
Role of SNAREs in Neurodegenerative Diseases


Abstract
:1. Introduction
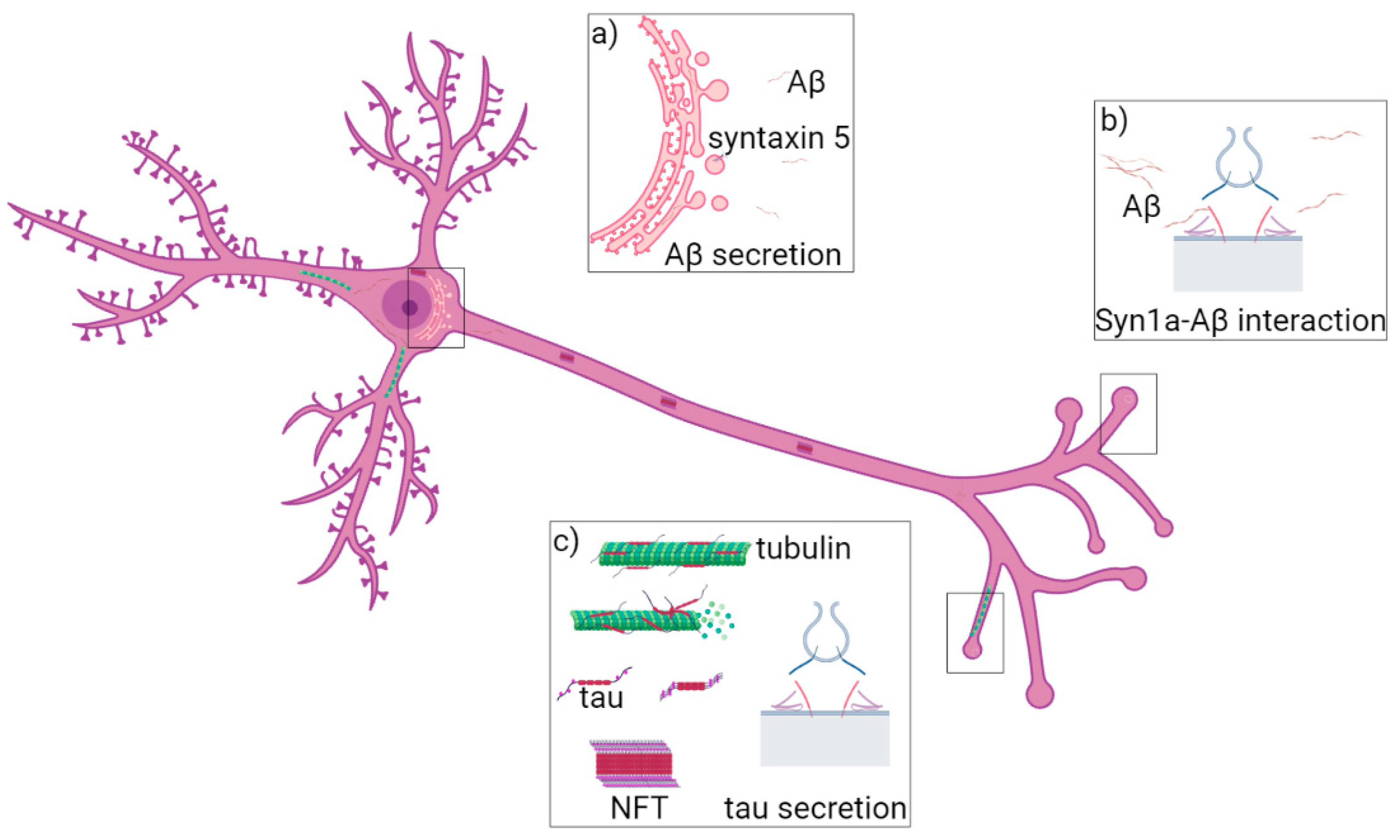
2. AD
2.1. Syntaxin-5 Is Involved in Aβ42 Accumulation
2.2. SNAP-25, Syntaxin-1a and VAMP2 Role in Neurodegeneration
2.3. Vamp8 and Secretion of Tau Protein
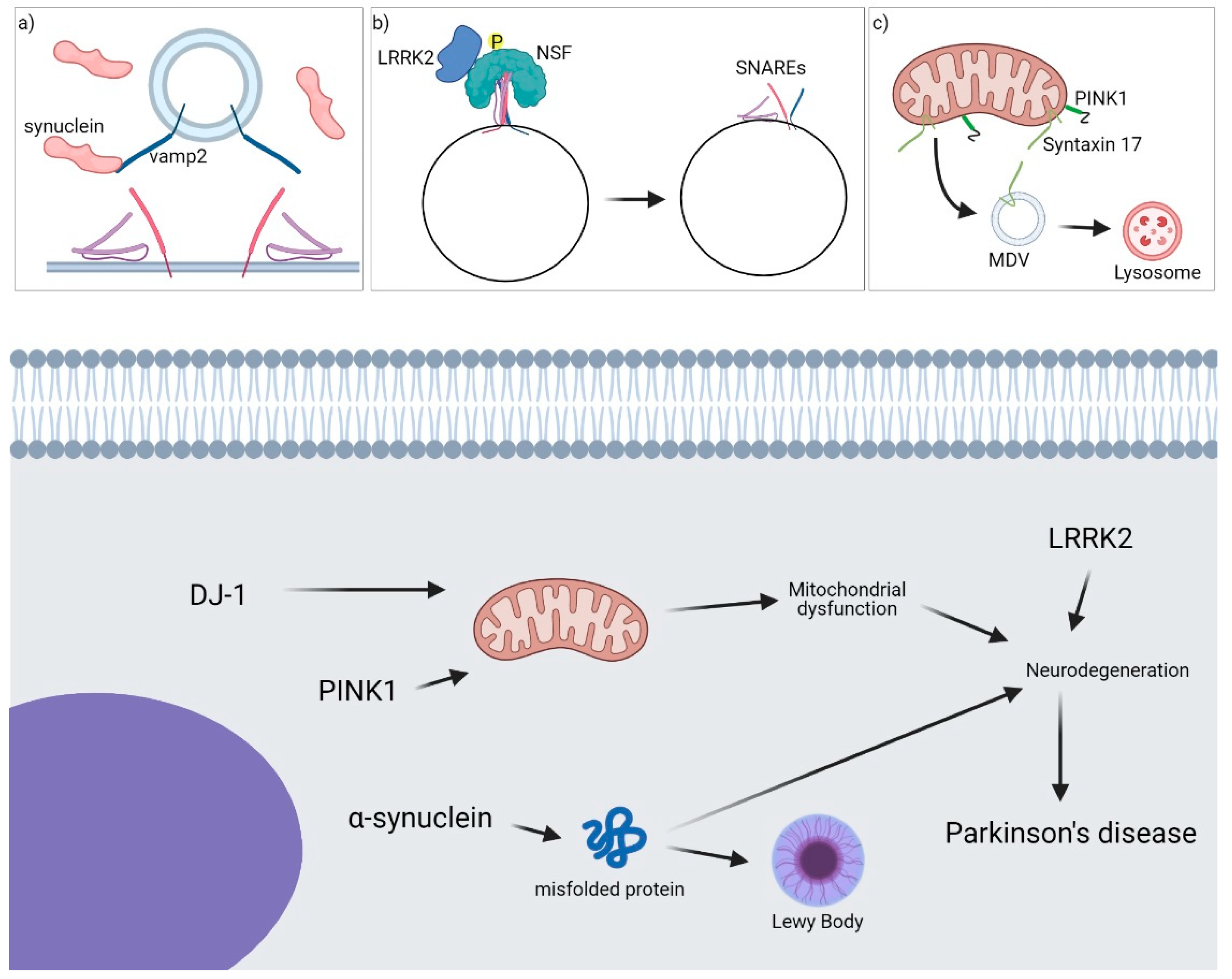
3. PD
3.1. Interaction between α-Syn and SNARE Proteins
3.2. Role of SNAREs in LRRK2-Mediated Synaptic Transmission
3.3. Role of Syntaxin-17 in PINK1/Parkin-Dependent Vesicle Transport Pathway
4. ALS
VAMP2 Role in Muscle Denervation and Astrocyte-Mediated Toxicity
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Morrison, B.M.; Shu, I.-W.; Wilcox, A.L.; Gordon, J.W.; Morrison, J.H. Early and Selective Pathology of Light Chain Neurofilament in the Spinal Cord and Sciatic Nerve of G86R Mutant Superoxide Dismutase Transgenic Mice. Exp. Neurol. 2000, 165, 207–220. [Google Scholar] [CrossRef]
- Dekkers, M.P.; Nikoletopoulou, V.; Barde, Y.A. Cell biology in neuroscience: Death of developing neurons: New insights and implications for connectivity. J. Cell Biol. 2013, 203, 385–393. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conneally, P.M. Huntington disease: Genetics and epidemiology. Am. J. Hum. Genet. 1984, 36, 506–526. [Google Scholar]
- Selvaraj, K.; Manickam, N.; Kumaran, E.; Thangadurai, K.; Elumalai, G.; Sekar, A.; Radhakrishnan, R.K.; Kandasamy, M. Deterioration of neuroregenerative plasticity in association with testicular atrophy and dysregulation of the hypothalamic-pituitary-gonadal (HPG) axis in Huntington’s disease: A putative role of the huntingtin gene in steroidogenesis. J. Steroid Biochem. Mol. Biol. 2020, 197, 105526. [Google Scholar] [CrossRef]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2016, 19, 1–10. [Google Scholar]
- Zhang, Y.; Xie, X.; Hu, J.; Afreen, K.S.; Zhang, C.-L.; Zhuge, Q.; Yang, J. Prospects of Directly Reprogrammed Adult Human Neurons for Neurodegenerative Disease Modeling and Drug Discovery: IN vs. iPSCs Models. Front. Neurosci. 2020, 14, 546484. [Google Scholar] [CrossRef]
- Ramroop, H.; Cruz, R. Electrodiagnostic Evaluation of Motor Neuron Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral Glycolysis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8924. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Davies, C.A.; Mann, D.M.; Sumpter, P.Q.; Yates, P.O. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 1987, 78, 151–164. [Google Scholar] [CrossRef]
