
Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells

 ,
,  , and
, and
Abstract
:1. Introduction
2. Purinergic P1 and P2 Receptors
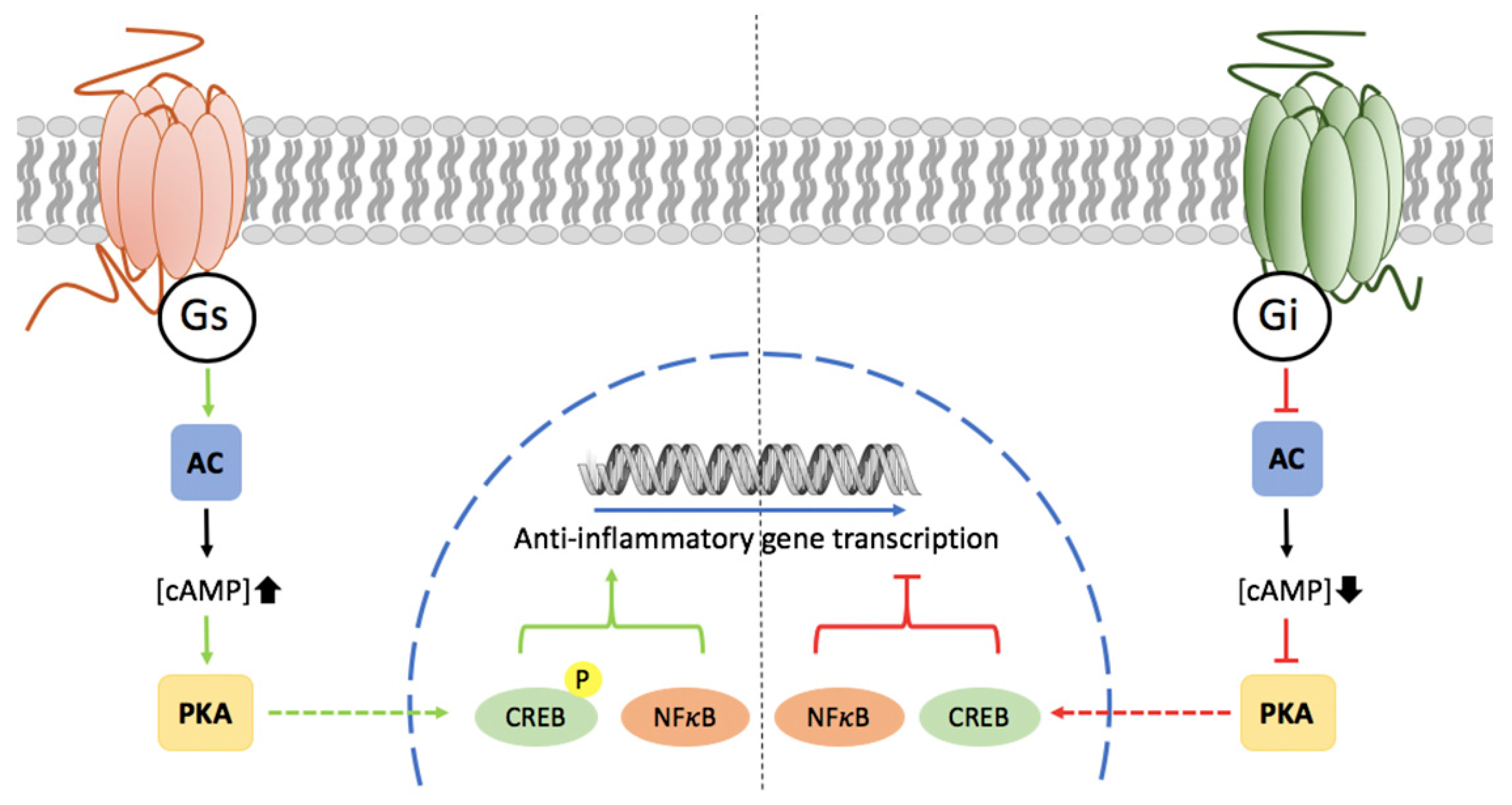
3. Potential of Adenosine Receptors (AR) as Therapeutic Targets
4. Neuron vs. Glia in Neurodegeneration
5. Microglia
“Based on limited numbers of markers, activated macrophages can be classified as classically activated (M1) macrophages that support microbicidal activity or alternatively activated (M2) macrophages that are not competent to eliminate pathogens”.
