
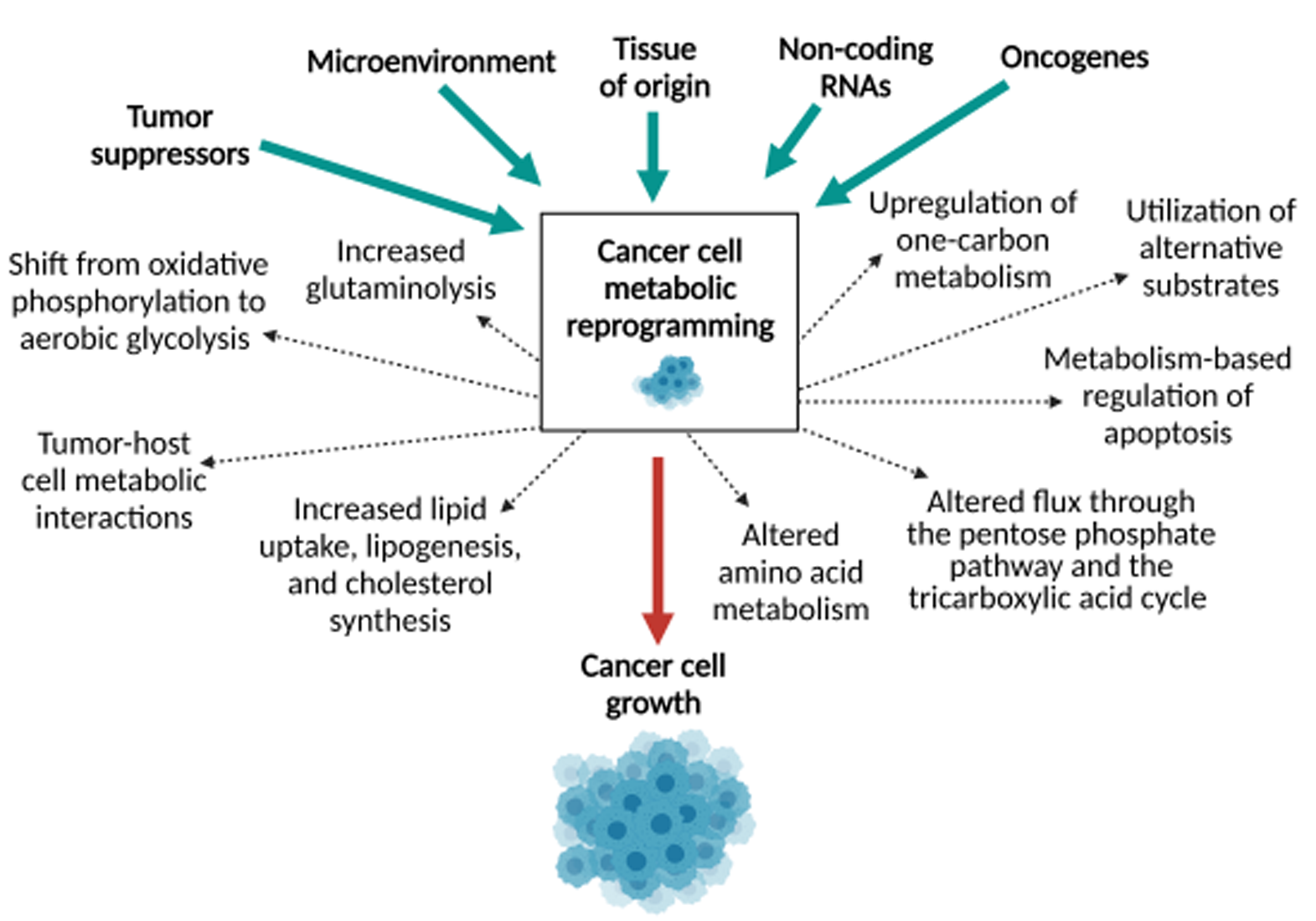
Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation



Abstract
:
1. Introduction
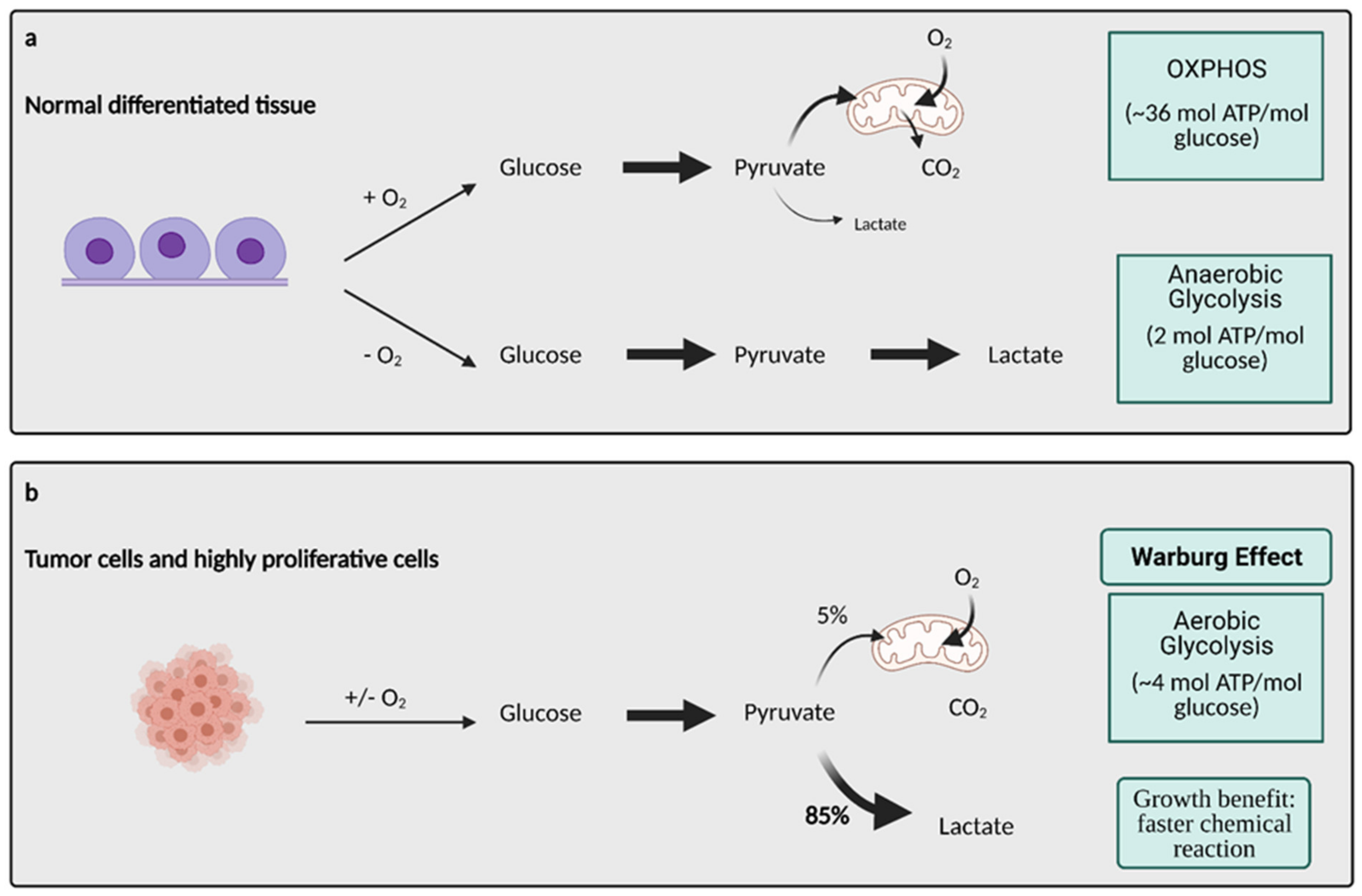
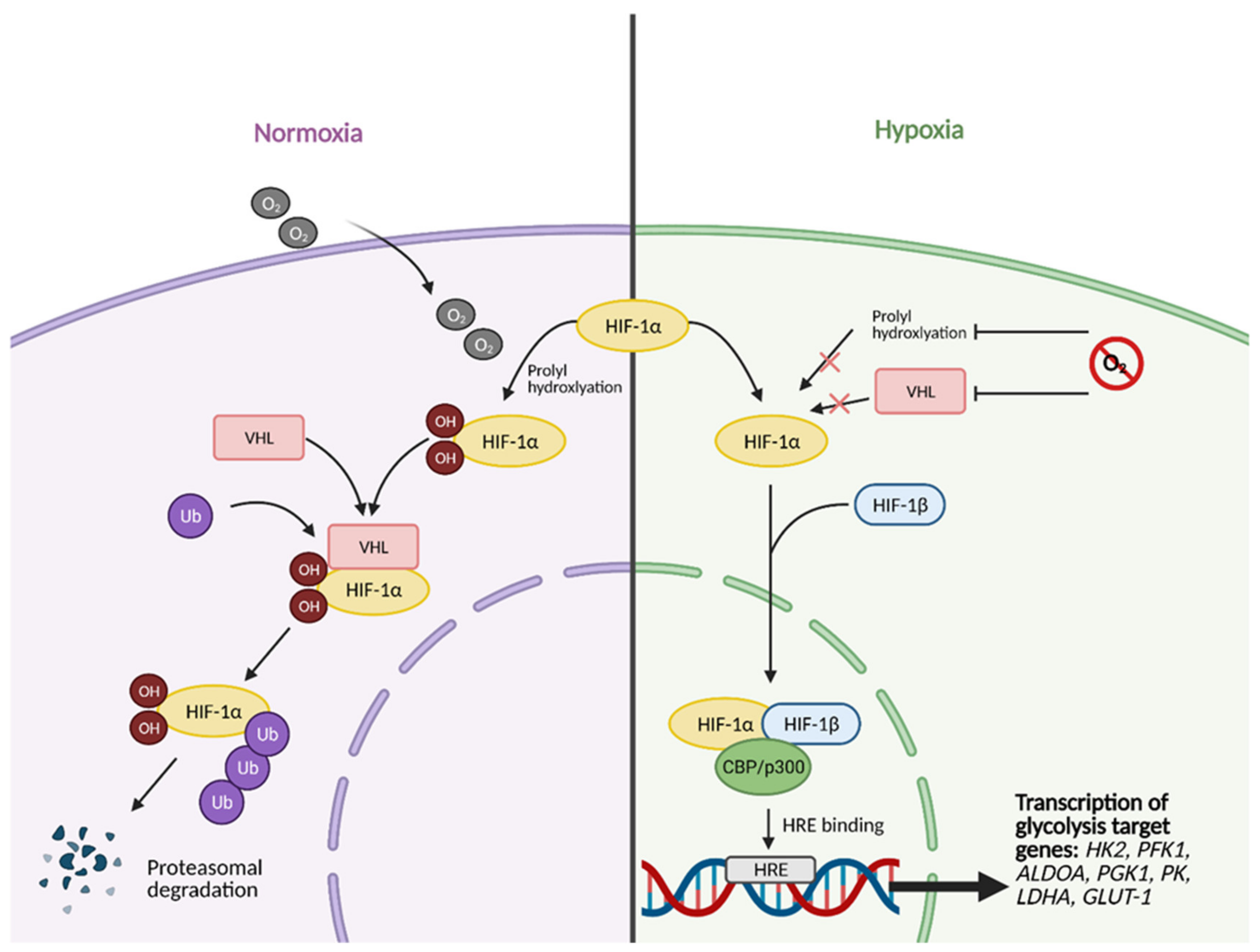
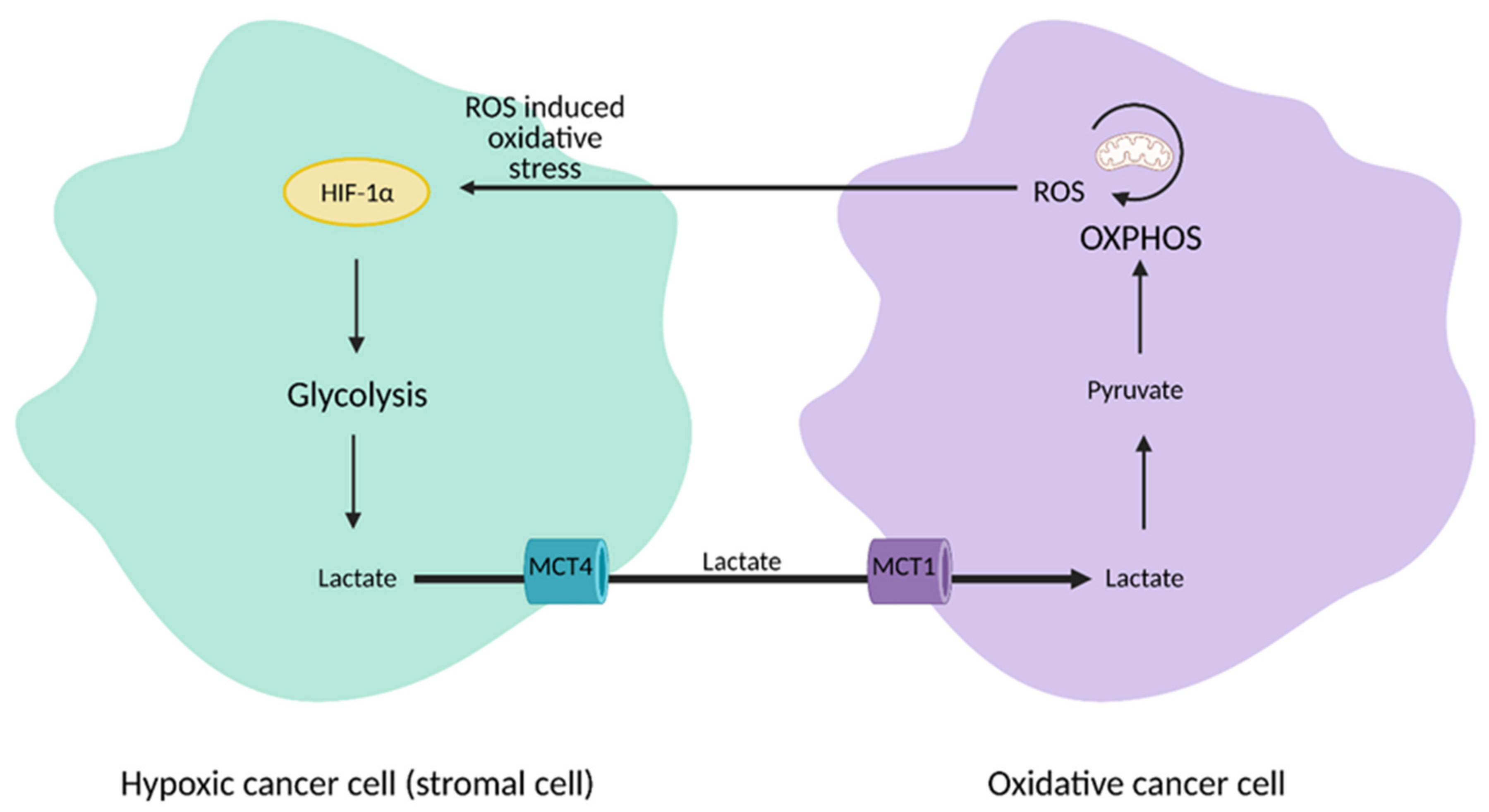
2. Glucose Metabolism and the Warburg Effect: A Century Later
3. Glutamine Metabolism
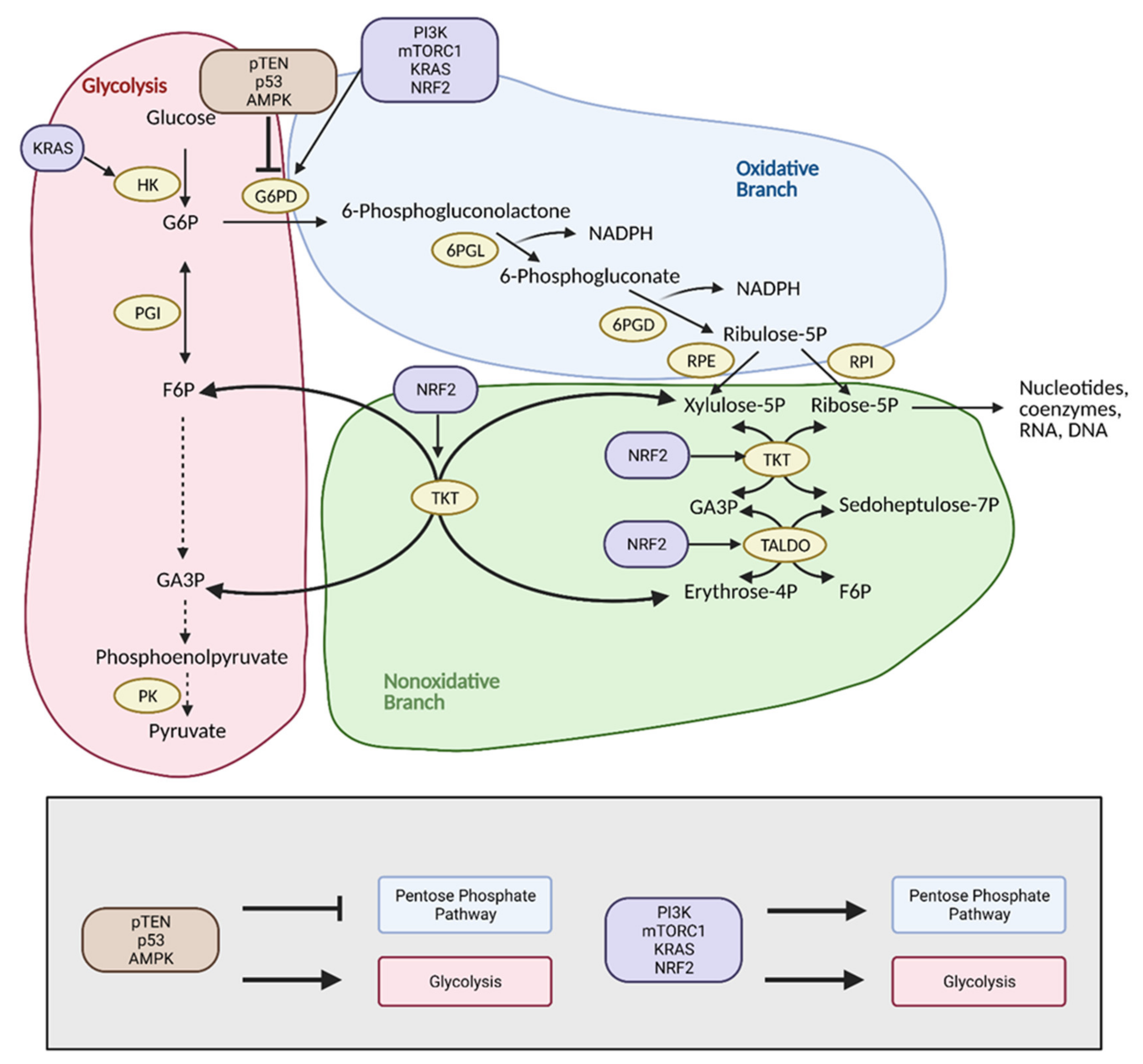
4. Pentose Phosphate Pathway
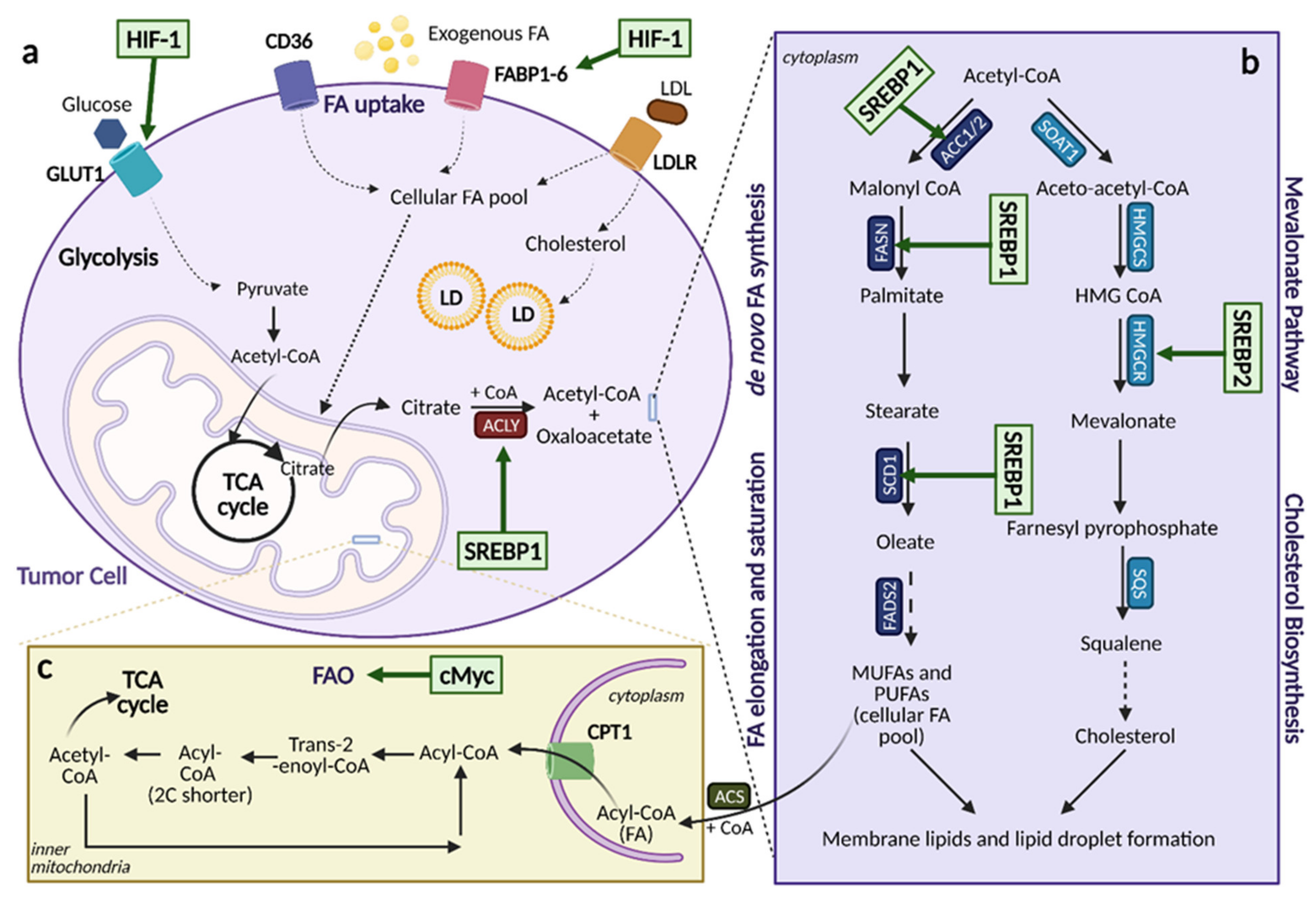
5. Lipid Metabolism
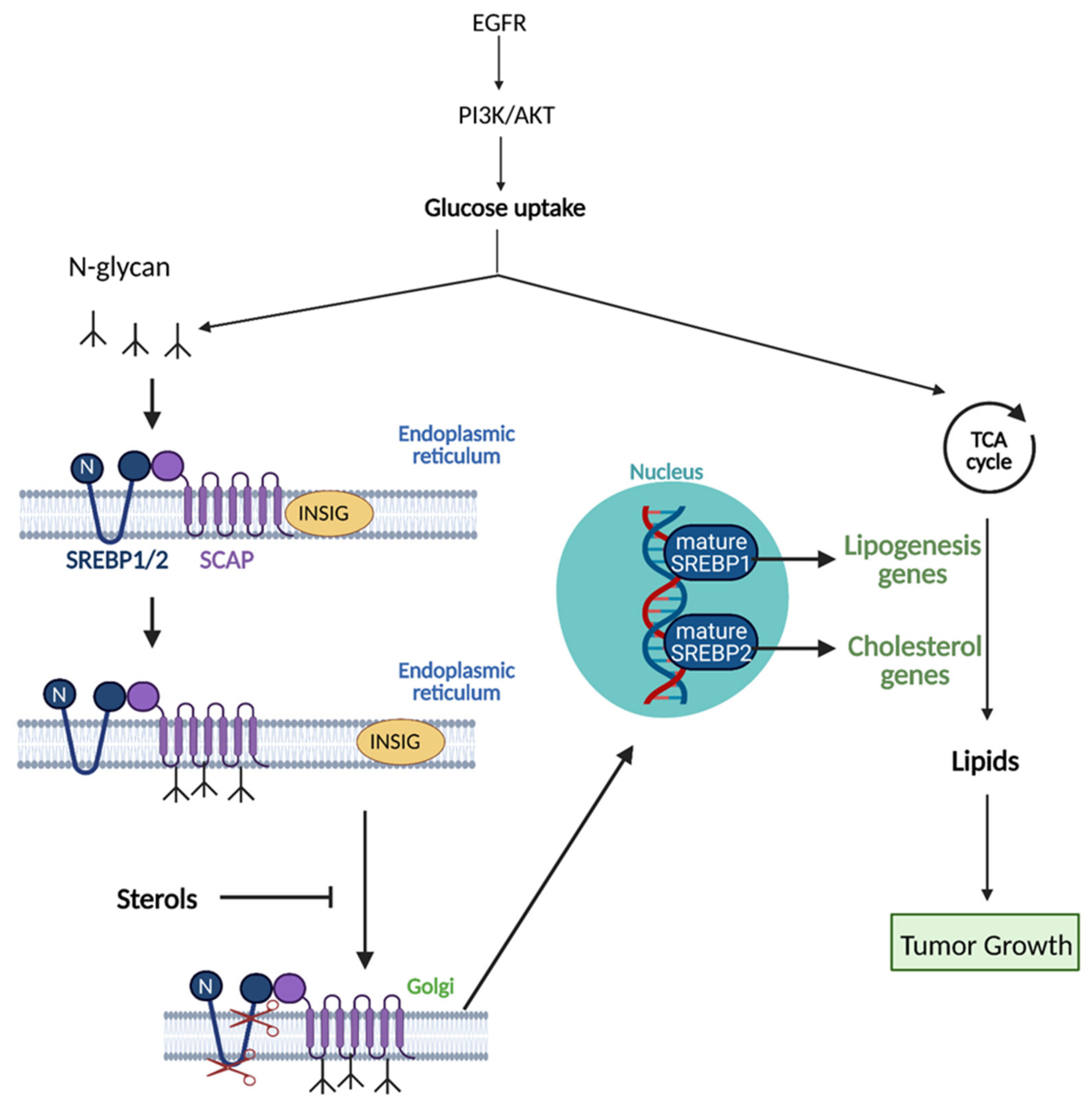
5.1. Lipid Acquisition: De Novo Lipogenesis and Lipid Uptake

5.2. Lipid Storage and Export
5.3. Lipolysis
5.4. Fatty Acid Oxidation
5.5. Mevalonate Pathway
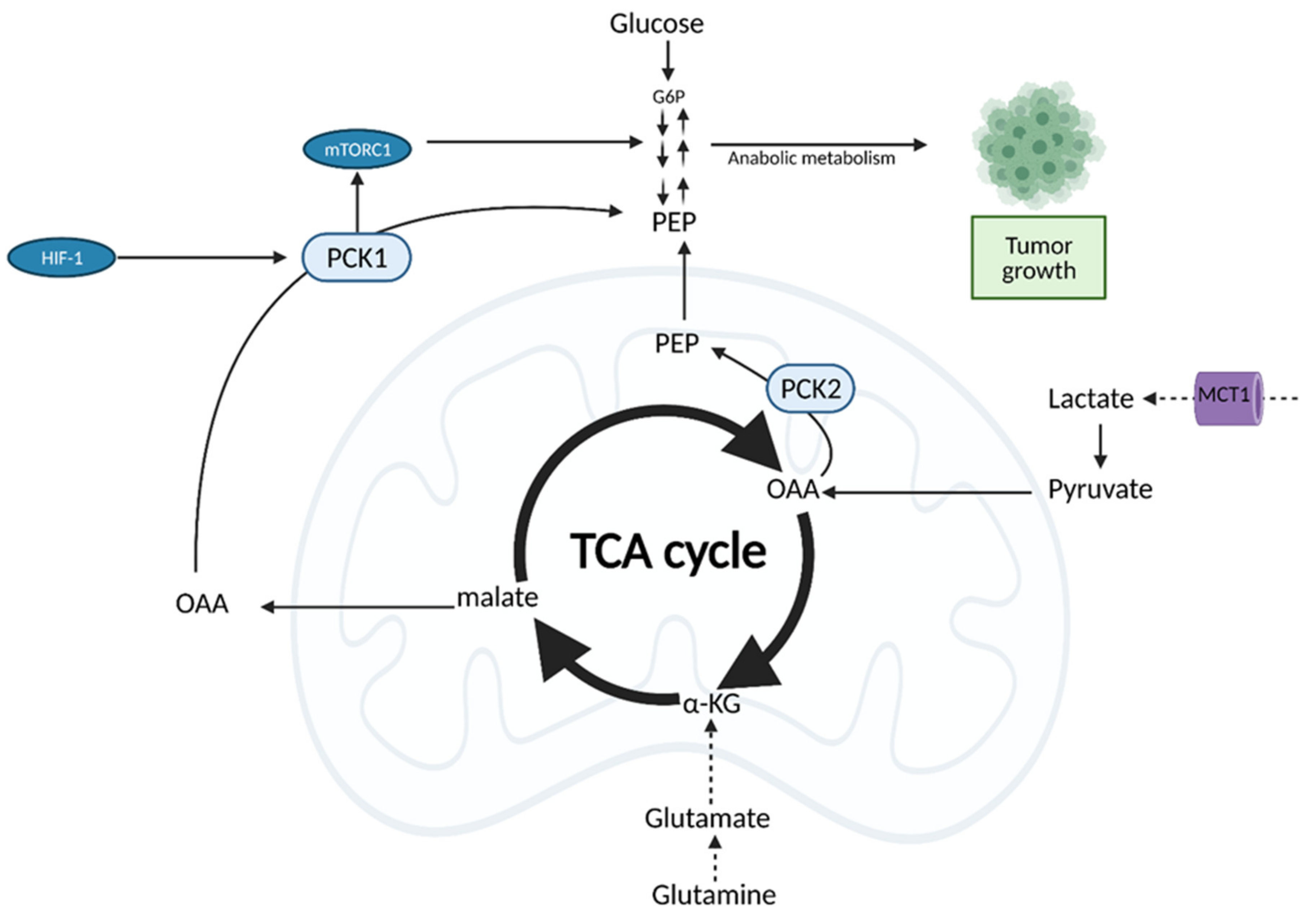
6. The Tricarboxylic Acid (TCA) Cycle
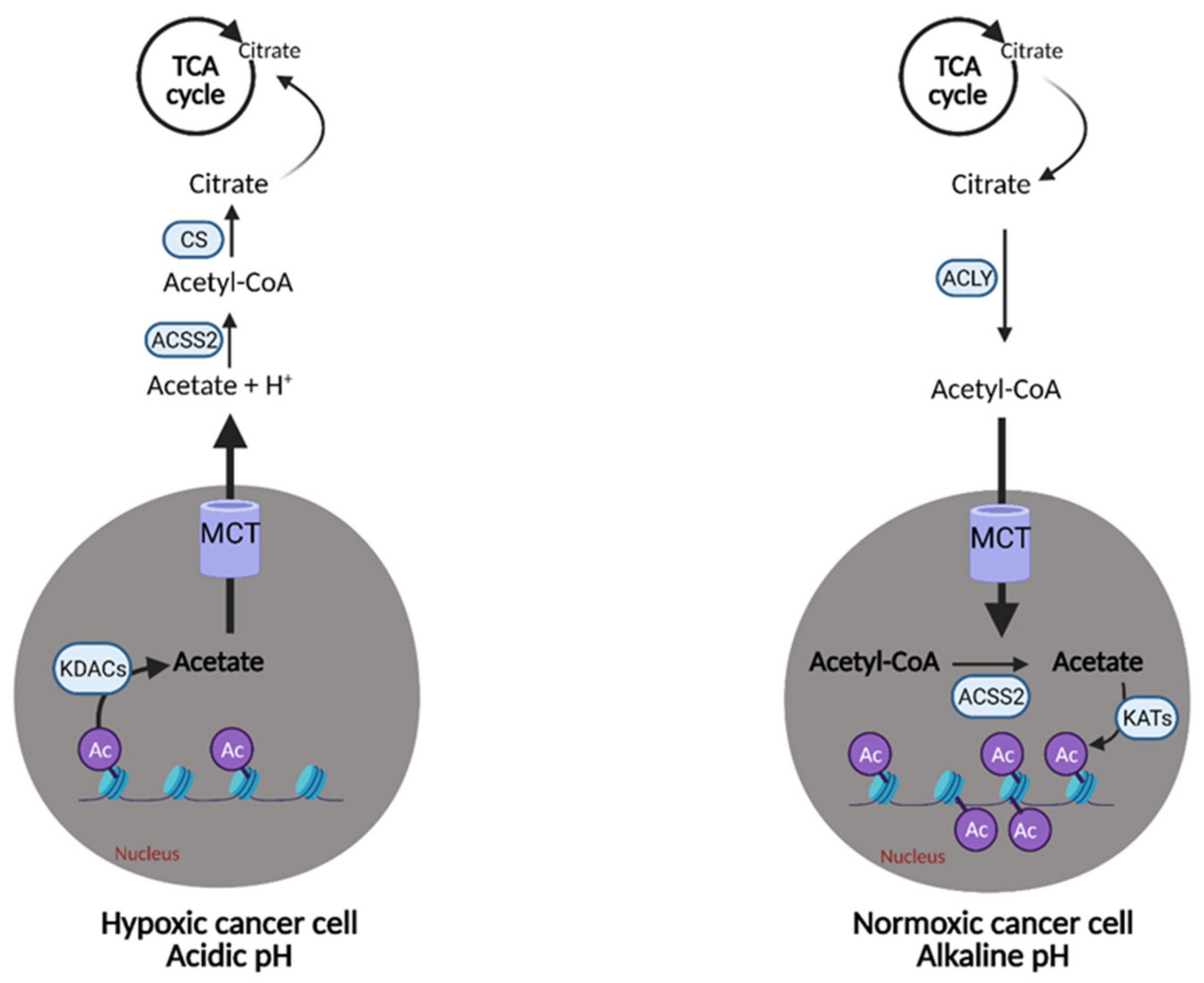
7. Acetate
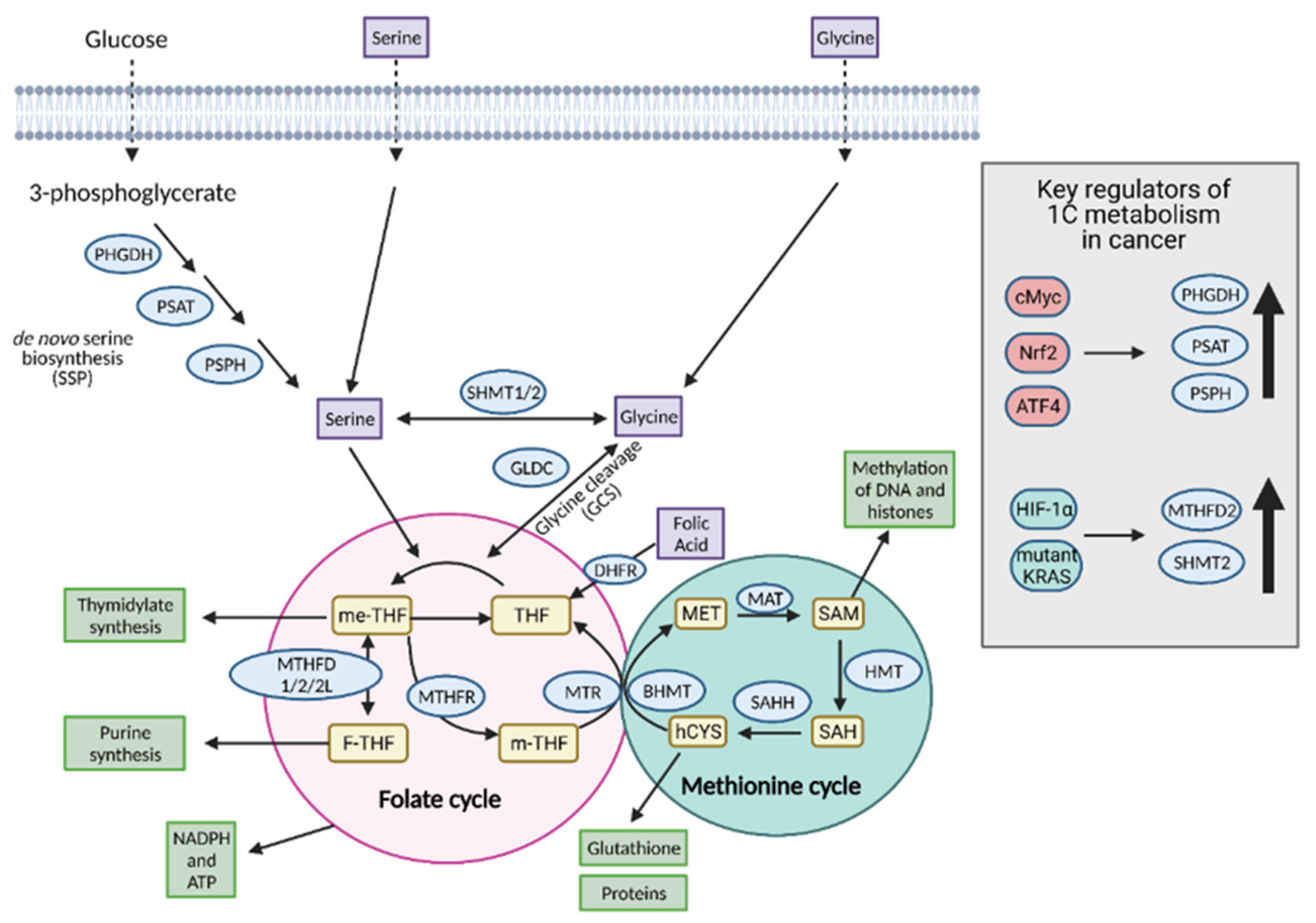
8. One-Carbon Metabolism
8.1. Input Molecules for 1C Metabolism
8.1.1. Serine
8.1.2. Glycine
8.1.3. Folate and Methionine Cycles
8.2. Molecules Produced as a Result of 1C Metabolism
8.2.1. Nucleotides
8.2.2. SAM
8.2.3. Glutathione
8.2.4. NADH/NADPH and ATP
9. Other NEAAs
9.1. Alanine
9.2. Aspartate
9.3. Asparagine
9.4. Cysteine
9.5. Proline
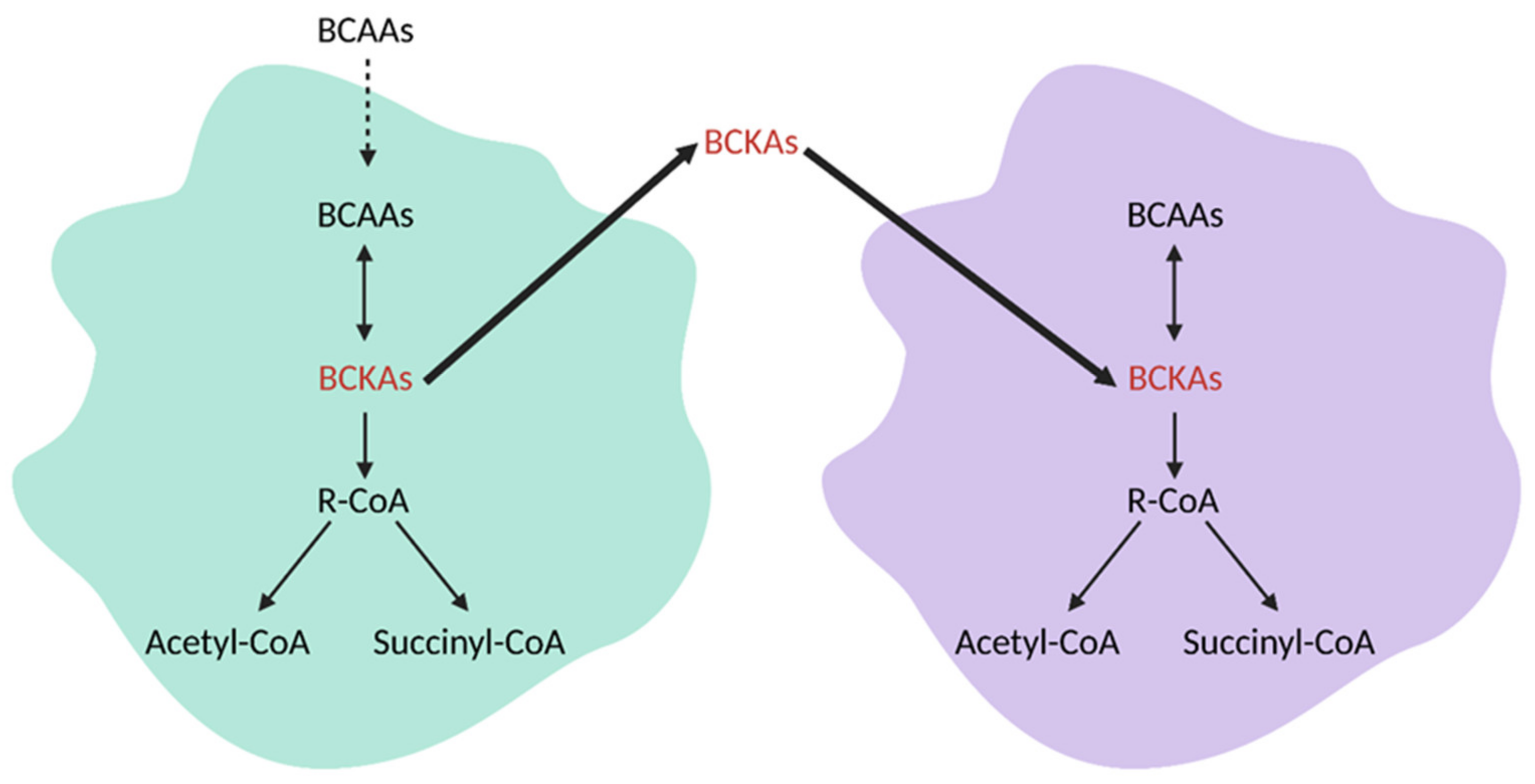
10. Branched Chain Amino Acid (BCAA) Metabolism
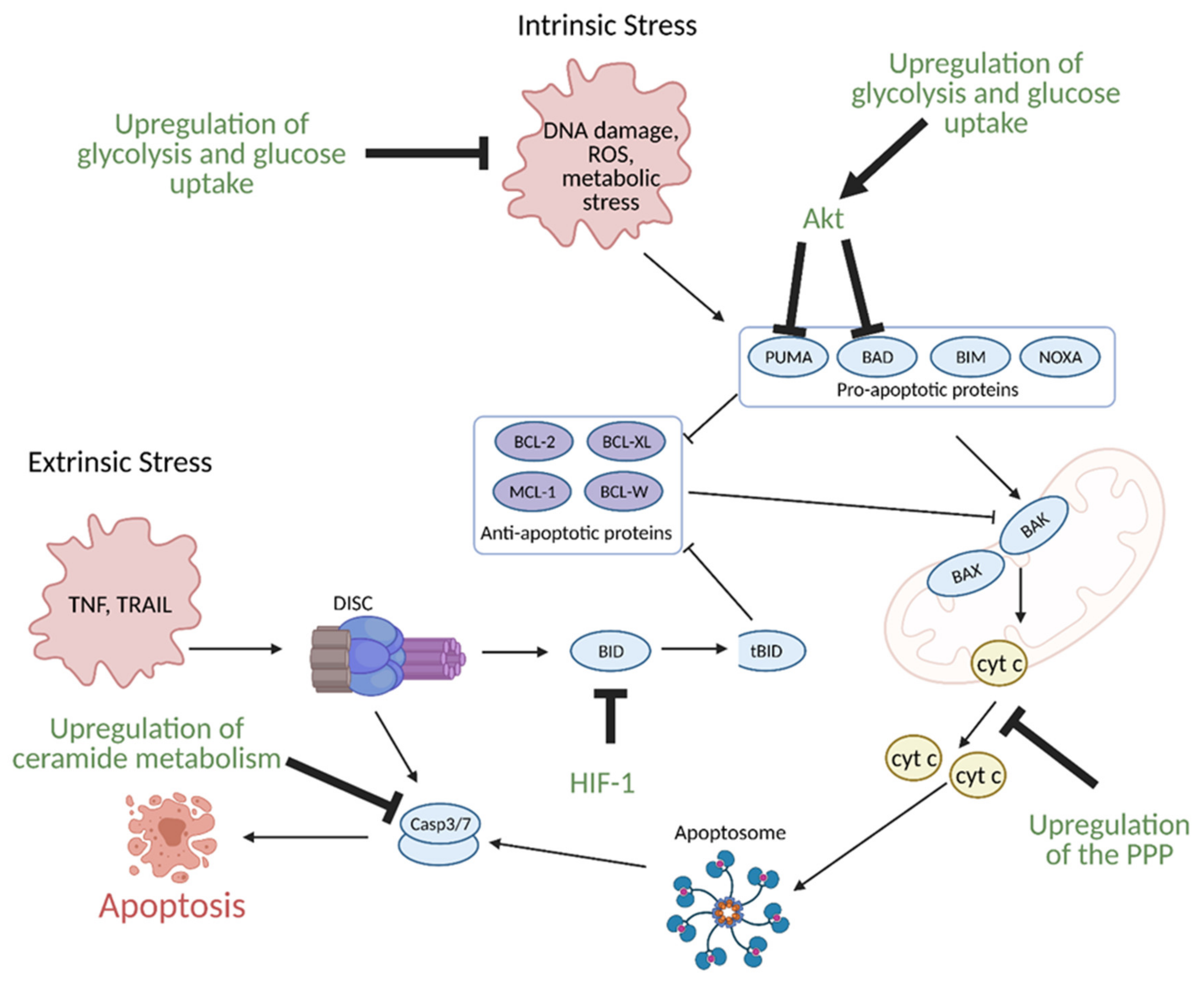
11. Regulation of Apoptosis by Metabolism
12. Role of Non-Coding RNAs in Cancer Cell Metabolism
13. Regulation of Cancer Growth via Tumor-Host Cell Metabolic Interactions
14. Anti-Cancer Drugs That Target Metabolism
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Romero-Garcia, S.; Lopez-Gonzalez, J.S.; Báez-Viveros, J.L.; Aguilar-Cazares, D.; Prado-Garcia, H. Tumor cell metabolism: An integral view. Cancer Biol. Ther. 2011, 12, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg effect 97 years after its discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Läsche, M.; Emons, G.; Gründker, C. Shedding new light on cancer metabolism: A metabolic tightrope between life and death. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Ghanavat, M.; Shahrouzian, M.; Zayeri, Z.D.; Banihashemi, S.; Kazemi, S.M.; Saki, N. Digging deeper through glucose metabolism and its regulators in cancer and metastasis. Life Sci. 2021, 264, 118603. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadeh, Z.; Çeşmeli, S.; Avcı, Ç.B. Crucial players in glycolysis: Cancer progress. Gene 2020, 726, 144158. [Google Scholar] [CrossRef]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer metabolism: Phenotype, signaling and therapeutic targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. p53 and glucose metabolism: An orchestra to be directed in cancer therapy. Pharmacol. Res. 2018, 131, 75–86. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic heterogeneity of cancer cells: An interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.-X.; Qian, D.Z.; Dai, M.-S. Molecular crosstalk between MYC and HIF in cancer. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Zhang, X.-P.; Liu, F.; Wang, W. Modeling the interplay between the HIF-1 and p53 pathways in hypoxia. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef]
- Bouchez, C.L.; Hammad, N.; Cuvellier, S.; Ransac, S.; Rigoulet, M.; Devin, A. The Warburg effect in yeast: Repression of mitochondrial metabolism is not a prerequisite to promote cell proliferation. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial oxidative phosphorylation in cutaneous melanoma. Br. J. Cancer 2020, 124, 115–123. [Google Scholar] [CrossRef]
- Nie, K.; Li, J.; He, X.; Wang, Y.; Zhao, Q.; Du, M.; Sun, H.; Wang, J.; Lyu, J.; Fang, H.; et al. COX6B2 drives metabolic reprogramming toward oxidative phosphorylation to promote metastasis in pancreatic ductal cancer cells. Oncogenesis 2020, 9. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the reverse Warburg effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. Erratum: From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef]
- Cacace, A.; Sboarina, M.; Vazeille, T.; Sonveaux, P. Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism. Oncogene 2016, 36, 2074–2084. [Google Scholar] [CrossRef]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Campos-Sandoval, J.A.; Santos-Jiménez, J.D.L.; Márquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef]
- Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef] [PubMed]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic rewiring faces tumor heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.W.; Cerione, R.A. Glutaminase: A hot spot for regulation of cancer cell metabolism? Oncotarget 2010, 1, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Daye, D.; Wellen, K.E. Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Semin. Cell Dev. Biol. 2012, 23, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; de Marzo, A.M.; van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Borodovsky, A.; Seltzer, M.J.; Riggins, G.J. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr. Opin. Oncol. 2012, 24, 83–89. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nakayama, Y.; Fukusaki, E.; Irino, Y. Glutamate production from ammonia via glutamate dehydrogenase 2 activity supports cancer cell proliferation under glutamine depletion. Biochem. Biophys. Res. Commun. 2018, 495, 761–767. [Google Scholar] [CrossRef]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Mates, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Lin, M.; Zhu, W.; Liang, Y.; Hong, X.; Zhao, Y.; Young, K.H.; Hu, W.; Feng, Z. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 2014, 5, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Huang, Y.; Zheng, J. STAT1 regulates human glutaminase 1 promoter activity through multiple binding sites in HIV-1 infected macrophages. PLoS ONE 2013, 8, e76581. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, Y.; Tian, C.; Taylor, L.; Curthoys, N.; Wang, Y.; Vernon, H.; Zheng, J. Interferon-α regulates glutaminase 1 promoter through STAT1 phosphorylation: Relevance to HIV-1 associated neurocognitive disorders. PLoS ONE 2012, 7, e32995. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, K.; Pan, C.Q.; Karlberg, T.; Balaji, G.; Uttamchandani, M.; Suresh, V.; Schuler, H.; Low, B.C.; Sivaraman, J. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Kodama, M.; Nakayama, K.I. A second Warburg-like effect in cancer metabolism: The metabolic shift of glutamine-derived nitrogen. BioEssays 2020, 42, 2000169. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nature Commun. 2020, 11. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2014, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors (review). Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The pentose phosphate pathway dynamics in cancer and its dependency on intracellular pH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The pentose phosphate pathway as a potential target for cancer therapy. Biomol. Ther. 2018, 26, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The pentose phosphate pathway and its involvement in cisplatin resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef]
- Hong, W.; Cai, P.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Inhibition of glucose-6-phosphate dehydrogenase reverses cisplatin resistance in lung cancer cells via the redox system. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Cossu, V.; Bonanomi, M.; Bauckneht, M.; Ravera, S.; Righi, N.; Miceli, A.; Morbelli, S.; Orengo, A.M.; Piccioli, P.; Bruno, S.; et al. Two high-rate pentose-phosphate pathways in cancer cells. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Schultz, H.; Kähler, D.; Branscheid, D.; Vollmer, E.; Zabel, P.; Goldmann, T. TKTL1 is overexpressed in a large portion of non-small cell lung cancer specimens. Diagn. Pathol. 2008, 3, 35. [Google Scholar] [CrossRef]
- Schmidt, M.; Voelker, H.-U.; Kapp, M.; Krockenberger, M.; Dietl, J.; Kammerer, U. Glycolytic phenotype in breast cancer: Activation of akt, up-regulation of GLUT1, TKTL1 and down-regulation of M2PK. J. Cancer Res. Clin. Oncol. 2009, 136, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, I.A.; Hennenlotter, J.; Stühler, V.; Kühs, U.; Scharpf, M.; Todenhöfer, T.; Stenzl, A.; Bedke, J. Transketolase like 1 (TKTL1) expression alterations in prostate cancer tumorigenesis. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 472.e421–472.e427. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guo, K.; Gao, D.; Kang, X.; Jiang, K.; Li, Y.; Sun, L.; Zhang, S.; Sun, C.; Liu, X.; et al. Identification of transaldolase as a novel serum biomarker for hepatocellular carcinoma metastasis using xenografted mouse model and clinic samples. Cancer Lett. 2011, 313, 154–166. [Google Scholar] [CrossRef]
- Xu, I.M.-J.; Lai, R.K.-H.; Lin, S.-H.; Tse, A.P.-W.; Chiu, D.K.-C.; Koh, H.-Y.; Law, C.-T.; Wong, C.-M.; Cai, Z.; Wong, C.C.-L.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef]
- Yagishita, Y.; Uruno, A.; Yamamoto, M. NRF2-mediated gene regulation and glucose homeostasis. In Molecular Nutrition Diabetes; Elsevier: Amsterdam, The Netherlands, 2016; pp. 331–348. [Google Scholar] [CrossRef]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2019, 122, 4–22. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Menendez, J.A. Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: Molecular mechanisms and therapeutic perspectives. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 381–391. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, M.-Y.; Park, S.W.; Moon, J.-S.; Koh, Y.-K.; Ahn, Y.-H.; Park, B.-W.; Kim, K.-S. Up-regulation of acetyl-CoA carboxylase α and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J. Biol. Chem. 2007, 282, 26122–26131. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Guo, D. SCAP links glucose to lipid metabolism in cancer cells. Mol. Cell. Oncol. 2016, 3, e1132120. [Google Scholar] [CrossRef]
- Bacci, M.; Lorito, N.; Smiriglia, A.; Morandi, A. Fat and Furious: Lipid Metabolism in Antitumoral Therapy Response and Resistence. Trends Cancer 2020, 7, 198–213. [Google Scholar]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed]
- Maan, M.; Peters, J.M.; Dutta, M.; Patterson, A.D. Lipid metabolism and lipophagy in cancer. Biochem. Biophys. Res. Commun. 2018, 504, 582–589. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef]
- Podgornik, H.; Sok, M.; Kern, I.; Marc, J.; Cerne, D. Lipoprotein lipase in non-small cell lung cancer tissue is highly expressed in a subpopulation of tumor-associated macrophages. Pathol. Res. Pract. 2013, 209, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Prieto, D.; Seija, N.; Uriepero, A.; Souto-Padron, T.; Oliver, C.; Irigoin, V.; Guillermo, C.; Navarrete, M.A.; Inés Landoni, A.; Dighiero, G.; et al. LPL protein in chronic lymphocytic leukaemia have different origins in mutated and unmutated patients. Advances for a new prognostic marker in CLL. Br. J. Haematol. 2018, 182, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S. Lipid metabolism in cancer cachexia. Ann. Palliat. Med. 2019, 8, 13–23. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.-L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Lin, K.; Zhao, Y.; Wu, Q.; Chen, D.; Wang, J.; Liang, Y.; Li, J.; Hu, J.; Wang, H.; et al. Adipocytes fuel gastric cancer omental metastasis via PITPNC1-mediated fatty acid metabolic reprogramming. Theranostics 2018, 8, 5452–5468. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2016, 19, 43–54. [Google Scholar] [CrossRef]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Ren, Y.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial-mesenchymal transition. Stem Cell Res. Ther. 2019, 10. [Google Scholar] [CrossRef]