- Guégan, C.; Przedborski, S. Programmed cell death in amyotrophic lateral sclerosis. J. Clin. Investig. 2003, 111, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Venderova, K.; Park, D.S. Programmed cell death in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009365. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, J.; Tian, J.; Robinson, A.C.; Davidson, Y.S.; Mann, D.M. Expression of one important chaperone protein, heat shock protein 27, in neurodegenerative diseases. Alzheimer’s Res. Ther. 2014, 6, 78. [Google Scholar] [CrossRef] [Green Version]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.-C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef]
- Barroeta-Espar, I.; Weinstock, L.D.; Perez-Nievas, B.G.; Meltzer, A.C.; Siao Tick Chong, M.; Amaral, A.C.; Murray, M.E.; Moulder, K.L.; Morris, J.C.; Cairns, N.J.; et al. Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis. 2019, 121, 327–337. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Zhao, T.; Li, X.-J.; Li, S. Mutant Huntingtin Impairs BDNF Release from Astrocytes by Disrupting Conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016, 36, 8790–8801. [Google Scholar] [CrossRef]
- Han, J.; Pluhackova, K.; Böckmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Holz, R.W.; Zimmerberg, J. Dynamic Relationship of the SNARE Complex with a Membrane. Biophys. J. 2019, 117, 627–630. [Google Scholar] [CrossRef]
- Rothman, J.E.; Krishnakumar, S.S.; Grushin, K.; Pincet, F. Hypothesis—Buttressed rings assemble, clamp, and release SNAREpins for synaptic transmission. FEBS Lett. 2017, 591, 3459–3480. [Google Scholar] [CrossRef]
- Südhof, T.C.; Rothman, J.E. Membrane Fusion: Grappling with SNARE and SM Proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhage, M.; Sørensen, J.B. SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron 2020, 107, 22–37. [Google Scholar] [CrossRef]
- Bowman, P.R.T.; Smith, G.L.; Gould, G.W. Cardiac SNARE Expression in Health and Disease. Front. Endocrinol. 2019, 10, 881. [Google Scholar] [CrossRef]
- Hong, W. SNAREs and traffic. Biochim. Biophys. Acta (BBA)-Bioenerg. 2005, 1744, 120–144. [Google Scholar] [CrossRef] [Green Version]
- Urbina, F.L.; Gupton, S.L. SNARE-Mediated Exocytosis in Neuronal Development. Front. Mol. Neurosci. 2020, 13, 133. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Sugiura, Y.; Han, W.; Gallardo, G.; Khvotchev, M.; Zhang, Y.; Kavalali, E.T.; Südhof, T.C.; Lin, W. Ubiquitin-Synaptobrevin Fusion Protein Causes Degeneration of Presynaptic Motor Terminals in Mice. J. Neurosci. 2015, 35, 11514–11531. [Google Scholar] [CrossRef] [Green Version]
- Baumert, M.; Maycox, P.R.; Navone, F.; De Camilli, P.; Jahn, R. Synaptobrevin: An integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J. 1989, 8, 379–384. [Google Scholar] [CrossRef]
- Elferink, L.; Trimble, W.S.; Scheller, R.H. Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J. Biol. Chem. 1989, 264, 11061–11064. [Google Scholar] [CrossRef]