6. Ischemic Preconditioning after Brain Ischemia
7. Microglia in Aging and in Neurodegenerative Diseases, Friend or Foe?
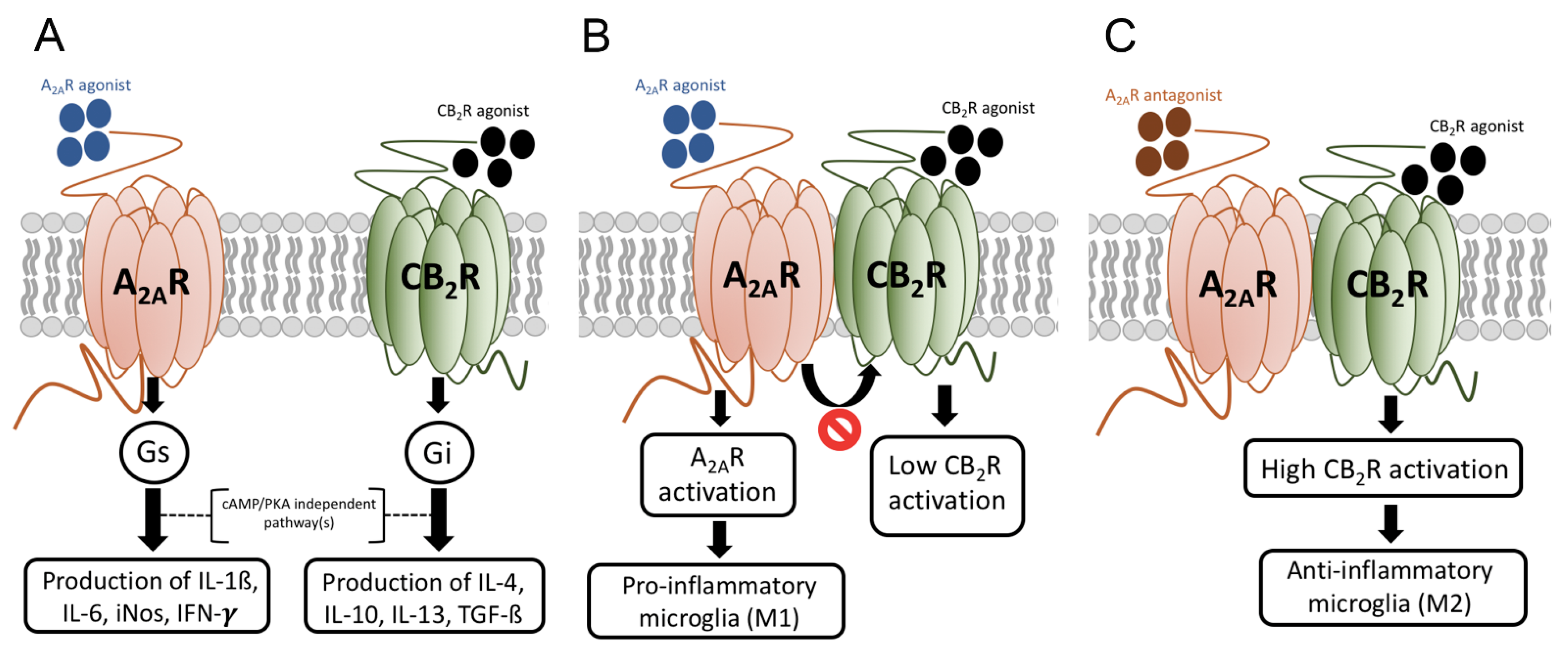
8. Skewing the M1/M2 Balance towards the Neuroprotective M2 Phenotype
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, B.; Teschemacher, A.G.; Kasparov, S. Astroglia as a cellular target for neuroprotection and treatment of neuro-psychiatric disorders. Glia 2017, 65, 1205–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Dipatre, P.L.; Gelman, B.B. Microglial cell activation in aging and Alzheimer disease: Partial linkage with neurofibrillary tangle burden in the hippocampus. J. Neuropathol. Exp. Neurol. 1997, 56, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, J.; Luber-Narod, J.; Styren, S.D.; Civin, W.H. Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 1988, 9, 339–349. [Google Scholar] [CrossRef]
- Streit, W.J.; Sparks, D.L. Activation of microglia in the brains of humans with heart disease and hypercholesterolemic rabbits. J. Mol. Med. 1997, 75, 130–138. [Google Scholar] [CrossRef]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of microglia in CNS inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Bonnekoh, P.; Miyazawa, T.; Oschlies, U.; Dux, E.; Hossmann, K.-A.; Kreutzberg, G. The microglial reaction in the rat hippocampus following global ischemia: Immuno-electron microscopy. Acta Neuropathol. 1992, 84, 588–595. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Barron, K.D. The microglial cell. A historical review. J. Neurol. Sci. 1995, 134, 57–68. [Google Scholar] [CrossRef]
- Banati, R.B.; Daniel, S.E.; Blunt, S.B. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 1998, 13, 221–227. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Otte-Höller, I.; Wesseling, P.; Van Nostrand, W.E.; Sorg, C.; de Waal, R.M.W.; Ruiter, D.J. A lysosomal marker for activated microglial cells involved in Alzheimer classic senile plaques. Acta Neuropathol. 1995, 90, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Alvarez, X.A.; Fernandez-Novoa, L.; Franco, A.; Mangues, R.; Pellicer, A.; Nishimura, T. Brain interleukin-1β in Alzheimer’s disease and vascular dementia. Methods Find. Exp. Clin. Pharmacol. 1994, 16, 141–151. [Google Scholar] [PubMed]
- Maat-Schieman, M.; Rozemuller, A.; Van Duinen, S.; Haan, J.; Eikelenboom, P.; Roos, R. Microglia in Diffuse Plaques in Hereditary Cerebral Hemorrhage with Amyloidosis (Dutch). An Immunohistochemical Study. J. Neuropathol. Exp. Neurol. 1994, 53, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain J. Neurol. 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [Green Version]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Fernández-Suárez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Mesquida-Veny, F.; Del Río, J.A.; Hervera, A. Macrophagic and microglial complexity after neuronal injury. Prog. Neurobiol. 2020, 101970. [Google Scholar] [CrossRef]
- Abdelaziz, M.H.; Abdelwahab, S.F.; Wan, J.; Cai, W.; Huixuan, W.; Jianjun, C.; Kumar, K.D.; Vasudevan, A.; Sadek, A.; Su, Z.; et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J. Transl. Med. 2020, 18, 1–12. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.-K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Amici, S.A.; Dong, J.; Guerau-De-Arellano, M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Nie, D.; Wang, L.-J.; Qiu, H.-C.; Ma, L.; Dong, M.-X.; Tu, W.-J.; Zhao, J. Microglial Polarization: Novel Therapeutic Strategy against Ischemic Stroke. Aging Dis. 2021, 12, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Gaire, B.P. Phytochemicals as regulators of microglia/macrophages activation in cerebral ischemia. Pharmacol. Res. 2021, 165, 105419. [Google Scholar] [CrossRef]
- Kobashi, S.; Terashima, T.; Katagi, M.; Nakae, Y.; Okano, J.; Suzuki, Y.; Urushitani, M.; Kojima, H. Transplantation of M2-Deviated Microglia Promotes Recovery of Motor Function after Spinal Cord Injury in Mice. Mol. Ther. 2020, 28, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Franco, R. Geoffrey Burnstock (1929–2020): The finest pharmacologist and an inspiring scientist. Purinergic Signal. 2021, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Muller, C.E.; Zimmermann, H. Obituary Geoff Burnstock (1929–2020). Naunyn-Schmiedebergs Arch. Pharmacol. 2020, 393, 1773–1776. [Google Scholar] [CrossRef]
- Abbracchio, M.P. Perspectives on Geoff Burnstock as researcher, teacher and friend. Biochem. Pharmacol. 2021, 187, 114395. [Google Scholar] [CrossRef]
- Burnstock, G. Physiology and Pathophysiology of Purinergic Neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Purinergic signalling: From discovery to current developments. Exp. Physiol. 2014, 99, 16–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.-J.; Luo, C.; Pu, F.-Q.; Zhu, J.-F.; Zhu, Z.-M. The role and pharmacological characteristics of ATP-gated ionotropic receptor P2X in cancer pain. Pharmacol. Res. 2020, 161, 105106. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176, S21–S141. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Unwin, R.; Inscho, E.W.; Leipziger, J.; Kishore, B.K. Extracellular Nucleotides and P2 Receptors in Renal Function. Physiol. Rev. 2020, 100, 211–269. [Google Scholar] [CrossRef]
- Wirsching, E.; Fauler, M.; Fois, G.; Frick, M. P2 Purinergic Signaling in the Distal Lung in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4973. [Google Scholar] [CrossRef]