- Göbel, A.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Cholesterol and beyond—The role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188351. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Clendening, J.W.; Pandyra, A.; Li, Z.; Boutros, P.C.; Martirosyan, A.; Lehner, R.; Jurisica, I.; Trudel, S.; Penn, L.Z. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood 2010, 115, 4787–4797. [Google Scholar] [CrossRef]
- Hassanabad, A.F. Current perspectives on statins as potential anti-cancer therapeutics: Clinical outcomes and underlying molecular mechanisms. Transl. Lung Cancer Res. 2019, 8, 692–699. [Google Scholar] [CrossRef]
- Cardaci, S.; Ciriolo, M.R. TCA cycle defects and cancer: When metabolism tunes redox state. Int. J. Cell Biol. 2012, 2012, 161837. [Google Scholar] [CrossRef]
- Scagliola, A.; Mainini, F.; Cardaci, S. The tricarboxylic acid cycle at the crossroad between cancer and immunity. Antioxid. Redox Signal. 2020, 32, 834–852. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Montal, E.D.; Dewi, R.; Bhalla, K.; Ou, L.; Hwang, B.J.; Ropell, A.E.; Gordon, C.; Liu, W.-J.; DeBerardinis, R.J.; Sudderth, J.; et al. PEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol. Cell 2015, 60, 571–583. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, C. Gluconeogenesis in cancer: Function and regulation of PEPCK, FBPase, and G6Pase. Trends Cancer 2019, 5, 30–45. [Google Scholar] [CrossRef]
- Montal, E.D.; Bhalla, K.; Dewi, R.E.; Ruiz, C.F.; Haley, J.A.; Ropell, A.E.; Gordon, C.; Haley, J.D.; Girnun, G.D. Inhibition of phosphoenolpyruvate carboxykinase blocks lactate utilization and impairs tumor growth in colorectal cancer. Cancer Metab. 2019, 7, 8. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang, G.J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Sajnani, K.; Islam, F.; Smith, R.A.; Gopalan, V.; Lam, A.K.-Y. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie 2017, 135, 164–172. [Google Scholar] [CrossRef]
- Leithner, K.; Hrzenjak, A.; Trötzmüller, M.; Moustafa, T.; Köfeler, H.C.; Wohlkoenig, C.; Stacher, E.; Lindenmann, J.; Harris, A.L.; Olschewski, A.; et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene 2015, 34, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Marquez, J.; Flores, J.; Kim, A.H.; Nyamaa, B.; Nguyen, A.T.T.; Park, N.; Han, J. Rescue of TCA cycle dysfunction for cancer therapy. J. Clin. Med. 2019, 8, 2161. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Cantley, L.C. Acetate fuels the cancer engine. Cell 2014, 159, 1492–1494. [Google Scholar] [CrossRef]
- Schug, Z.T.; Vande, V.J.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Shuvalov, O.; Petukhov, A.; Daks, A.; Fedorova, O.; Vasileva, E.; Barlev, N.A. One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 2017, 8, 23955–23977. [Google Scholar] [CrossRef]
- Snell, K. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv. Enzym. Regul. 1984, 22, 325–400. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Vander, H.M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef]
- Vié, N.; Copois, V.; Bascoul-Mollevi, C.; Denis, V.; Bec, N.; Robert, B.; Fraslon, C.; Conseiller, E.; Molina, F.; Larroque, C.; et al. Overexpression of phosphoserine aminotransferase PSAT1 stimulates cell growth and increases chemoresistance of colon cancer cells. Mol. Cancer 2008, 7, 14. [Google Scholar] [CrossRef]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Bachelor, M.A.; Lu, Y.; Owens, D.M. l-3-phosphoserine phosphatase (PSPH) regulates cutaneous squamous cell carcinoma proliferation independent of l-serine biosynthesis. J. Dermatol. Sci. 2011, 63, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2019, 219. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Bruns, H.; Kazanavicius, D.; Schultze, D.; Saeedi, M.A.; Yamanaka, K.; Strupas, K.; Schemmer, P. Glycine inhibits angiogenesis in colorectal cancer: Role of endothelial cells. Amino Acids 2016, 48, 2549–2558. [Google Scholar] [CrossRef]
- Pan, S.; Fan, M.; Liu, Z.; Li, X.; Wang, H. Serine, glycine and one-carbon metabolism in cancer (review). Int. J. Oncol. 2020. [Google Scholar] [CrossRef]
- Rosenzweig, A.; Blenis, J.; Gomes, A.P. Beyond the Warburg effect: How do cancer cells regulate one-carbon metabolism? Front. Cell Dev. Biol. 2018, 6, 90. [Google Scholar] [CrossRef]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Tedeschi, P.M.; Bertino, J.R. Overexpression of the mitochondrial folate and glycine-serine pathway: A new determinant of methotrexate selectivity in tumors. Cancer Res. 2013, 73, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P. Methionine dependence of cancer. Biomolecules 2020, 10, 568. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Borrego, S.L.; Fahrmann, J.; Datta, R.; Stringari, C.; Grapov, D.; Zeller, M.; Chen, Y.; Wang, P.; Baldi, P.; Gratton, E.; et al. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metab. 2016, 4, 9. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.D.; Eich, M.L.; Varambally, S. Dysregulation of de novo nucleotide biosynthetic pathway enzymes in cancer and targeting opportunities. Cancer Lett. 2020, 470, 134–140. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017, 36, 252–259. [Google Scholar] [CrossRef]
- Choi, B.-H.; Coloff, J.L. The diverse functions of non-essential amino acids in cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef]
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metab. 2018, 28, 706–720.e706. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Danial, N.N. Grasping for aspartate in tumour metabolism. Nat. Cell Biol. 2018, 20, 738–739. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef]