- Archer, B.T., III; Ozçelik, T.; Jahn, R.; Francke, U.; Südhof, T.C. Structures and chromosomal localizations of two human genes encoding synaptobrevins 1. J. Biol. Chem. 1990, 265, 17267–17273. [Google Scholar] [CrossRef]
- Deák, F.; Shin, O.-H.; Kavalali, E.T.; Südhof, T.C. Structural Determinants of Synaptobrevin 2 Function in Synaptic Vesicle Fusion. J. Neurosci. 2006, 26, 6668–6676. [Google Scholar] [CrossRef] [Green Version]
- Sampo, B.; Kaech, S.; Kunz, S.; Banker, G. Two Distinct Mechanisms Target Membrane Proteins to the Axonal Surface. Neuron 2003, 37, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, N.; Sun, T.; Xu, J.; Chiang, H.-C.; Shin, W.; Wu, L.-G. The SNARE Proteins SNAP25 and Synaptobrevin Are Involved in Endocytosis at Hippocampal Synapses. J. Neurosci. 2013, 33, 9169–9175. [Google Scholar] [CrossRef]
- Xu, J.; Brewer, K.D.; Perez-Castillejos, R.; Rizo, J. Subtle Interplay between synaptotagmin and complexin binding to the SNARE complex. J. Mol. Biol. 2013, 425, 3461–3475. [Google Scholar] [CrossRef] [Green Version]
- Haberman, A.; Williamson, W.R.; Epstein, D.; Wang, D.; Rina, S.; Meinertzhagen, I.A.; Hiesinger, P.R. The synaptic vesicle SNARE neuronal Synaptobrevin promotes endolysosomal degradation and prevents neurodegeneration. J. Cell Biol. 2012, 196, 261–276. [Google Scholar] [CrossRef] [Green Version]
- Cornille, F.; Deloye, F.; Fournié-Zaluski, M.-C.; Roques, B.P.; Poulain, B. Inhibition of Neurotransmitter Release by Synthetic Proline-rich Peptides Shows That the N-terminal Domain of Vesicle-associated Membrane Protein/Synaptobrevin Is Critical for Neuro-exocytosis. J. Biol. Chem. 1995, 270, 16826–16832. [Google Scholar] [CrossRef] [Green Version]
- Pham, E.; Crews, L.; Ubhi, K.; Hansen, L.; Adame, A.; Cartier, A.; Salmon, D.; Galasko, D.; Michael, S.; Savas, J.N.; et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010, 277, 3051–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingazov, E.R.; Ugrumov, M.V. Gene expression of proteins of the vesicle cycle in the striatum and motor cortex under functional failure of nigrostriatal system. Dokl. Biochem. Biophys. 2016, 470, 313–315. [Google Scholar] [CrossRef]
- Hernandez-Zimbron, L.F.; Rivas-Arancibia, S. Syntaxin 5 Overexpression and beta-Amyloid 1-42 Accumulation in Endoplasmic Reticulum of Hippocampal Cells in Rat Brain Induced by Ozone Exposure. Biomed. Res. Int. 2016, 2016, 2125643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLelland, G.-L.; Lee, S.A.; McBride, H.M.; Fon, E.A. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 2016, 214, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Suga, K.; Saito, A.; Mishima, T.; Akagawa, K. ER and Golgi stresses increase ER-Golgi SNARE Syntaxin5: Implications for organelle stress and betaAPP processing. Neurosci. Lett. 2015, 604, 30–35. [Google Scholar] [CrossRef]