- Martin, M.; Aran, J.M.; Colomer, D.; Huguet, J.; Centelles, J.J.; Vives-Corrons, J.L.; Franco, R. Surface adenosine deaminase: A novel B-cell marker in chronic lymphocytic leukemia. Hum. Immunol. 1995, 42, 265–273. [Google Scholar] [CrossRef]
- Centelles, J.J.; Franco, R.; Canela, E.I.; Bozal, J. Kinetics of the 5′-nucleotidase and the adenosine deaminase in subcellular fractions of rat brain. Neurochem. Res. 1986, 11, 471–479. [Google Scholar] [CrossRef]
- Martín, M.; Huguet, J.; Centelles, J.J.; Franco, R. Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex. Possible role of adenosine deaminase as costimulatory molecule. J. Immunol. 1995, 155, 4630–4643. [Google Scholar]
- Aran, J.M.; Colomer, D.; Matutes, E.; Vives-Corrons, J.L.; Franco, R. Presence of adenosine deaminase on the surface of mononuclear blood cells: Immunochemical localization using light and electron microscopy. J. Histochem. Cytochem. 1991, 39, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Canela, E.I.; Bozal, J. Heterogeneous localization of some purine enzymes in subcellular fractions of rat brain and cerebellum. Neurochem. Res. 1986, 11, 423–435. [Google Scholar] [CrossRef]
- Kameoka, J.; Tanaka, T.; Nojima, Y.; Schlossman, S.; Morimoto, C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 1993, 261, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Alnouri, M.W.; Jepards, S.; Casari, A.; Schiedel, A.C.; Hinz, S.; Müller, C.E. Selectivity is species-dependent: Characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 2015, 11, 389–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabet, M.; Mallol, J.; Lluis, C.; Franco, R. Calcium mobilization in Jurkat cells via A2b adenosine receptors. Br. J. Pharmacol. 1997, 122, 1075–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, S.; Lacher, S.K.; Seibt, B.F.; Müller, C.E. BAY60-6583 Acts as a Partial Agonist at Adenosine A2B Receptors. J. Pharmacol. Exp. Ther. 2014, 349, 427–436. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. CGTP Collaborators The concise guide to Pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174, S17–S129. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Baler, R.; Bouvier, M.; Caron, M.G.; Devi, L.A.; Durroux, T.; Fuxe, K.; George, S.R.; Javitch, J.A.; Lohse, M.J.; et al. Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 2009, 5, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic Control of Striatal Glutamatergic Neurotransmission by Adenosine A1-A2A Receptor Heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Navarro, G.; Cordomí, A.; Zelman-Femiak, M.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Cortes, A.; Casadó, V.; Mallol, J.; et al. Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with Gi and Gs. BMC Biol. 2016, 14, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Cordomí, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Pérez-Benito, L.; Ferre, S.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 24. [Google Scholar] [CrossRef]
- Cristóvão-Ferreira, S.; Navarro, G.; Brugarolas, M.; Pérez-Capote, K.; Vaz, S.H.; Fattorini, G.; Conti, F.; Lluis, C.; Ribeiro, J.A.; McCormick, P.J.; et al. A1R-A2AR heteromers coupled to Gs and G i/o proteins modulate GABA transport into astrocytes. Purinergic Signal. 2013, 9, 433–449. [Google Scholar] [CrossRef] [Green Version]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, Y.-C.; De Filippo, E.; Danish, A.; Lacher, S.K.; Cervinkova, B.; Rafehi, M.; et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, E.; Hinz, S.; Pellizzari, V.; Deganutti, G.; El-Tayeb, A.; Navarro, G.; Franco, R.; Moro, S.; Schiedel, A.C.; Müller, C.E. A2A and A2B adenosine receptors: The extracellular loop 2 determines high (A2A) or low affinity (A2B) for adenosine. Biochem. Pharmacol. 2020, 172, 113718. [Google Scholar] [CrossRef]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jiménez, J.; Raïch, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; de Medina, V.S.; et al. Potentiation of cannabinoid signaling in microglia by adenosine A2A receptor antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef]
- Carriba, P.; Ortiz, O.; Patkar, K.; Justinova, Z.; Stroik, J.; Themann, A.; Müller, C.; Woods, A.S.; Hope, B.T.; Ciruela, F.; et al. Striatal Adenosine A2A and Cannabinoid CB1 Receptors Form Functional Heteromeric Complexes that Mediate the Motor Effects of Cannabinoids. Neuropsychopharmacology 2007, 32, 2249–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinna, A.; Bonaventura, J.; Farré, D.; Sánchez, M.; Simola, N.; Mallol, J.; Lluís, C.; Costa, G.; Baqi, Y.; Müller, C.E.; et al. l-DOPA disrupts adenosine A2A–cannabinoid CB1–dopamine D2 receptor heteromer cross-talk in the striatum of hemiparkinsonian rats: Biochemical and behavioral studies. Exp. Neurol. 2014, 253, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Hinz, S.; Navarro, G.; Franco, R.; Müller, C.E.; Fuxe, K. Understanding the Role of Adenosine A2AR Heteroreceptor Complexes in Neurodegeneration and Neuroinflammation. Front. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Kui, L.; Demetrios, T.; Gong, X.; Tang, M. A Glimmer of Hope: Maintain Mitochondrial Homeostasis to Mitigate Alzheimer’s Disease. Aging Dis. 2020, 11, 1260–1275. [Google Scholar] [CrossRef]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Zottola, A.C.P.; Serrat, R.; Puente, N.; Grandes, P.; Marsicano, G.; Hebert-Chatelain, E. Functional Analysis of Mitochondrial CB1 Cannabinoid Receptors (mtCB1) in the Brain. Biofilms 2017, 593, 143–174. [Google Scholar] [CrossRef]