- Terry, A.R.; Hay, N. Fuelling cancer cells. Nat. Rev. Endocrinol. 2019, 15, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Serpa, J. Cysteine as a carbon source, a hot spot in cancer cells survival. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef]
- Li, J.T.; Yin, M.; Wang, D.; Wang, J.; Lei, M.Z.; Zhang, Y.; Liu, Y.; Zhang, L.; Zou, S.W.; Hu, L.P.; et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2020, 22, 167–174. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell. Mol. Life Sci. 2021, 78, 195–206. [Google Scholar] [CrossRef]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef]
- Wang, P.; Wu, S.; Zeng, X.; Zhang, Y.; Zhou, Y.; Su, L.; Lin, W. BCAT1 promotes proliferation of endometrial cancer cells through reprogrammed BCAA metabolism. Int. J. Clin. Exp. Pathol. 2018, 11, 5536–5546. [Google Scholar] [PubMed]
- Xue, P.; Zeng, F.; Duan, Q.; Xiao, J.; Liu, L.; Yuan, P.; Fan, L.; Sun, H.; Malyarenko, O.S.; Lu, H.; et al. BCKDK of BCAA catabolism cross-talking with the MAPK pathway promotes tumorigenesis of colorectal cancer. EBioMedicine 2017, 20, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Vander, H.M.G. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Chapter two—Metabolic regulation of apoptosis in cancer. In International Review of Cell and Molecular Biology; Jeon, K.W., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 327, pp. 43–87. [Google Scholar]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 2007, 26, 2027–2038. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. HIF-1-regulated glucose metabolism: A key to apoptosis resistance? Cell Cycle 2007, 6, 790–792. [Google Scholar] [CrossRef]
- Yu, C.; Minemoto, Y.; Zhang, J.; Liu, J.; Tang, F.; Bui, T.N.; Xiang, J.; Lin, A. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol. Cell 2004, 13, 329–340. [Google Scholar] [CrossRef]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Nganga, R.; Oleinik, N.; Ogretmen, B. Chapter one—Mechanisms of ceramide-dependent cancer cell death. In Advances in Cancer Research; Chalfant, C.E., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 140, pp. 1–25. [Google Scholar]
- Ségui, B.; Andrieu-Abadie, N.; Jaffrézou, J.-P.; Benoist, H.; Levade, T. Sphingolipids as modulators of cancer cell death: Potential therapeutic targets. Biochim. Biophys. Acta Biomembr. 2006, 1758, 2104–2120. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Li, Z.; Sun, X. Non-coding RNAs operate in the crosstalk between cancer metabolic reprogramming and metastasis. Front. Oncol. 2020, 10, 810. [Google Scholar] [CrossRef]
- Slack, F.; Chinnaiyan, A. The role of non-coding RNAs in oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Shankaraiah, R.C.; Veronese, A.; Sabbioni, S.; Negrini, M. Non-coding RNAs in the reprogramming of glucose metabolism in cancer. Cancer Lett. 2018, 419, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, L.; Cao, Y.; Chen, S.; Cao, J.; Wu, D.; Chen, J.; Xiong, H.; Pan, Z.; Qiu, F.; et al. Overexpression of lncRNA IGFBP4-1 reprograms energy metabolism to promote lung cancer progression. Mol. Cancer 2017, 16, 154. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef]
- Lau, A.N.; Vander, H.M.G. Metabolism in the tumor microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40. [Google Scholar] [CrossRef]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic heterogeneity in human lung tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Kennedy, K.M.; Scarbrough, P.M.; Ribeiro, A.; Richardson, R.; Yuan, H.; Sonveaux, P.; Landon, C.D.; Chi, J.-T.; Pizzo, S.; Schroeder, T.; et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS ONE 2013, 8, e75154. [Google Scholar] [CrossRef]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: A metabolic survival role for tumor-associated stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Singer, K.; Cheng, W.C.; Kreutz, M.; Ho, P.C.; Siska, P.J. Immunometabolism in cancer at a glance. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef]
- Vergati, M.; Krasniqi, E.; Monte, G.D.; Riondino, S.; Vallone, D.; Guadagni, F.; Ferroni, P.; Roselli, M. Ketogenic diet and other dietary intervention strategies in the treatment of cancer. Curr. Med. Chem. 2017, 24, 1170–1185. [Google Scholar] [CrossRef]
- Mohanti, B.K.; Rath, G.K.; Anantha, N.; Kannan, V.; Das, B.S.; Chandramouli, B.A.R.; Banerjee, A.K.; Das, S.; Jena, A.; Ravichandran, R.; et al. Improving cancer radiotherapy with 2-deoxy-d-glucose: Phase I/II clinical trials on human cerebral gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 103–111. [Google Scholar] [CrossRef]
- Stein, M.; Lin, H.; Jeyamohan, C.; Dvorzhinski, D.; Gounder, M.; Bray, K.; Eddy, S.; Goodin, S.; White, E.; DiPaola, R.S. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 2010, 70, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: From bench side to bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.M.; Mohamed, W.G.; Seddik, M.-A.H.; Ahmed, A.-S.A.; Mahmoud, A.G.; Amer, W.H.; Helmy, N.M.M.; Hamed, A.R.; Ahmed, N.S.; Abd-Allah, A.A.-R. Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: A concise literature review and case study. Chin. J. Cancer 2014, 33, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Chapiro, J.; Sur, S.; Savic, L.J.; Ganapathy-Kanniappan, S.; Reyes, J.; Duran, R.; Thiruganasambandam, S.C.; Moats, C.R.; Lin, M.; Luo, W.; et al. Systemic delivery of microencapsulated 3-bromopyruvate for the therapy of pancreatic cancer. Clin. Cancer Res. 2014, 20, 6406. [Google Scholar] [CrossRef] [PubMed]
- De Marinis, F.; Rinaldi, M.; Ardizzoni, A.; Bruzzi, P.; Pennucci, M.C.; Portalone, L.; D’Aprile, M.; Ripanti, P.; Romano, F.; Belli, M.; et al. The role of vindesine and lonidamine in the treatment of elderly patients with advanced non-small cell lung cancer: A phase III randomized fonicap trial. Tumori J. 1999, 85, 177–182. [Google Scholar] [CrossRef]
