruitment. Am. J. Pathol. 2001, 158, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.J.; Perry, V.H.; Pitossi, F.J.; Butchart, A.G.; Chertoff, M.; Waters, S.; Dempster, R.; Anthony, D.C. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am. J. Pathol. 2005, 166, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Losy, J.; Zaremba, J.; Skrobanski, P. CXCL1 (GRO-alpha) chemokine in acute ischaemic stroke patients. Folia Neuropathol. 2005, 43, 97–102. [Google Scholar]
- Rahbar, A.; Cederarv, M.; Wolmer-Solberg, N.; Tammik, C.; Stragliotto, G.; Peredo, I.; Fornara, O.; Xu, X.; Dzabic, M.; Taher, C.; et al. Enhanced neutrophil activity is associated with shorter time to tumor progression in glioblastoma patients. Oncoimmunology 2016, 5, e1075693. [Google Scholar] [CrossRef] [Green Version]
- Zaremba, J.; Losy, J. Interleukin-12 in acute ischemic stroke patients. Folia Neuropathol. 2006, 44, 59–66. [Google Scholar]
- Li, X.; Huang, M.; Zhao, R.; Zhao, C.; Liu, Y.; Zou, H.; Chen, L.; Guan, Y.; Zhang, Y.A. Intravenously Delivered Allogeneic Mesenchymal Stem Cells Bidirectionally Regulate Inflammation and Induce Neurotrophic Effects in Distal Middle Cerebral Artery Occlusion Rats Within the First 7 Days After Stroke. Cell Physiol. Biochem. 2018, 46, 1951–1970. [Google Scholar] [CrossRef]
- Gao, F.; Chiu, S.M.; Motan, D.A.; Zhang, Z.; Chen, L.; Ji, H.L.; Tse, H.F.; Fu, Q.L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef] [Green Version]
- Adamski, M.G.; Li, Y.; Wagner, E.; Yu, H.; Seales-Bailey, C.; Soper, S.A.; Murphy, M.; Baird, A.E. Expression profile based gene clusters for ischemic stroke detection. Genomics 2014, 104, 163–169. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.C.; Delgado-Esteban, M.; Rodriguez-Hernandez, I.; Sobrino, T.; Perez de la Ossa, N.; Reverte, S.; Bolanos, J.P.; Gonzalez-Sarmiento, R.; Castillo, J.; Almeida, A. The human Tp53 Arg72Pro polymorphism explains different functional prognosis in stroke. J. Exp. Med. 2011, 208, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Timasheva, Y.R.; Nasibullin, T.R.; Mustafina, O.E. The CXCR2 Gene Polymorphism Is Associated with Stroke in Patients with Essential Hypertension. Cereb. Dis. Extra 2015, 5, 124–131. [Google Scholar] [CrossRef]
- Dorward, D.A.; Lucas, C.D.; Chapman, G.B.; Haslett, C.; Dhaliwal, K.; Rossi, A.G. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am. J. Pathol. 2015, 185, 1172–1184. [Google Scholar] [CrossRef] [Green Version]
- Cussell, P.J.G.; Gomez Escalada, M.; Milton, N.G.N.; Paterson, A.W.J. The N-formyl peptide receptors: Contemporary roles in neuronal function and dysfunction. Neural. Regen Res. 2020, 15, 1191–1198. [Google Scholar] [CrossRef]
- Zheng, Y.; Humphry, M.; Maguire, J.J.; Bennett, M.R.; Clarke, M.C. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 2013, 38, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Zhu, Z.; Xu, T.; Zhong, C.; Wang, A.; Xie, X.; Peng, Y.; Peng, H.; Li, Q.; Ju, Z.; et al. Plasma S100A8/A9 Concentrations and Clinical Outcomes of Ischemic Stroke in 2 Independent Multicenter Cohorts. Clin. Chem. 2020, 66, 706–717. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [Green Version]
- Heizmann, C.W.; Fritz, G.; Schafer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci. 2002, 7, d1356–d1368. [Google Scholar]