- Valenzuela, R.; Costa-Besada, M.A.; Iglesias-Gonzalez, J.; Perez-Costas, E.; Villar-Cheda, B.; Garrido-Gil, P.; Melendez-Ferro, M.; Soto-Otero, R.; Lanciego, J.L.; Henrion, D.; et al. Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 2016, 7, e2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.-H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of Coffee and Caffeine Intake With the Risk of Parkinson Disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Cunha, R.A. Cafeína, receptores de adenosina, memoria y enfermedad de Alzheimer. Med. Cli. 2008, 131, 790–795. (In Spanish) [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Ribeiro, J.A.; Sebastião, A.M. Caffeine and Adenosine. J. Alzheimers Dis. 2010, 20, S3–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr. Gerontol. Int. 2013, 14, 430–439. [Google Scholar] [CrossRef]
- Liu, R.; Guo, X.; Park, Y.; Huang, X.; Sinha, R.; Freedman, N.D.; Hollenbeck, A.R.; Blair, A.; Chen, H. Caffeine Intake, Smoking, and Risk of Parkinson Disease in Men and Women. Am. J. Epidemiol. 2012, 175, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk Factors for Alzheimer’s Disease: A Prospective Analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciúncula, L.O. Caffeine Consumption Prevents Memory Impairment, Neuronal Damage, and Adenosine A2A Receptors Upregulation in the Hippocampus of a Rat Model of Sporadic Dementia. J. Alzheimers Dis. 2013, 34, 509–518. [Google Scholar] [CrossRef]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.A.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.A.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A Receptor Blockade Prevents Synaptotoxicity and Memory Dysfunction Caused by—Amyloid Peptides via p38 Mitogen-Activated Protein Kinase Pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a Protective Factor in Dementia and Alzheimer’s Disease. J. Alzheimers Dis. 2010, 20, S167–S174. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife Coffee and Tea Drinking and the Risk of Late-Life Dementia: A Population-Based CAIDE Study. J. Alzheimers Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife Healthy-Diet Index and Late-Life Dementia and Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. Extra 2011, 1, 103–112. [Google Scholar] [CrossRef]
- Pedata, F.; Pugliese, A.M.; Melani, A.; Gianfriddo, M. Introduction: A2A receptors in neuroprotection of dopaminergic neurons. Neurology 2003, 61, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Navarro, G. Adenosine A2A Receptor Antagonists in Neurodegenerative Diseases: Huge Potential and Huge Challenges. Front. Psychiatry 2018, 9, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Franco, R. Café y salud mental. Aten. Primaria 2009, 41, 578–581. (In Spanish) [Google Scholar] [CrossRef] [Green Version]
- Abbas, M.M.; Xu, Z.; Tan, L.C. Epidemiology of Parkinson’s Disease-East Versus West. Mov. Disord. Clin. Pr. 2018, 5, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Drury, A.N.; Szent-Györgyi, A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart1. J. Physiol. 1929, 68, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.M.; Tsuboi, K.K.; Zeig, N.J. Effect of Nucleosides on Acute Left Ventricular Failure in the Isolated Dog Heart. Circ. Res. 1959, 7, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berne, R.M. The role of adenosine in the regulation of coronary blood flow. Circ. Res. 1980, 47, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.M.; Berne, R.M. Coronary Vasodilator Properties of Purine and Pyrimidine Derivatives. Circ. Res. 1956, 4, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Llach, A.; Molina, C.E.; Prat-Vidal, C.; Fernandes, J.; Casadó, V.; Ciruela, F.; Lluís, C.; Franco, R.; Cinca, J.; Hove-Madsen, L. Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur. Heart J. 2010, 32, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Hove-Madsen, L.; Prat-Vidal, C.; Llach, A.; Ciruela, F.; Casadó, V.; Lluis, C.; Bayes-Genis, A.; Cinca, J.; Franco, R. Reply: Does the adenosine A2A receptor stimulate the ryanodine receptor? Cardiovasc. Res. 2007, 73, 249–250. [Google Scholar] [CrossRef]
- Hove-Madsen, L.; Prat-Vidal, C.; Llach, A.; Ciruela, F.; Casadó, V.; Lluis, C.; Bayes-Genis, A.; Cinca, J.; Franco, R. Adenosine A2A receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc. Res. 2006, 72, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Oñatibia-Astibia, A.; Martínez-Pinilla, E. Health benefits of methylxanthines in cacao and chocolate. Nutrients 2013, 5, 4159–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oñatibia-Astibia, A.; Franco, R.; Martínez-Pinilla, E. Health benefits of methylxanthines in neurodegenerative diseases. Mol. Nutr. Food Res. 2017, 61, 1600670. [Google Scholar] [CrossRef]
- Oñatibia-Astibia, A.; Martínez-Pinilla, E.; Franco, R. The potential of methylxanthine-based therapies in pediatric respiratory tract diseases. Respir. Med. 2016, 112, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Postuma, R.B.; Montplaisir, J.Y.; Pelletier, A.; Dauvilliers, Y.; Oertel, W.; Iranzo, A.; Ferini-Strambi, L.; Arnulf, I.; Hogl, B.; Manni, R.; et al. Environmental risk factors for REM sleep behavior disorder: A multicenter case-control study. Neurology 2012, 79, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-F. Chapter 7. Caffeine and Parkinson’s Disease: From Molecular Targets to Epidemiology and Clinical Trials. In Coffee; Royal Society of Chemistry (RSC): London, UK, 2019; pp. 171–195. [Google Scholar]