- Bustos, V.; Pulina, M.V.; Bispo, A.; Lam, A.; Flajolet, M.; Gorelick, F.S.; Greengard, P. Phosphorylated Presenilin 1 decreases beta-amyloid by facilitating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 7148–7153. [Google Scholar] [CrossRef] [Green Version]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [Green Version]
- Almandoz-Gil, L.; Persson, E.; Lindström, V.; Ingelsson, M.; Erlandsson, A.; Bergstrom, J. In Situ Proximity Ligation Assay Reveals Co-Localization of Alpha-Synuclein and SNARE Proteins in Murine Primary Neurons. Front. Neurol. 2018, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Darios, F.; Ruiperez, V.; Lopez, I.; Villanueva, J.; Gutierrez, L.M.; Davletov, B. Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 2010, 11, 528–533. [Google Scholar] [CrossRef] [Green Version]
- Law, C.; Profes, M.S.; Levesque, M.; Kaltschmidt, J.A.; Verhage, M.; Kania, A. Normal Molecular Specification and Neurodegenerative Disease-Like Death of Spinal Neurons Lacking the SNARE-Associated Synaptic Protein Munc18. J. Neurosci. 2016, 36, 561–576. [Google Scholar] [CrossRef] [Green Version]
- Santos, T.C.; Wierda, K.; Broeke, J.H.; Toonen, R.F.; Verhage, M. Early Golgi Abnormalities and Neurodegeneration upon Loss of Presynaptic Proteins Munc18-1, Syntaxin-1, or SNAP. J. Neurosci. 2017, 37, 4525–4539. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, J.; Kim, H.Y.; Ryoo, N.; Lee, S.; Kim, Y.; Rhim, H.; Shin, Y.K. Amyloid-beta Oligomers May Impair SNARE-Mediated Exocytosis by Direct Binding to Syntaxin 1a. Cell Rep. 2015, 12, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.K.; Anderson, H.; Yu, G.; Robertson, A.G.; Allen, S.J.; Tyler, S.J.; Naylor, R.L.; Mason, G.; Wilcock, G.W.; Roche, P.; et al. Identification of syntaxin 1A as a novel binding protein for presenilin. Mol. Brain Res. 2000, 78, 100–107. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Reitböck, P.; Anichtchik, O.; Bellucci, A.; Iovino, M.; Ballini, C.; Fineberg, E.; Ghetti, B.; Della Corte, L.; Spano, P.; Tofaris, G.K.; et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 2010, 133, 2032–2044. [Google Scholar] [CrossRef]
- Agliardi, C.; Meloni, M.; Guerini, F.R.; Zanzottera, M.; Bolognesi, E.; Baglio, F.; Clerici, M. Oligomeric alpha-Syn and SNARE complex proteins in peripheral extracellular vesicles of neural origin are biomarkers for Parkinson’s disease. Neurobiol. Dis. 2021, 148, 105185. [Google Scholar] [CrossRef]