- Doumas, S.; Kolokotronis, A.; Stefanopoulos, P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect. Immun. 2005, 73, 1271–1274. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Veltkamp, R.; Gill, D. Clinical Trials of Immunomodulation in Ischemic Stroke. Neurotherapeutics 2016, 13, 791–800. [Google Scholar] [CrossRef]
- Downes, C.E.; Crack, P.J. Neural injury following stroke: Are Toll-like receptors the link between the immune system and the CNS? Br. J. Pharmacol. 2010, 160, 1872–1888. [Google Scholar] [CrossRef] [Green Version]
- Raicevic, G.; Rouas, R.; Najar, M.; Stordeur, P.; Boufker, H.I.; Bron, D.; Martiat, P.; Goldman, M.; Nevessignsky, M.T.; Lagneaux, L. Inflammation modifies the pattern and the function of Toll-like receptors expressed by human mesenchymal stromal cells. Hum. Immunol. 2010, 71, 235–244. [Google Scholar] [CrossRef]
- Najar, M.; Krayem, M.; Meuleman, N.; Bron, D.; Lagneaux, L. Mesenchymal Stromal Cells and Toll-Like Receptor Priming: A Critical Review. Immune Netw. 2017, 17, 89–102. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.S.; Arbour, N.; Manusow, J.; Montgrain, V.; Blain, M.; McCrea, E.; Shapiro, A.; Antel, J.P. TLR signaling tailors innate immune responses in human microglia and astrocytes. J. Immunol. 2005, 175, 4320–4330. [Google Scholar] [CrossRef] [Green Version]
- Anthoney, N.; Foldi, I.; Hidalgo, A. Toll and Toll-like receptor signalling in development. Development 2018, 145, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Werling, D.; Jungi, T.W. TOLL-like receptors linking innate and adaptive immune response. Vet. Immunol. Immunopathol. 2003, 91, 1–12. [Google Scholar] [CrossRef]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, G.; Harhausen, D.; Schepers, C.; Hoffmann, O.; Rohr, C.; Prinz, V.; Konig, J.; Lehrach, H.; Nietfeld, W.; Trendelenburg, G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem. Biophys. Res. Commun. 2007, 359, 574–579. [Google Scholar] [CrossRef]
- Li, D.; Pan, X.; Zhao, J.; Chi, C.; Wu, G.; Wang, Y.; Liao, S.; Wang, C.; Ma, J.; Pan, J. Bone Marrow Mesenchymal Stem Cells Suppress Acute Lung Injury Induced by Lipopolysaccharide Through Inhibiting the TLR2, 4/NF-kappaB Pathway in Rats with Multiple Trauma. Shock 2016, 45, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.L.; Chen, T.H.; Wu, Y.H.; Chen, G.A.; Weng, T.H.; Tseng, P.H.; Hsieh, S.L.; Fu, S.L.; Lin, C.H.; Chen, C.J.; et al. A novel TLR2-triggered signalling crosstalk synergistically intensifies TNF-mediated IL-6 induction. J. Cell Mol. Med. 2014, 18, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- Loiarro, M.; Ruggiero, V.; Sette, C. Targeting TLR/IL-1R signalling in human diseases. Mediators Inflamm. 2010, 2010, 674363. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hematti, P. Mesenchymal stem cell-educated macrophages: A novel type of alternatively activated macrophages. Exp. Hematol 2009, 37, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Raffaghello, L.; Bianchi, G.; Bertolotto, M.; Montecucco, F.; Busca, A.; Dallegri, F.; Ottonello, L.; Pistoia, V. Human mesenchymal stem cells inhibit neutrophil apoptosis: A model for neutrophil preservation in the bone marrow niche. Stem Cells 2008, 26, 151–162. [Google Scholar] [CrossRef]