- Ferré, S.; Ciruela, F.; Canals, M.; Marcellino, D.; Burgueno, J.; Casadó, V.; Hillion, J.; Torvinen, M.; Fanelli, F.; de Benedetti, P.; et al. Adenosine A2A-dopamine D2 receptor–receptor heteromers. Targets for neuro-psychiatric disorders. Park. Relat. Disord. 2004, 10, 265–271. [Google Scholar] [CrossRef]
- Tanganelli, S.; Nielsen, K.S.; Ferraro, L.; Antonelli, T.; Kehr, J.; Franco, R.; Ferré, S.; Agnati, L.; Fuxe, K.; Scheel-Krüger, J. Striatal plasticity at the network level. Focus on adenosine A2A and D2 interactions in models of Parkinson’s Disease. Park. Relat. Disord. 2004, 10, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Ferré, S.; Agnati, L.; Torvinen, M.; Ginés, S.; Hillion, J.; Casadó, V.; Lledó, P.; Zoli, M.; Lluis, C.; et al. Evidence for adenosine/dopamine receptor interactions: Indications for heteromerization. Neuropsychopharmacology 2000, 23, S50–S59. [Google Scholar] [CrossRef]
- Fuxe, K.; Ferré, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and Dopamine D2 Heteromeric Receptor Complexes and Their Function. J. Mol. Neurosci. 2005, 26, 209–220. [Google Scholar] [CrossRef]
- Ginés, S.; Hillion, J.; Torvinen, M.; Le Crom, S.; Casadó, V.; Canela, E.I.; Rondin, S.; Lew, J.Y.; Watson, S.; Zoli, M.; et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 8606–8611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torvinen, M.; Ginés, S.; Hillion, J.; Latini, S.; Canals, M.; Ciruela, F.; Bordoni, F.; Staines, W.; Pedata, F.; Agnati, L.; et al. Interactions among adenosine deaminase, adenosine A1 receptors and dopamine D1 receptors in stably cotransfected fibroblast cells and neurons. Neuroscience 2002, 113, 709–719. [Google Scholar] [CrossRef]
- Agnati, L.F.; Leo, G.; Vergoni, A.-V.; Martınez, E.; Hockemeyer, J.; Lluís, C.; Franco, R.; Fuxe, K.; Ferre, S. Neuroprotective effect of L-DOPA co-administered with the adenosine A2A receptor agonist CGS 21680 in an animal model of Parkinson’s disease. Brain Res. Bull. 2004, 64, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Boia, R.; Elvas, F.; Madeira, M.H.; Aires, I.D.; Rodrigues-Neves, A.C.; Tralhão, P.; Szabó, E.C.; Baqi, Y.; Müller, C.E.; Tomé, Â.R.; et al. Treatment with A2A receptor antagonist KW6002 and caffeine intake regulate microglia reactivity and protect retina against transient ischemic damage. Cell Death Dis. 2017, 8, e3065. [Google Scholar] [CrossRef] [Green Version]
- Jenner, P. An Overview of Adenosine A2A Receptor Antagonists in Parkinson’s Disease. Int. Rev. Neurobiol. 2014, 119, 71–86. [Google Scholar] [CrossRef]
- Ferreira, D.G.; Batalha, V.L.; Vicente Miranda, H.; Coelho, J.E.; Gomes, R.; Gonçalves, F.Q.; Real, J.I.; Rino, J.; Albino-Teixeira, A.; Cunha, R.A.; et al. Adenosine A2A Receptors Modulate α-Synuclein Aggregation and Toxicity. Cereb. Cortex 2015, 27, bhv268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-F. Adenosine Receptor Control of Cognition in Normal and Disease. Int. Rev. Neurobiol. 2014, 119, 257–307. [Google Scholar] [CrossRef]
- Franco, R.; Martínez-Pinilla, E.; Navarro, G.; Zamarbide, M. Potential of GPCRs to modulate MAPK and mTOR pathways in Alzheimer’s disease. Prog. Neurobiol. 2017, 149–150, 21–38. [Google Scholar] [CrossRef]
- Rebola, N.; Simões, A.P.; Canas, P.M.; Tomé, A.R.; Andrade, G.M.; Barry, C.E.; Agostinho, P.M.; Lynch, M.A.; Cunha, R.A. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J. Neurochem. 2011, 117, 100–111. [Google Scholar] [CrossRef]
- Armentero, M.T.; Pinna, A.; Ferré, S.; Lanciego, J.L.; Müller, C.E.; Franco, R. Past, present and future of A(2A) adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol. Ther. 2011, 132, 280–299. [Google Scholar] [CrossRef] [Green Version]
- Nobre, H.V., Jr.; de Cunha Andrade, G.M.; de Vasconcelos, L.M.; Magalhães, H.I.F.; Neto, R.N.O.; Maia, F.D.; de Moraes, M.O.; Leal, L.K.A.M.; de Barros Viana, G.S. Caffeine and CSC, adenosine A2A antagonists, offer neuroprotection against 6-OHDA-induced neurotoxicity in rat mesencephalic cells. Neurochem. Int. 2010, 56, 51–58. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Cattabeni, F. Brain adenosine receptors as targets for therapeutic intervention in neurodegenerative diseases. Ann. N. Y. Acad. Sci. 1999, 890, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Leonelli, M.; Torrão, A.; Britto, L. Unconventional neurotransmitters, neurodegeneration and neuroprotection. Braz. J. Med. Biol. Res. 2009, 42, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-F.; Sonsalla, P.K.; Pedata, F.; Melani, A.; Domenici, M.R.; Popoli, P.; Geiger, J.; Lopes, L.V.; de Mendonça, A. Adenosine A2A receptors and brain injury: Broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog. Neurobiol. 2007, 83, 310–331. [Google Scholar] [CrossRef]
- Mizuno, Y.; Kondo, T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Mizuno, Y. Japanese Istradefylline Study Group A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin. Neuropharmacol. 2015, 38, 41–46. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. Methylxanthines, inflammation, and cancer: Fundamental mechanisms. Handb. Exp. Pharmacol. 2011, 200, 469–481. [Google Scholar]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1α driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitkovsky, M.V. Lessons from the A2A Adenosine Receptor Antagonist-Enabled Tumor Regression and Survival in Patients with Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 16–19. [Google Scholar] [CrossRef]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Hotson, A.N.; Miller, R.A. Targeting the A2AR in cancer; early lessons from the clinic. Curr. Opin. Pharmacol. 2020, 53, 126–133. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. The birth of oligodendrocytes in the anatomical and neuropathological literature: The seminal contribution of Pío del Río-Hortega. Clin. Neuropathol. 2012, 31, 435–436. [Google Scholar] [CrossRef]