- Berruti, A.; Bitossi, R.; Gorzegno, G.; Bottini, A.; Alquati, P.; Matteis, A.D.; Nuzzo, F.; Giardina, G.; Danese, S.; Lena, M.D.; et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: Final results of a phase III study with a factorial design. J. Clin. Oncol. 2002, 20, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wei, L.; Liu, Y.; Ding, Y.; Liu, X.; Zhang, X.; Wang, X.; Yao, Y.; Lu, J.; Wang, Q.; et al. Gen-27, a newly synthesized flavonoid, inhibits glycolysis and induces cell apoptosis via suppression of hexokinase II in human breast cancer cells. Biochem. Pharmacol. 2017, 125, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, S.; Zhao, Y.; Huang, S.; Zhao, J. Molecular docking and molecular dynamics simulation analyses of urea with ammoniated and ammoxidized lignin. J. Mol. Graph. Model. 2017, 71, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, F.; Lu, J.; Xia, Y.; He, L.; Chen, K.; Li, J.; Li, S.; Liu, T.; Zheng, Y.; et al. By reducing hexokinase 2, resveratrol induces apoptosis in HCC cells addicted to aerobic glycolysis and inhibits tumor growth in mice. Oncotarget 2015, 6, 13703–13717. [Google Scholar] [CrossRef] [PubMed]
- Boocock, D.J.; Faust, G.E.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J.; et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1246–1252. [Google Scholar] [CrossRef]
- Li, W.; Hao, J.; Zhang, L.; Cheng, Z.; Deng, X.; Shu, G. Astragalin reduces hexokinase 2 through increasing miR-125b to inhibit the proliferation of hepatocellular carcinoma cells in vitro and in vivo. J. Agric. Food Chem. 2017, 65, 5961–5972. [Google Scholar] [CrossRef]
- Xu, D.; Jin, J.; Yu, H.; Zhao, Z.; Ma, D.; Zhang, C.; Jiang, H. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J. Exp. Clin. Cancer Res. 2017, 36, 44. [Google Scholar] [CrossRef]
- Flaig, T.W.; Gustafson, D.L.; Su, L.J.; Zirrolli, J.A.; Crighton, F.; Harrison, G.S.; Pierson, A.S.; Agarwal, R.; Glodé, L.M. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Investig. New Drugs 2007, 25, 139–146. [Google Scholar] [CrossRef]
- Gunnink, L.K.; Alabi, O.D.; Kuiper, B.D.; Gunnink, S.M.; Schuiteman, S.J.; Strohbehn, L.E.; Hamilton, K.E.; Wrobel, K.E.; Louters, L.L. Curcumin directly inhibits the transport activity of GLUT1. Biochimie 2016, 125, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-H.; Ho, C.-T.; Chen, Z.-F.; Chen, L.-C.; Whang-Peng, J.; Lin, T.-N.; Ho, Y.-S. The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter. J. Food Drug Anal. 2018, 26, 221–231. [Google Scholar] [CrossRef]
- Lin, S.-T.; Tu, S.-H.; Yang, P.-S.; Hsu, S.-P.; Lee, W.-H.; Ho, C.-T.; Wu, C.-H.; Lai, Y.-H.; Chen, M.-Y.; Chen, L.-C. Apple polyphenol phloretin inhibits colorectal cancer cell growth via inhibition of the type 2 glucose transporter and activation of p53-mediated signaling. J. Agric. Food Chem. 2016, 64, 6826–6837. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Hurren, R.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; Minden, M.D.; Bilan, P.J.; et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008, 7, 3546–3555. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Rahier, N.J.; Molinier, N.; Long, C.; Deshmukh, S.K.; Kate, A.S.; Ranadive, P.; Verekar, S.A.; Jiotode, M.; Lavhale, R.R.; Tokdar, P.; et al. Anticancer activity of koningic acid and semisynthetic derivatives. Bioorgan. Med. Chem. 2015, 23, 3712–3721. [Google Scholar] [CrossRef]
- Kumagai, S.; Narasaki, R.; Hasumi, K. Glucose-dependent active ATP depletion by koningic acid kills high-glycolytic cells. Biochem. Biophys. Res. Commun. 2008, 365, 362–368. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S. Evolution of GAPDH as a druggable target of tumor glycolysis? Expert Opin. Ther. Targets 2018, 22, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A phase I study of CPI-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Buyse, M.E.; Alistar, A.T.; Rocha, L.C.M.; Luther, S.; Pardee, T.S.; van Cutsem, E. A phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncol. 2019, 15, 3189–3196. [Google Scholar] [CrossRef] [PubMed]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef]
- Shen, Y.C.; Ou, D.L.; Hsu, C.; Lin, K.L.; Chang, C.Y.; Lin, C.Y.; Liu, S.H.; Cheng, A.L. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer 2013, 108, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Xiong, X.; Huang, G.; Liu, J.; Sheng, S.; Wang, H.; Qin, W. Dichloroacetate enhances adriamycin-induced hepatoma cell toxicity in vitro and in vivo by increasing reactive oxygen species levels. PLoS ONE 2014, 9, e92962. [Google Scholar] [CrossRef]
- Chu, Q.S.-C.; Sangha, R.; Spratlin, J.; Vos, L.J.; Mackey, J.R.; McEwan, A.J.B.; Venner, P.; Michelakis, E.D. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Lippard, S.J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. USA 2009, 106, 22199–22204. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes Taxol-resistant cancer cells to Taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: An in vivo study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Marchiq, I.; Pouysségur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.Α.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef]
- Amancio, C. The PKB/AKT pathway in cancer. Curr. Pharm. Des. 2010, 16, 34–44. [Google Scholar] [CrossRef]