- Suga, K.; Tomiyama, T.; Mori, H.; Akagawa, K. Syntaxin 5 interacts with presenilin holoproteins, but not with their N- or C-terminal fragments, and affects β-amyloid peptide production. Biochem. J. 2004, 381, 619–628. [Google Scholar] [CrossRef]
- Rendón, W.O.; Martínez-Alonso, E.; Tomás, M.; Martínez-Martínez, N.; Martínez-Menárguez, J.A. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem. Cell Biol. 2012, 139, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Tomas, M.; Martinez-Alonso, E.; Martinez-Martinez, N.; Cara-Esteban, M.; Martinez-Menarguez, J.A. Fragmentation of the Golgi complex of dopaminergic neurons in human substantia nigra: New cytopathological findings in Parkinson’s disease. Histol. Histopathol. 2020, 18270. [Google Scholar] [CrossRef]
- Beilina, A.; Bonet-Ponce, L.; Kumaran, R.; Kordich, J.J.; Ishida, M.; Mamais, A.; Kaganovich, A.; Saez-Atienzar, S.; Gershlick, D.C.; Roosen, D.A.; et al. The Parkinson’s Disease Protein LRRK2 Interacts with the GARP Complex to Promote Retrograde Transport to the trans-Golgi Network. Cell Rep. 2020, 31, 107614. [Google Scholar] [CrossRef]
- Zhang, H.; Initiative, T.A.D.N.; Therriault, J.; Kang, M.S.; Ng, K.P.; Pascoal, T.A.; Rosa-Neto, P.; Gauthier, S. Cerebrospinal fluid synaptosomal-associated protein 25 is a key player in synaptic degeneration in mild cognitive impairment and Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Agliardi, C.; Guerini, F.R.; Zanzottera, M.; Bianchi, A.; Nemni, R.; Clerici, M. SNAP-25 in Serum Is Carried by Exosomes of Neuronal Origin and Is a Potential Biomarker of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 5792–5798. [Google Scholar] [CrossRef]
- Yun, H.J.; Park, J.; Ho, D.H.; Kim, H.; Kim, C.-H.; Oh, H.; Ga, I.; Seo, H.; Chang, S.; Son, I.; et al. LRRK2 phosphorylates Snapin and inhibits interaction of Snapin with SNAP-25. Exp. Mol. Med. 2013, 45, e36. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.K.; Choi, M.G.; Kim, J.Y.; Yang, Y.; Lai, Y.; Kweon, D.H.; Lee, N.K.; Shin, Y.K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.-G.; Kim, M.J.; Kim, D.-G.; Yu, R.; Jang, Y.-N.; Oh, W.-J. Sequestration of synaptic proteins by alpha-synuclein aggregates leading to neurotoxicity is inhibited by small peptide. PLoS ONE 2018, 13, e0195339. [Google Scholar] [CrossRef]
- Lou, X.; Kim, J.; Hawk, B.J.; Shin, Y.-K. α-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 2017, 474, 2039–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of α-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Kim, S.; Varkey, J.; Lou, X.; Song, J.K.; Diao, J.; Langen, R.; Shin, Y.-K. Nonaggregated α-Synuclein Influences SNARE-Dependent Vesicle Docking via Membrane Binding. Biochemistry 2014, 53, 3889–3896. [Google Scholar] [CrossRef]