- Del Rio-Hortega, P. Studies on neuroglia: Glia with very few processes (oligodendroglia) by Pío del Río-Hortega. 1921. Clin. Neuropathol. 2012, 31, 440–459. [Google Scholar] [PubMed]
- Iglesias-Rozas, J.R.; Garrosa, M. The discovery of oligodendroglia cells by Rio-Hortega: His original articles. 1921. Clin. Neuropathol. 2012, 31, 437–439. [Google Scholar]
- Oehmichen, M. Are Resting and/or Reactive Microglia Macrophages? Immunobiology 1982, 161, 246–254. [Google Scholar] [CrossRef]
- Mège, J.-L.; Mehraj, V.; Capo, C. Macrophage polarization and bacterial infections. Curr. Opin. Infect. Dis. 2011, 24, 230–234. [Google Scholar] [CrossRef]
- Matthews, M. Microglia and reactive “M” cells of degenerating central nervous system: Does similar morphology and function imply a common origin? Cell Tissue Res. 1974, 148, 477–491. [Google Scholar] [CrossRef]
- Matthews, M.A.; Kruger, L. Electron microscopy of non-neuronal cellular changes accompanying neural degeneration in thalamic nuclei of the rabbit. II. Reactive elements within the neuropil. J. Comp. Neurol. 1973, 148, 313–345. [Google Scholar] [CrossRef]
- Delgado, M.; Leceta, J.; Ganea, I. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit the production of inflammatory mediators by activated microglia. J. Leukoc. Biol. 2003, 73, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Ganea, D. Neuroprotective effect of vasoactive intestinal peptide (VIP) in a mouse model of Parkinson’s disease by blocking microglial activation. FASEB J. 2003, 17, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Nunan, R.; Sivasathiaseelan, H.; Khan, D.; Zaben, M.; Gray, W. Microglial VPAC1R mediates a novel mechanism of neuroimmune-modulation of hippocampal precursor cells via IL-4 release. Glia 2014, 62, 1313–1327. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit the MEKK1/MEK4/JNK signaling pathway in endotoxin-activated microglia. Biochem. Biophys. Res. Commun. 2002, 293, 771–776. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Delgado, M. Vasoactive intestinal peptide inhibits cyclooxygenase-2 expression in activated macrophages, microglia, and dendritic cells. Brain. Behav. Immun. 2008, 22, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M. Inhibition of interferon (IFN) gamma-induced Jak-STAT1 activation in microglia by vasoactive intestinal peptide: Inhibitory effect on CD40, IFN-induced protein-10, and inducible nitric-oxide synthase expression. J. Biol. Chem. 2003, 278, 27620–27629. [Google Scholar] [CrossRef] [Green Version]
- Saura, J.; Angulo, E.; Ejarque, A.; Casado, V.; Tusell, J.M.; Moratalla, R.; Chen, J.-F.; Schwarzschild, M.A.; Lluis, C.; Franco, R.; et al. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J. Neurochem. 2005, 95, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Angulo, E.; Casadó, V.; Mallol, J.; Canela, E.I.; Viñals, F.; Ferrer, I.; Lluis, C.; Franco, R. A1 Adenosine Receptors Accumulate in Neurodegenerative Structures in Alzheimer’s Disease and Mediate Both Amyloid Precursor Protein Processing and Tau Phosphorylation and Translocation. Brain Pathol. 2006, 13, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Pacher, P.; Vizi, E.S.; Illes, P. Adenosine receptor signaling in the brain immune system. Trends Pharmacol. Sci. 2005, 26, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Fiebich, B.L.; Biber, K.; Lieb, K.; Van Calker, D.; Berger, M.; Bauer, J.; Gebicke-Haerter, P.J. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2A-receptors. Glia 1996, 18, 152–180. [Google Scholar] [CrossRef]
- Merighi, S.; Bencivenni, S.; Vincenzi, F.; Varani, K.; Borea, P.A.; Gessi, S. A 2B adenosine receptors stimulate IL-6 production in primary murine microglia through p38 MAPK kinase pathway. Pharmacol. Res. 2017, 117, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L.; Greco, A.; Potenza, R.L.; Pezzola, A.; Blum, D.; Bantubungi, K.; Popoli, P. Effects of the Adenosine A2A Receptor Antagonist SCH 58621 on Cyclooxygenase-2 Expression, Glial Activation, and Brain-Derived Neurotrophic Factor Availability in a Rat Model of Striatal Neurodegeneration. J. Neuropathol. Exp. Neurol. 2007, 66, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; Le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P. Neuronal death in the development of the vertebrate nervous system. Trends Neurosci. 1985, 8, 345–349. [Google Scholar] [CrossRef]
- Wasterlain, C.G.; Niquet, J.; Thompson, K.W.; Baldwin, R.; Liu, H.; Sankar, R.; Mazarati, A.M.; Naylor, D.; Katsumori, H.; Suchomelova, L.; et al. Seizure-induced neuronal death in the immature brain. Prog. Brain Res. 2002, 135, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Represa, A.; Niquet, J.; Pollard, H.; Ben-Ari, Y. Cell death, gliosis, and synaptic remodeling in the hippocampus of epileptic rats. J. Neurobiol. 1995, 26, 413–425. [Google Scholar] [CrossRef]