- Kindler, H.L.; Wroblewski, K.; Wallace, J.A.; Hall, M.J.; Locker, G.; Nattam, S.; Agamah, E.; Stadler, W.M.; Vokes, E.E. Gemcitabine plus sorafenib in patients with advanced pancreatic cancer: A phase II trial of the University of Chicago Phase II consortium. Investig. New Drugs 2012, 30, 382–386. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Di Magno, L.; Manni, S.; di Pastena, F.; Coni, S.; Macone, A.; Cairoli, S.; Sambucci, M.; Infante, P.; Moretti, M.; Petroni, M.; et al. Phenformin inhibits hedgehog-dependent tumor growth through a complex I-independent redox/corepressor module. Cell Rep. 2020, 30, 1735–1752.e1737. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta 2016, 1866, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhang, Q.; Pan, J.; Lee, Y.; Ouari, O.; Hardy, M.; Zielonka, M.; Myers, C.R.; Zielonka, J.; Weh, K.; et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat. Commun. 2019, 10, 2205. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Shestov, A.A.; Worth, A.J.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Glickson, J.D.; Blair, I.A. Inhibition of mitochondrial complex II by the anticancer agent lonidamine. J. Biol. Chem. 2016, 291, 42–57. [Google Scholar] [CrossRef]
- Oudard, S.; Carpentier, A.; Banu, E.; Fauchon, F.; Celerier, D.; Poupon, M.F.; Dutrillaux, B.; Andrieu, J.M.; Delattre, J.Y. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J. Neurooncol. 2003, 63, 81–86. [Google Scholar] [CrossRef]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef]
- Diepart, C.; Karroum, O.; Magat, J.; Feron, O.; Verrax, J.; Calderon, P.B.; Grégoire, V.; Leveque, P.; Stockis, J.; Dauguet, N.; et al. Arsenic trioxide treatment decreases the oxygen consumption rate of tumor cells and radiosensitizes solid tumors. Cancer Res. 2012, 72, 482–490. [Google Scholar] [CrossRef]
- Moncada, P.S. Nitric oxide and oxygen: Actions and interactions in health and disease. Redox Biol. 2015, 5, 421. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Y.; Seo, W.; Zhang, R.; Lu, C.; Wang, Y.; Luo, L.; Paul, B.; Yan, W.; Saxena, D.; et al. Targeting cellular metabolism to reduce head and neck cancer growth. Sci. Rep. 2019, 9, 4995. [Google Scholar] [CrossRef]
- Masamha, C.P.; LaFontaine, P. Molecular targeting of glutaminase sensitizes ovarian cancer cells to chemotherapy. J. Cell. Biochem. 2018, 119, 6136–6145. [Google Scholar] [CrossRef]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef]
- Guo, L.; Zhou, B.; Liu, Z.; Xu, Y.; Lu, H.; Xia, M.; Guo, E.; Shan, W.; Chen, G.; Wang, C. Blockage of glutaminolysis enhances the sensitivity of ovarian cancer cells to PI3K/mTOR inhibition involvement of STAT3 signaling. Tumour Biol. 2016, 37, 11007–11015. [Google Scholar] [CrossRef]
- Reis, L.M.D.; Adamoski, D.; Souza, R.O.O.; Ascenção, C.F.R.; de Oliveira, K.R.S.; Corrêa-da-Silva, F.; de Sá Patroni, F.M.; Dias, M.M.; Consonni, S.R.; de Moraes-Vieira, P.M.M.; et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 2019, 294, 9342–9357. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.S.; da Costa Rosa, M.; Stumpo, V.; Rais, R.; Slusher, B.S.; Riggins, G.J. The glutamine antagonist prodrug JHU-083 slows malignant glioma growth and disrupts mTOR signaling. Neurooncol. Adv. 2021, 3, vdaa149. [Google Scholar] [CrossRef] [PubMed]
- Nyce, J.W. Autoinflammatory reaction in dogs treated for cancer via G6PD inhibition. Case Rep. Vet. Med. 2017, 2017, 4275305. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Jiang, C.; Feng, Y.; Chen, R.; Lin, X.; Zhang, Z.; Han, L.; Chen, X.; Li, H.; Guo, Y.; et al. Effects of G6PD activity inhibition on the viability, ROS generation and mechanical properties of cervical cancer cells. Biochim. Biophys. Acta 2016, 1863, 2245–2254. [Google Scholar] [CrossRef]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef]
- Köhler, E.; Barrach, H.; Neubert, D. Inhibition of NADP dependent oxidoreductases by the 6-aminonicotinamide analogue of NADP. FEBS Lett. 1970, 6, 225–228. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Dheeraj, A.; Agarwal, C.; Schlaepfer, I.R.; Raben, D.; Singh, R.; Agarwal, R.; Deep, G. A novel approach to target hypoxic cancer cells via combining β-oxidation inhibitor etomoxir with radiation. Hypoxia 2018, 6, 23–33. [Google Scholar] [CrossRef]
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R. Molecular pathways: Fatty acid synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef]
- Shiragami, R.; Murata, S.; Kosugi, C.; Tezuka, T.; Yamazaki, M.; Hirano, A.; Yoshimura, Y.; Suzuki, M.; Shuto, K.; Koda, K. Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int. J. Oncol. 2013, 43, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Chávez-Blanco, A.; Domínguez-Gómez, G.; Taja-Chayeb, L.; Morales-Barcenas, R.; Trejo-Becerril, C.; Perez-Cardenas, E.; Gonzalez-Fierro, A.; Dueñas-González, A. Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy. Expert Opin. Investig. Drugs 2018, 27, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Lai, B.S.; Anderson, V.E. Inhibition of triclosan to fatty acid synthase from goose uropygial glands and human breast cancer cells in vitro. Ai Zheng 2003, 22, 270–273. [Google Scholar]
- Liu, B.; Wang, Y.; Fillgrove, K.L.; Anderson, V.E. Triclosan inhibits enoyl-reductase of type I fatty acid synthase in vitro and is cytotoxic to MCF-7 and SKBr-3 breast cancer cells. Cancer Chemother. Pharmacol. 2002, 49, 187–193. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a selective FASN inhibitor, shows potent anti-tumor activity in the MMTV-Neu model of HER2(+) breast cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Cuyàs, E.; Gumuzio, J.; Bosch-Barrera, J.; Leis, O.; Martin, Á.G.; Menendez, J.A. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014, 5, 8306–8316. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.P.; Fargnoli, J.; Henley, B.J.; Yang, W.P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef]
- Król, S.K.; Kiełbus, M.; Rivero-Müller, A.; Stepulak, A. Comprehensive review on betulin as a potent anticancer agent. Biomed. Res. Int. 2015, 2015, 584189. [Google Scholar] [CrossRef]