- Brown, E.E.; Blauwendraat, C.; Trinh, J.; Rizig, M.; Nalls, M.A.; Leveille, E.; Ruskey, J.A.; Jonvik, H.; Tan, M.M.; Bandres-Ciga, S.; et al. Analysis of DNM3 and VAMP4 as genetic modifiers of LRRK2 Parkinson’s disease. Neurobiol. Aging 2021, 97, 148.e17–148.e24. [Google Scholar] [CrossRef]
- Pilliod, J.; Desjardins, A.; Pernègre, C.; Jamann, H.; Larochelle, C.; Fon, E.A.; Leclerc, N. Clearance of intracellular tau protein from neuronal cells via VAMP8-induced secretion. J. Biol. Chem. 2020, 295, 17827–17841. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Minakaki, G.; Keramioti, M.V.; Xylaki, M.; Balafas, E.; Chrysanthou-Piterou, M.; Kloukina, I.; Vekrellis, K. GABA transmission via ATP-dependent K+channels regulates α-synuclein secretion in mouse striatum. Brain 2016, 139, 871–890. [Google Scholar] [CrossRef] [Green Version]
- Benskey, M.J.; Perez, R.G.; Manfredsson, F.P. The contribution of alpha synuclein to neuronal survival and function—Implications for Parkinson’s disease. J. Neurochem. 2016, 137, 331–359. [Google Scholar] [CrossRef]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2019, 10, 1312. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Colnaghi, L.; Rondelli, D.; Muzi-Falconi, M.; Sertic, S. Tau and DNA Damage in Neurodegeneration. Brain Sci. 2020, 10, 946. [Google Scholar] [CrossRef]
- Guerini, F.R.; Farina, E.; Costa, A.S.; Baglio, F.; Saibene, F.L.; Margaritella, N.; Calabrese, E.; Zanzottera, M.; Bolognesi, E.; Nemni, R.; et al. ApoE and SNAP-25 Polymorphisms Predict the Outcome of Multidimensional Stimulation Therapy Rehabilitation in Alzheimer’s Disease. Neurorehabil. Neural Repair 2016, 30, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Antonin, W.; Dulubova, I.; Araç, D.; Pabst, S.; Plitzner, J.; Rizo, J.; Jahn, R. The N-terminal Domains of Syntaxin 7 and vti1b Form Three-helix Bundles That Differ in Their Ability to Regulate SNARE Complex Assembly. J. Biol. Chem. 2002, 277, 36449–36456. [Google Scholar] [CrossRef] [Green Version]
- Behrendorff, N.; Dolai, S.; Hong, W.; Gaisano, H.Y.; Thorn, P. Vesicle-associated Membrane Protein 8 (VAMP8) Is a SNARE (Soluble N-Ethylmaleimide-sensitive Factor Attachment Protein Receptor) Selectively Required for Sequential Granule-to-granule Fusion. J. Biol. Chem. 2011, 286, 29627–29634. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef]