- Hornykiewicz, O. Die topische Lokalization und des Verhalten von Noradrenalin und Dopamin (3-Hydroxytyramin) in der Substantia nigra der normalen und Parkinson Kranken Menschen. Wien. Klin. Wochschr. 1963, 75, 309–312. (In German) [Google Scholar]
- Holzer, G.; Hornykiewicz, O. Über den Dopamin-(Hydroxytyramin-)Stoffwechsel im Gehirn der Ratte. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1959, 237, 27–33. (In German) [Google Scholar] [CrossRef]
- Hornykiewicz, O. The action of dopamine on the arterial blood pressure of the guinea-pig. Br. J. Pharmacol. Chemother. 1958, 13, 91–94. [Google Scholar] [CrossRef] [Green Version]
- Birkmayer, W.; Hornykiewicz, O. The L-dihydroxyphenylalanine (L-DOPA) effect in Parkinson’s syndrome in man: On the pathogenesis and treatment of Parkinson akinesis. Arch. Psychiatr. Nervenkr. Z Gesamte Neurol. Psychiatr. 1962, 203, 560–574. (In German) [Google Scholar] [CrossRef]
- Birkmayer, W.; Hornykiewicz, O. Additional experimental studies on L-DOPA in Parkinson’s syndrome and reserpine parkinsonism. Arch. Psychiatr. Nervenkr. 1964, 206, 367–381. [Google Scholar] [CrossRef]
- Li, Y.L.; He, R.R. Protective effect of preconditioning on ischemic heart and characterization of adenosine receptors in ischemic rabbit hearts. Zhongguo Yao Li Xue Bao Acta Pharmacol. Sin. 1995, 16, 505–508. [Google Scholar]
- Liu, G.S.; Thornton, J.; Van Winkle, D.M.; Stanley, A.W.; Olsson, R.A.; Downey, J.M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 1991, 84, 350–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urmaliya, V.B.; Pouton, C.W.; Ledent, C.; Short, J.L.; White, P.J. Cooperative Cardioprotection Through Adenosine A1 and A2A Receptor Agonism in Ischemia-Reperfused Isolated Mouse Heart. J. Cardiovasc. Pharmacol. 2010, 56, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kato, H.; Nakata, N.; Kogure, K. Protection of rat hippocampus against ischemic neuronal damage by pretreatment with sublethal ischemia. Brain Res. 1992, 586, 121–124. [Google Scholar] [CrossRef]
- Sweeney, M.I. Neuroprotective Effects of Adenosine in Cerebral Ischemia: Window of Opportunity. Neurosci. Biobehav. Rev. 1997, 21, 207–217. [Google Scholar] [CrossRef]
- Ishida, T.; Yarimizu, K.; Gute, D.C.; Korthuis, R.J. Mechanisms of ischemic preconditioning. Shock 1997, 8, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Péerez-Pinzón, M.; Mumford, P.; Rosenthal, M.; Sick, T. Anoxic preconditioning in hippocampal slices: Role of adenosine. Neuroscience 1996, 75, 687–694. [Google Scholar] [CrossRef]
- Reshef, A.; Sperling, O.; Zoref-Shani, E. Preconditioning of primary rat neuronal cultures against ischemic injury: Characterization of the ‘time window of protection’. Brain Res. 1996, 741, 252–257. [Google Scholar] [CrossRef]
- Kato, H.; Kogure, K.; Araki, T.; Itoyama, Y. Astroglial and microglial reactions in the gerbil hippocampus with induced ischemic tolerance. Brain Res. 1994, 664, 69–76. [Google Scholar] [CrossRef]
- Kato, H.; Kogure, K.; Araki, T.; Itoyama, Y. Graded expression of immunomolecules on activated microglia in the hippocampus following ischemia in a rat model of ischemic tolerance. Brain Res. 1995, 694, 85–93. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Maslov, L.N.; Singh, N.; Jaggi, A.S. Remote ischemic preconditioning-induced neuroprotection in cerebral ischemia-reperfusion injury: Preclinical evidence and mechanisms. Eur. J. Pharmacol. 2020, 883, 173380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.-M.; Li, W.-B.; Li, Q.-J.; Liu, H.-Q.; Feng, R.-F.; Zhao, H.-G. A short cerebral ischemic preconditioning up-regulates adenosine receptors in the hippocampal CA1 region of rats. Neurosci. Res. 2004, 48, 397–404. [Google Scholar] [CrossRef]
- Roth, S. Endogenous neuroprotection in the retina. Brain Res. Bull. 2004, 62, 461–466. [Google Scholar] [CrossRef]
- Pugliese, A.M.; Latini, S.; Corradetti, R.; Pedata, F. Brief, repeated, oxygen-glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: Role of adenosine receptors. Br. J. Pharmacol. 2003, 140, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Guo, M.; Wang, X.; Zhao, Y.; Zhao, Q.; Ding, H.; Dong, Q.; Cui, M. Ischemic preconditioning with a ketogenic diet improves brain ischemic tolerance through increased extracellular adenosine levels and hypoxia-inducible factors. Brain Res. 2017, 1667, 11–18. [Google Scholar] [CrossRef]
- Yun, J.; Li, J.; Zuo, Z. Transferred inter-cell ischemic preconditioning-induced neuroprotection may be mediated by adenosine A1 receptors. Brain Res. Bull. 2014, 103, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Jia, H.X.; Li, S.Q.; Zhang, X.; Wu, Y.J.; Feng, L.; Liu, X.L.; Sun, X.C.; Li, W. Bin The role of adenosine in up-regulation of p38 MAPK and ERK during limb ischemic preconditioning-induced brain ischemic tolerance. Brain Res. 2019, 1707, 172–183. [Google Scholar] [CrossRef]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Dai, S.-S.; Zhou, Y.-G. Adenosine 2A receptor: A crucial neuromodulator with bidirectional effect in neuroinflammation and brain injury. Rev. Neurosci. 2011, 22, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Pedata, F.; Pugliese, A.M.; Coppi, E.; Dettori, I.; Maraula, G.; Cellai, L.; Melani, A. Adenosine A2A receptors modulate acute injury and neuroinflammation in brain ischemia. Mediators Inflamm. 2014, 2014, 805198. [Google Scholar] [CrossRef] [Green Version]