- Mashima, T.; Oh-hara, T.; Sato, S.; Mochizuki, M.; Sugimoto, Y.; Yamazaki, K.; Hamada, J.; Tada, M.; Moriuchi, T.; Ishikawa, Y.; et al. p53-defective tumors with a functional apoptosome-mediated pathway: A new therapeutic target. J. Natl. Cancer Inst. 2005, 97, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, J.; Liang, Z.; He, F.; Yang, L.; Li, P.; Jiang, Y.; Wang, B.; Zhou, C.; Wang, Y.; et al. Simvastatin and atorvastatin inhibit DNA replication licensing factor MCM7 and effectively suppress RB-deficient tumors growth. Cell Death Dis. 2017, 8, e2673. [Google Scholar] [CrossRef] [PubMed]
- Borgquist, S.; Bjarnadottir, O.; Kimbung, S.; Ahern, T.P. Statins: A role in breast cancer therapy? J. Intern. Med. 2018, 284, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov. Med. 2016, 21, 373–380. [Google Scholar]
- Megías-Vericat, J.E.; Ballesta-López, O.; Barragán, E.; Montesinos, P. IDH1-mutated relapsed or refractory AML: Current challenges and future prospects. Blood Lymphat. Cancer 2019, 9, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Palmisiano, N.; Mantzaris, I.; Mims, A.; DiNardo, C.; Silverman, L.R.; Wang, E.S.; Fiedler, W.; Baldus, C.; Schwind, S.; et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: Phase I study results. Leukemia 2020, 34, 2903–2913. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.; Bähr, O.; Schuler, M.; Rohrberg, K.; Chawla, S.P.; Janku, F.; Schiff, D.; Heinemann, V.; Narita, Y.; Lenz, H.J.; et al. Phase I assessment of safety and therapeutic activity of BAY1436032 in patients with IDH1-mutant solid tumors. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-based design and identification of FT-2102 (olutasidenib), a potent mutant-selective IDH1 inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Tallman, M.S.; de Botton, S.; Kantarjian, H.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Dogra, R.; Bhatia, R.; Shankar, R.; Bansal, P.; Rawal, R.K. Enasidenib: First mutant IDH2 inhibitor for the treatment of refractory and relapsed acute myeloid leukemia. Anticancer Agents Med. Chem. 2018, 18, 1936–1951. [Google Scholar] [CrossRef] [PubMed]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Nguyen, T.T.T.; Shang, E.; Siegelin, M.D. Novel IDH1-targeted glioma therapies. CNS Drugs 2019, 33, 1155–1166. [Google Scholar] [CrossRef]
- Dekhne, A.S.; Shah, K.; Ducker, G.S.; Katinas, J.M.; Wong-Roushar, J.; Nayeen, M.J.; Doshi, A.; Ning, C.; Bao, X.; Frühauf, J.; et al. Novel pyrrolo[3,2-d]pyrimidine compounds target mitochondrial and cytosolic one-carbon metabolism with broad-spectrum antitumor efficacy. Mol. Cancer Ther. 2019, 18, 1787–1799. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lu, Y.X.; Chen, D.L.; Zuo, Z.X.; Liu, Z.X.; Wu, Q.N.; Mo, H.Y.; Wang, Z.X.; Wang, D.S.; Pu, H.Y.; et al. Modulation of redox homeostasis by inhibition of MTHFD2 in colorectal cancer: Mechanisms and therapeutic implications. J. Natl. Cancer Inst. 2019, 111, 584–596. [Google Scholar] [CrossRef]
- Fu, C.; Sikandar, A.; Donner, J.; Zaburannyi, N.; Herrmann, J.; Reck, M.; Wagner-Döbler, I.; Koehnke, J.; Müller, R. The natural product carolacton inhibits folate-dependent C1 metabolism by targeting FolD/MTHFD. Nat. Commun. 2017, 8, 1529. [Google Scholar] [CrossRef]
- Okimoto, T.; Kotani, H.; Iida, Y.; Koyanagi, A.; Tanino, R.; Tsubata, Y.; Isobe, T.; Harada, M. Pemetrexed sensitizes human lung cancer cells to cytotoxic immune cells. Cancer Sci. 2020, 111, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Azuma, K.; Yamanaka, T.; Sugawara, S.; Yoshioka, H.; Wakuda, K.; Atagi, S.; Iwamoto, Y.; Hayashi, H.; Okamoto, I.; et al. Randomized phase III study of continuation maintenance bevacizumab with or without pemetrexed in advanced nonsquamous non-small-cell lung cancer: COMPASS (WJOG5610L). J. Clin. Oncol. 2020, 38, 793–803. [Google Scholar] [CrossRef]
- Rossi, G.; Alama, A.; Genova, C.; Rijavec, E.; Tagliamento, M.; Biello, F.; Coco, S.; dal Bello, M.G.; Boccardo, S.; Grossi, F. The evolving role of pemetrexed disodium for the treatment of non-small cell lung cancer. Expert Opin. Pharmacother. 2018, 19, 1969–1976. [Google Scholar] [CrossRef] [PubMed]
- Rana, R.M.; Rampogu, S.; Abid, N.B.; Zeb, A.; Parate, S.; Lee, G.; Yoon, S.; Kim, Y.; Kim, D.; Lee, K.W. In silico study identified methotrexate analog as potential inhibitor of drug resistant human dihydrofolate reductase for cancer therapeutics. Molecules 2020, 25, 3510. [Google Scholar] [CrossRef]
- Neradil, J.; Pavlasova, G.; Sramek, M.; Kyr, M.; Veselska, R.; Sterba, J. DHFR-mediated effects of methotrexate in medulloblastoma and osteosarcoma cells: The same outcome of treatment with different doses in sensitive cell lines. Oncol. Rep. 2015, 33, 2169–2175. [Google Scholar] [CrossRef]
- Lluch, A.; Barrios, C.H.; Torrecillas, L.; Ruiz-Borrego, M.; Bines, J.; Segalla, J.; Guerrero-Zotano, Á.; García-Sáenz, J.A.; Torres, R.; de la Haba, J.; et al. Phase III trial of adjuvant capecitabine after standard neo/adjuvant chemotherapy in patients with early triple-negative breast cancer (GEICAM/2003-11_CIBOMA/2004-01). J. Clin. Oncol. 2020, 38, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.S.; Godara, A.; Byrne, M.M.; Saif, M.W. Capecitabine for the treatment of pancreatic cancer. Expert Opin. Pharmacother. 2019, 20, 399–409. [Google Scholar] [CrossRef]
- Sharma, R.; Hoskins, J.M.; Rivory, L.P.; Zucknick, M.; London, R.; Liddle, C.; Clarke, S.J. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphisms and toxicity to capecitabine in advanced colorectal cancer patients. Clin. Cancer Res. 2008, 14, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Gotanda, K.; Hirota, T.; Matsumoto, N.; Ieiri, I. MicroRNA-433 negatively regulates the expression of thymidylate synthase (TYMS) responsible for 5-fluorouracil sensitivity in HeLa cells. BMC Cancer 2013, 13, 369. [Google Scholar] [CrossRef]