- Brooks, D.J. Imaging Familial and Sporadic Neurodegenerative Disorders Associated with Parkinsonism. Neurotherapeutics 2021, 1–19. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2003, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [Green Version]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proukakis, C.; Houlden, H.; Schapira, A.H. Somatic alpha-synuclein mutations in Parkinson’s disease: Hypothesis and preliminary data. Mov. Disord. 2013, 28, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibáñez, P.; Bonnet, A.M.; Débarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Dürr, A.; Brice, A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Juhász, G. A mitochondrial-derived vesicle HOPS to endolysosomes using Syntaxin. J. Cell Biol. 2016, 214, 241–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouaud, T.; Corbille, A.G.; Leclair-Visonneau, L.; de Guilhem de Lataillade, A.; Lionnet, A.; Preterre, C.; Damier, P.; Derkinderen, P. Pathophysiology of Parkinson’s disease: Mitochondria, alpha-synuclein and much more. Rev. Neurol. 2021, 177, 260–271. [Google Scholar] [CrossRef]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, G. Potential of extracellular vesicles in the Parkinson’s disease—Pathological mediators and biomarkers. Neurochem. Int. 2021, 144, 104974. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H.; Yang, Z.; Oltedal, L.; Davanger, S.; Hay, J.C. Intramolecular protein-protein and protein-lipid interactions control the conformation and subcellular targeting of neuronal Ykt. J. Cell Sci. 2004, 117, 4495–4508. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H.; Zinsser, S.; Rhee, Y.; Vik-Mo, E.O.; Davanger, S.; Hay, J.C. Mammalian Ykt6 Is a Neuronal SNARE Targeted to a Specialized Compartment by its Profilin-like Amino Terminal Domain. Mol. Biol. Cell 2003, 14, 698–720. [Google Scholar] [CrossRef] [Green Version]
- Belluzzi, E.; Gonnelli, A.; Cirnaru, M.-D.; Marte, A.; Plotegher, N.; Russo, I.; Civiero, L.; Cogo, S.; Carrion, M.P.; Franchin, C.; et al. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol. Neurodegener. 2016, 11, 1. [Google Scholar] [CrossRef]
- Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nat. Cell Biol. 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Salvi, S.; Ialongo, T.; Marongiu, R.; Elia, A.E.; Caputo, V.; Romito, L.; Albanese, A.; Dallapiccola, B.; Bentivoglio, A.R. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 2004, 56, 336–341. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nat. Cell Biol. 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Arasaki, K.; Shimizu, H.; Mogari, H.; Nishida, N.; Hirota, N.; Furuno, A.; Kudo, Y.; Baba, M.; Baba, N.; Cheng, J.; et al. A Role for the Ancient SNARE Syntaxin 17 in Regulating Mitochondrial Division. Dev. Cell 2015, 32, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Strong, M.J.; Donison, N.S.; Volkening, K. Alterations in Tau Metabolism in ALS and ALS-FTSD. Front. Neurol. 2020, 11, 598907. [Google Scholar] [CrossRef]
- Yousefian-Jazi, A.; Seol, Y.; Kim, J.; Ryu, H.L.; Lee, J.; Ryu, H. Pathogenic Genome Signatures That Damage Motor Neurons in Amyotrophic Lateral Sclerosis. Cells 2020, 9, 2687. [Google Scholar] [CrossRef]