- Ortiz, L.A.; Dutreil, M.; Fattman, C.; Pandey, A.C.; Torres, G.; Go, K.; Phinney, D.G. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc. Natl. Acad. Sci. USA 2007, 104, 11002–11007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abumaree, M.H.; Al Jumah, M.A.; Kalionis, B.; Jawdat, D.; Al Khaldi, A.; Abomaray, F.M.; Fatani, A.S.; Chamley, L.W.; Knawy, B.A. Human placental mesenchymal stem cells (pMSCs) play a role as immune suppressive cells by shifting macrophage differentiation from inflammatory M1 to anti-inflammatory M2 macrophages. Stem Cell Rev. Rep. 2013, 9, 620–641. [Google Scholar] [CrossRef]
- Gosain, A.; DiPietro, L.A. Aging and wound healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef]
- Gerdoni, E.; Gallo, B.; Casazza, S.; Musio, S.; Bonanni, I.; Pedemonte, E.; Mantegazza, R.; Frassoni, F.; Mancardi, G.; Pedotti, R.; et al. Mesenchymal stem cells effectively modulate pathogenic immune response in experimental autoimmune encephalomyelitis. Ann. Neurol. 2007, 61, 219–227. [Google Scholar] [CrossRef]
- Chen, L.; Tredget, E.E.; Wu, P.Y.; Wu, Y. Paracrine factors of mesenchymal stem cells recruit macrophages and endothelial lineage cells and enhance wound healing. PLoS ONE 2008, 3, e1886. [Google Scholar] [CrossRef] [Green Version]
- Freytes, D.O.; Kang, J.W.; Marcos-Campos, I.; Vunjak-Novakovic, G. Macrophages modulate the viability and growth of human mesenchymal stem cells. J. Cell Biochem. 2013, 114, 220–229. [Google Scholar] [CrossRef]
- Wei, X.; Yang, X.; Han, Z.P.; Qu, F.F.; Shao, L.; Shi, Y.F. Mesenchymal stem cells: A new trend for cell therapy. Acta Pharmacol. Sin. 2013, 34, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Kalladka, D.; Muir, K.W. Brain repair: Cell therapy in stroke. Stem Cells Cloning 2014, 7, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Park, J.S. Usage of Human Mesenchymal Stem Cells in Cell-based Therapy: Advantages and Disadvantages. Dev. Reprod. 2017, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dong, X.; Tian, M.; Liu, C.; Wang, K.; Li, L.; Liu, Z.; Liu, J. Stem cell-based therapies for ischemic stroke: A systematic review and meta-analysis of clinical trials. Stem Cell Res. Ther. 2020, 11, 252. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Linstow, C.U.; Hindkjær, S.M.; Nielsen, P.V.; Degn, M.; Lambertsen, K.L.; Finsen, B.; Clausen, B.H. Bone Marrow-Derived IL-1Ra Increases TNF Levels Poststroke. Cells 2021, 10, 956. https://doi.org/10.3390/cells10040956
von Linstow CU, Hindkjær SM, Nielsen PV, Degn M, Lambertsen KL, Finsen B, Clausen BH. Bone Marrow-Derived IL-1Ra Increases TNF Levels Poststroke. Cells. 2021; 10(4):956. https://doi.org/10.3390/cells10040956
Chicago/Turabian Stylevon Linstow, Christian Ulrich, Sofie Mozart Hindkjær, Pernille Vinther Nielsen, Matilda Degn, Kate Lykke Lambertsen, Bente Finsen, and Bettina Hjelm Clausen. 2021. "Bone Marrow-Derived IL-1Ra Increases TNF Levels Poststroke" Cells 10, no. 4: 956. https://doi.org/10.3390/cells10040956
APA Stylevon Linstow, C. U., Hindkjær, S. M., Nielsen, P. V., Degn, M., Lambertsen, K. L., Finsen, B., & Clausen, B. H. (2021). Bone Marrow-Derived IL-1Ra Increases TNF Levels Poststroke. Cells, 10(4), 956. https://doi.org/10.3390/cells10040956