- Madeira, M.H.; Boia, R.; Elvas, F.; Martins, T.; Cunha, R.A.; Ambrósio, A.F.; Santiago, A.R. Selective A2A receptor antagonist prevents microglia-mediated neuroinflammation and protects retinal ganglion cells from high intraocular pressure–induced transient ischemic injury. Transl. Res. 2016, 169, 112–128. [Google Scholar] [CrossRef]
- Atef, R.M.; Agha, A.M.; Abdel-Rhaman, A.R.A.; Nassar, N.N. The Ying and Yang of Adenosine A1 and A2A Receptors on ERK1/2 Activation in a Rat Model of Global Cerebral Ischemia Reperfusion Injury. Mol. Neurobiol. 2018, 55, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Bessis, A.; Béchade, C.; Bernard, D.; Roumier, A. Microglial control of neuronal death and synaptic properties. Glia 2006, 55, 233–238. [Google Scholar] [CrossRef]
- Graeber, M.B. Changing Face of Microglia. Science 2010, 330, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Conde, J.R.; Streit, W.J. Microglia in the Aging Brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef]
- Schuitemaker, A.; Van Der Doef, T.F.; Boellaard, R.; Van Der Flier, W.M.; Yaqub, M.; Windhorst, A.D.; Barkhof, F.; Jonker, C.; Kloet, R.W.; Lammertsma, A.A.; et al. Microglial activation in healthy aging. Neurobiol. Aging 2012, 33, 1067–1072. [Google Scholar] [CrossRef]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, Í.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia. Brain Behav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Aymerich, M.S.; Rojo-Bustamante, E.; Molina, C.; Celorrio, M.; Sánchez-Arias, J.A.; Franco, R. Neuroprotective Effect of JZL184 in MPP(+)-Treated SH-SY5Y Cells Through CB2 Receptors. Mol. Neurobiol. 2015, 53, 2312–2319. [Google Scholar] [CrossRef]
- Scotter, E.L.; Abood, M.E.; Glass, M. The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br. J. Pharmacol. 2010, 160, 480–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán, M.; Sánchez, C.; Galve-Roperh, I. Control of the cell survival/death decision by cannabinoids. J. Mol. Med. 2000, 78, 613–625. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J. The biomedical challenge of neurodegenerative disorders: An opportunity for cannabinoid-based therapies to improve on the poor current therapeutic outcomes. Br. J. Pharmacol. 2019, 176, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Morales, P.; Rodríguez-Cueto, C.; Fernández-Ruiz, J.; Jagerovic, N.; Franco, R. Targeting Cannabinoid CB2 Receptors in the Central Nervous System. Medicinal Chemistry Approaches with Focus on Neurodegenerative Disorders. Front. Neurosci. 2016, 10, 406. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Olives, C.; Rivas-Santisteban, R.; Lillo, J.; Navarro, G.; Franco, R. Recent Advances in the Potential of Cannabinoids for Neuroprotection in Alzheimer’s, Parkinson’s, and Huntington’s Diseases. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2021; Volume 1264, pp. 81–92. [Google Scholar]
- Gyoneva, S.; Orr, A.G.; Traynelis, S.F. Differential regulation of microglial motility by ATP/ADP and adenosine. Park. Relat. Disord. 2009, 15, S195–S199. [Google Scholar] [CrossRef]
- Gomes, C.; Ferreira, R.; George, J.; Sanches, R.; Rodrigues, D.I.; Gonçalves, N.; Cunha, R.A. Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor-dependent manner: A2A receptor blockade prevents BDNF release and proliferation of microglia. J. Neuroinflamm. 2013, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Santiago, A.R.; Baptista, F.I.; Santos, P.F.; Cristóvão, G.; Ambrósio, A.F.; Cunha, R.A.; Gomes, C.A. Role of microglia adenosine A2A receptors in retinal and brain neurodegenerative diseases. Mediat. Inflamm. 2014, 2014, 465694. [Google Scholar] [CrossRef] [PubMed]
- Hammarberg, C.; Schulte, G.; Fredholm, B.B. Evidence for functional adenosine A3 receptors in microglia cells. J. Neurochem. 2003, 86, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Martí Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine Receptors as Neuroinflammation Modulators: Role of A1 Agonists and A2A Antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| mRNA Transcript Expression Levels (Scaled Tags Per Million) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cerebral Cortex | Olfactory Region | Hippocampal Formation | Amygdala | Basal Ganglia | Thalamus | Midbrain | Pons and Medula | Cerebellum | Corpus Callosum | Spinal Cord | Pituitary Gland | |
| A1R | 139.6 | 83.6 | 92.2 | 79.4 | 149.2 | 100.7 | 139.3 | 143.9 | 74.2 | 147.8 | 100.7 | 2.7 |
| A2AR | 9.4 | 3.6 | 5.8 | 3.9 | 53.4 | 13.7 | 3.0 | 6.0 | 3.0 | 3.6 | 0.8 | 1.1 |
| A2BR | 14.3 | 8.9 | 14.5 | 12.8 | 15.5 | 4.9 | 0.9 | 11.3 | 13.7 | 3.9 | 7.9 | 1.9 |
| A3R | 27.3 | 9.9 | 31.9 | 33.9 | 47.9 | 47.9 | 61.3 | 50.9 | 6.6 | 32.6 | 98.4 | 7.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, R.; Lillo, A.; Rivas-Santisteban, R.; Reyes-Resina, I.; Navarro, G. Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells. Cells 2021, 10, 1124. https://doi.org/10.3390/cells10051124
Franco R, Lillo A, Rivas-Santisteban R, Reyes-Resina I, Navarro G. Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells. Cells. 2021; 10(5):1124. https://doi.org/10.3390/cells10051124
Chicago/Turabian StyleFranco, Rafael, Alejandro Lillo, Rafael Rivas-Santisteban, Irene Reyes-Resina, and Gemma Navarro. 2021. "Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells" Cells 10, no. 5: 1124. https://doi.org/10.3390/cells10051124
APA StyleFranco, R., Lillo, A., Rivas-Santisteban, R., Reyes-Resina, I., & Navarro, G. (2021). Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells. Cells, 10(5), 1124. https://doi.org/10.3390/cells10051124