- Redler, R.L.; Dokholyan, N.V. The Complex Molecular Biology of Amyotrophic Lateral Sclerosis (ALS). Prog. Mol. Biol. Transl. Sci. 2012, 107, 215–262. [Google Scholar] [CrossRef] [Green Version]
- Iacoangeli, A.; Initiative, A.D.N.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Philips, T.; Rothstein, J. Glial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| SNARE Type | Name of the SNARE Protein (Alternative Name) | Neurodegenerative Disease | References |
|---|---|---|---|
| Qa (syntaxins) | Syntaxin-1 (HPC-1) | AD, PD, ALS | [30,40,41,47,48,49,50,51,52,53,54,55] |
| Syntaxin-2 (Epimorphin) | |||
| Syntaxin-3 | |||
| Syntaxin-4 | |||
| Syntaxin-5 | AD, PD | [42,44,56,57,58] | |
| Syntaxin-7 | |||
| Syntaxin-11 | |||
| Syntaxin-13 (Syntaxin-12) | |||
| Syntaxin-16 | |||
| Syntaxin-17 | AD, PD | [43,45] | |
| Syntaxin-18 | |||
| Qb | GS-27 | ||
| GS-28 | |||
| Vti1a | |||
| Vti1b | |||
| Qc | BET1 | AD | [44] |
| GS-15 | |||
| Slt1 | |||
| Syntaxin-6 | PD | [59] | |
| Syntaxin-8 | |||
| Syntaxin-10 | |||
| Qb-c | SNAP-23 (Syndet) | ||
| SNAP-25 | AD; PD; ALS | [30,40,43,47,48,50,53,60,61,62] | |
| SNAP-29 (GS-32) | |||
| R | VAMP1 (Synaptobrevin 1) | ||
| VAMP2 (Syb 2) | AD; PD; ALS | [30,40,47,48,53,55,63,64,65,66,67,68] | |
| VAMP3 (Cellubrevin) | |||
| VAMP4 | PD | [59,69] | |
| VAMP5 | |||
| VAMP7 (Ti-VAMP) | PD | [43] | |
| VAMP8 (Endobrevin) | AD, PD | [45,70,71,72] | |
| YKT6P | PD | [46] | |
| SEC22b (ERS-24) | PD | [46] | |
| Unclassified | SEC22a | ||
| SEC22c | |||
| SEC20 (Bnip1) |
| Neurodegenerative Disease | Molecule Affected | Function |
|---|---|---|
| AD | Aβ | Aβ accumulation, secretion, plaque formation |
| AD | Tau | Cytoplasmic transport, NFTs |
| AD | Munc 18-1 | Regulation of SNARE complex formation, exocytosis and neurotransmission |
| AD | presenilin 1 | Regulation of Aβ production |
| PD | α-synuclein | Synaptic modulatory protein |
| PD | LRRK2 | Synaptic vesicle fusion |
| PD | NSF | Regulation of SNARE complex disassembly |
| ALS | SOD1 | Superoxide dismutase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margiotta, A. Role of SNAREs in Neurodegenerative Diseases. Cells 2021, 10, 991. https://doi.org/10.3390/cells10050991
Margiotta A. Role of SNAREs in Neurodegenerative Diseases. Cells. 2021; 10(5):991. https://doi.org/10.3390/cells10050991
Chicago/Turabian StyleMargiotta, Azzurra. 2021. "Role of SNAREs in Neurodegenerative Diseases" Cells 10, no. 5: 991. https://doi.org/10.3390/cells10050991
APA StyleMargiotta, A. (2021). Role of SNAREs in Neurodegenerative Diseases. Cells, 10(5), 991. https://doi.org/10.3390/cells10050991