- Atkinson, M.R.; Murray, A.W. Inhibition of pruine phosphoribosyltransferases of ehrlich ascites-tumour cells by 6-mercaptopurine. Biochem. J. 1965, 94, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Adamson, P.C.; Balis, F.M.; Arndt, C.A.; Holcenberg, J.S.; Narang, P.K.; Murphy, R.F.; Gillespie, A.J.; Poplack, D.G. Intrathecal 6-mercaptopurine: Preclinical pharmacology, phase I/II trial, and pharmacokinetic study. Cancer Res. 1991, 51, 6079–6083. [Google Scholar] [PubMed]
- Munshi, P.N.; Lubin, M.; Bertino, J.R. 6-thioguanine: A drug with unrealized potential for cancer therapy. Oncologist 2014, 19, 760–765. [Google Scholar] [CrossRef]
- Vora, A.; Mitchell, C.D.; Lennard, L.; Eden, T.O.; Kinsey, S.E.; Lilleyman, J.; Richards, S.M. Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: A randomised trial. Lancet 2006, 368, 1339–1348. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | State of Development | References |
|---|---|---|---|
| 2-deoxyglucose (2-DG) | Hexokinase 2 (HK2) | Phase II | [171,172] |
| 3-bromopyruvate (3BP) | Phase I | [173,174,175] | |
| Lonidamine | Phase II | [176,177] | |
| Genistein-27 | Preclinical | [178] | |
| Benserazide | Preclinical | [179] | |
| Resveratrol | Phase I | [180,181] | |
| Astragalin | Preclinical | [182] | |
| Chrysin | Preclinical | [183] | |
| Silybin | Glucose transporters (GLUTs) | Phase I | [184] |
| Cytochalasin B | Preclinical | [185] | |
| Phloretin | Preclinical | [186,187] | |
| Fasentin | Preclinical | [188] | |
| STF-31 | Preclinical | [189] | |
| WZB117 | Preclinical | [190,191] | |
| Ritonavir | Preclinical | [192] | |
| Koningic acid | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Preclinical | [193,194] |
| Iodoacetate | Preclinical | [195] | |
| CPI-613 | Pyruvate dehydrogenase (PDH)/α-ketoglutarate dehydrogenase | Phase III | [196,197,198] |
| Dichloroacetate (DCA) | Pyruvate dehydrogenase kinase (PDK) | Phase I | [199,200,201,202] |
| Mitaplatin (cisplatin and DCA fusion) | Phase I | [203] | |
| Oxamate | Lactate dehydrogenase (LDHA) | Preclinical | [204] |
| FX11 | Preclinical | [205] | |
| α-Cyano-4-hydroxycinnamic acid | Preclinical | [206] | |
| Cinnamate | Monocarboxylate transporters (MCTs) | Preclinical | [207] |
| AZD3965 | Phase I | [208,209] | |
| Afuresertib | PI3K/Akt | Phase I | [210,211,212] |
| Uprosertib | Phase I | ||
| Ipatasertib | Phase I | ||
| Sorafenib | Phase II | [213] | |
| Metformin | Complex I, oxidative phosphorylation (OXPHOS) | Preclinical and clinical studies | [214] |
| Phenformin | Preclinical and clinical studies | [215] | |
| Lonidamine | Complex II (OXPHOS) | Phase II | [216,217,218,219] |
| Atovaquone | Complex III (OXPHOS) | Early Phase I | [220] |
| Arsenic trioxide | Complex IV (OXPHOS) | Phase III | [22,221] |
| Nitric oxide | Preclinical and clinical studies | [222] | |
| BPTES | Glutaminase (GLS1) | Preclinical | [223,224] |
| CB-839 | Phase II | [225,226,227] | |
| JHU-083 | Preclinical | [228] | |
| Dehydroepiandrosterone (DHEA) | Glucose-6-phosphate dehydrogenase (G6PD) | Phase I | [229,230] |
| Polydatin | Preclinical | [231] | |
| 6-aminonicotinamide (6-AN) | 6-phosphogluconate dehydrogenase (6GPD) | Preclinical | [232] |
| Etomoxir | Carnitine palmitoyl transferase 1 (CPT1) | Retired from phase II clinical trials for diabetes and heart failures | [233,234] |
| TVB-2640 | Fatty acid synthase (FASN) | Phase II | [235,236] |
| Cerulenin | Preclinical | [237] | |
| Orlistat | Preclinical | [238] | |
| GSK2194069 | Preclinical | [236] | |
| Triclosan | Discontinued for safety issues | [239,240] | |
| Fasnall | Preclinical | [241] | |
| SB-204990 | ATP-citrate lyase (ACLY) | Preclinical | [242] |
| Soraphen A | Acetyl-CoA carboxylase (ACC) | Preclinical | [243,244] |
| BZ36 | Stearoyl-CoA desaturase (SCD) | Preclinical | [245] |
| A939572 | Preclinical | [246] | |
| Fatostatin | Sterol regulatory element-binding protein (SREBP) | Preclinical | [247] |
| Betulin | Preclinical | [248] | |
| Triacscin C | Acetyl-CoA synthase (ACS) | Preclinical | [249] |
| Statins | 3-hydroxy-methylglutaryl-CoA reductase (HMGCR) | Preclinical and clinical studies | [250,251] |
| AG-120 (ivosidenib) | Mutant isocitrate dehydrogenase 1 (IDH1) | Phase III | [252,253] |
| IDH305 | Mutant IDH2 | Phase II | [254,255,256] |
| BAY1436032 | Mutant IDH1/2 | Phase I | [257,258,259] |
| FT-2102 | Phase II | [260] | |
| AG-221 (enasidenib) | Phase III | [261,262,263] | |
| AG-881 | Phase III | [264,265] | |
| AGF347 | Serine hydroxymethyltransferase 1/2 (SHMT1/2) | Preclinical | [266] |
| LY345899 | Methylene tetrahydrofolate dehydrogenase 2 (MTHFD2) | Preclinical | [267] |
| Carolacton | MTHFD1/2 | Preclinical | [268] |
| LY231514/MTA/pemetrexed | Dihydrofolate reductase (DHFR), thymidylate synthase (TS), glycinamide ribonucleotide formyltransferase (GARFT) | Phase IV | [269,270,271] |
| Amethopterin/MTX/methotrexate | TS, DHFR | Phase IV | [272,273] |
| Capecitabine | TS | Phase IV | [274,275,276] |
| 5-Fluorouracil | Phase III | [277] | |
| 6-Mercaptopurine | Phosphoribosyl pyrophosphate amidotransferase (PPAT) | Phase III | [278,279] |
| 6-Thioguanine | Phase III | [280,281] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. https://doi.org/10.3390/cells10051056
Schiliro C, Firestein BL. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells. 2021; 10(5):1056. https://doi.org/10.3390/cells10051056
Chicago/Turabian StyleSchiliro, Chelsea, and Bonnie L. Firestein. 2021. "Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation" Cells 10, no. 5: 1056. https://doi.org/10.3390/cells10051056
APA StyleSchiliro, C., & Firestein, B. L. (2021). Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells, 10(5), 1056. https://doi.org/10.3390/cells10051056