
The Orai1-AC8 Interplay: How Breast Cancer Cells Escape from Orai1 Channel Inactivation




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Store-Operated Ca2+ Entry
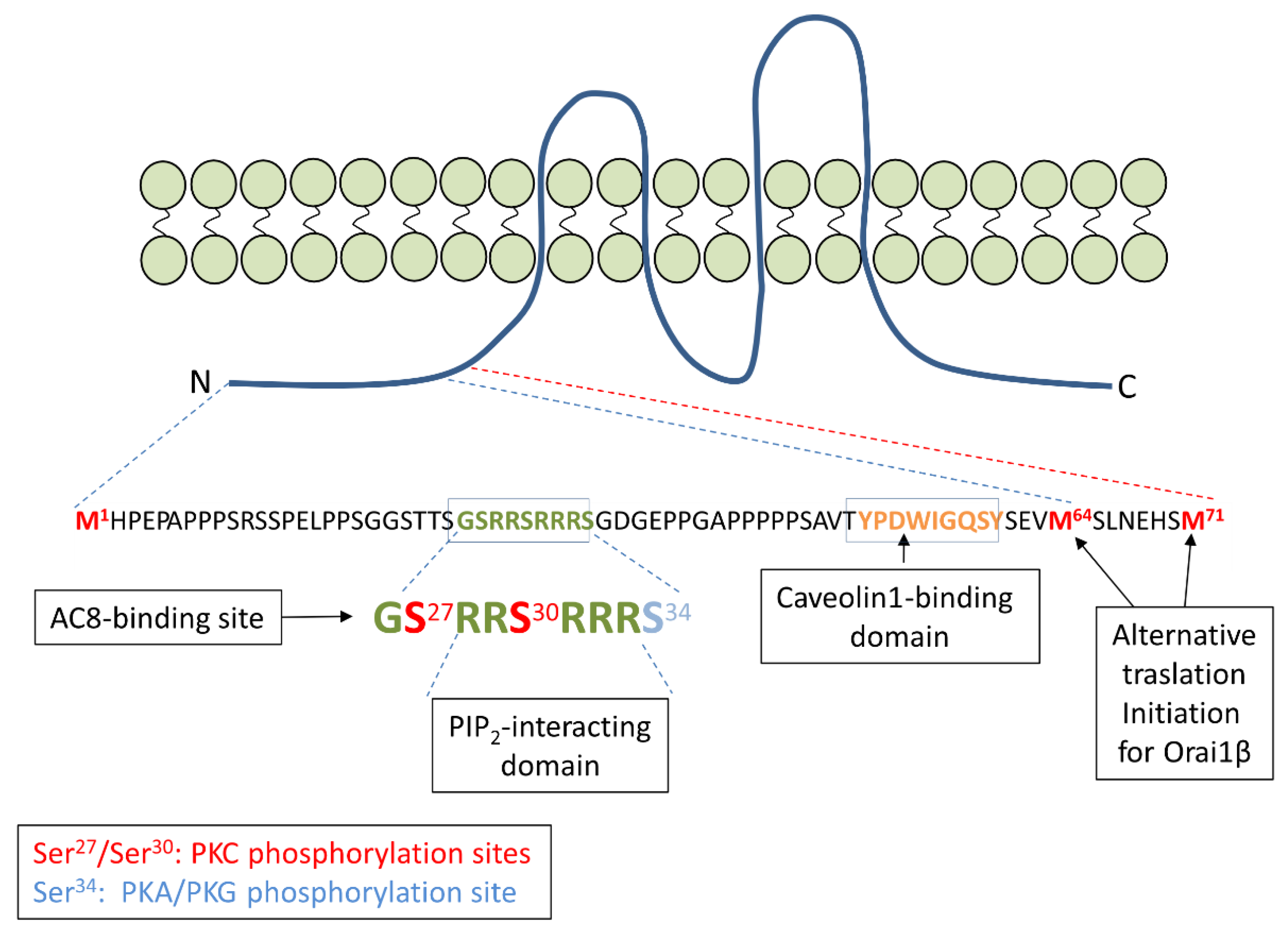
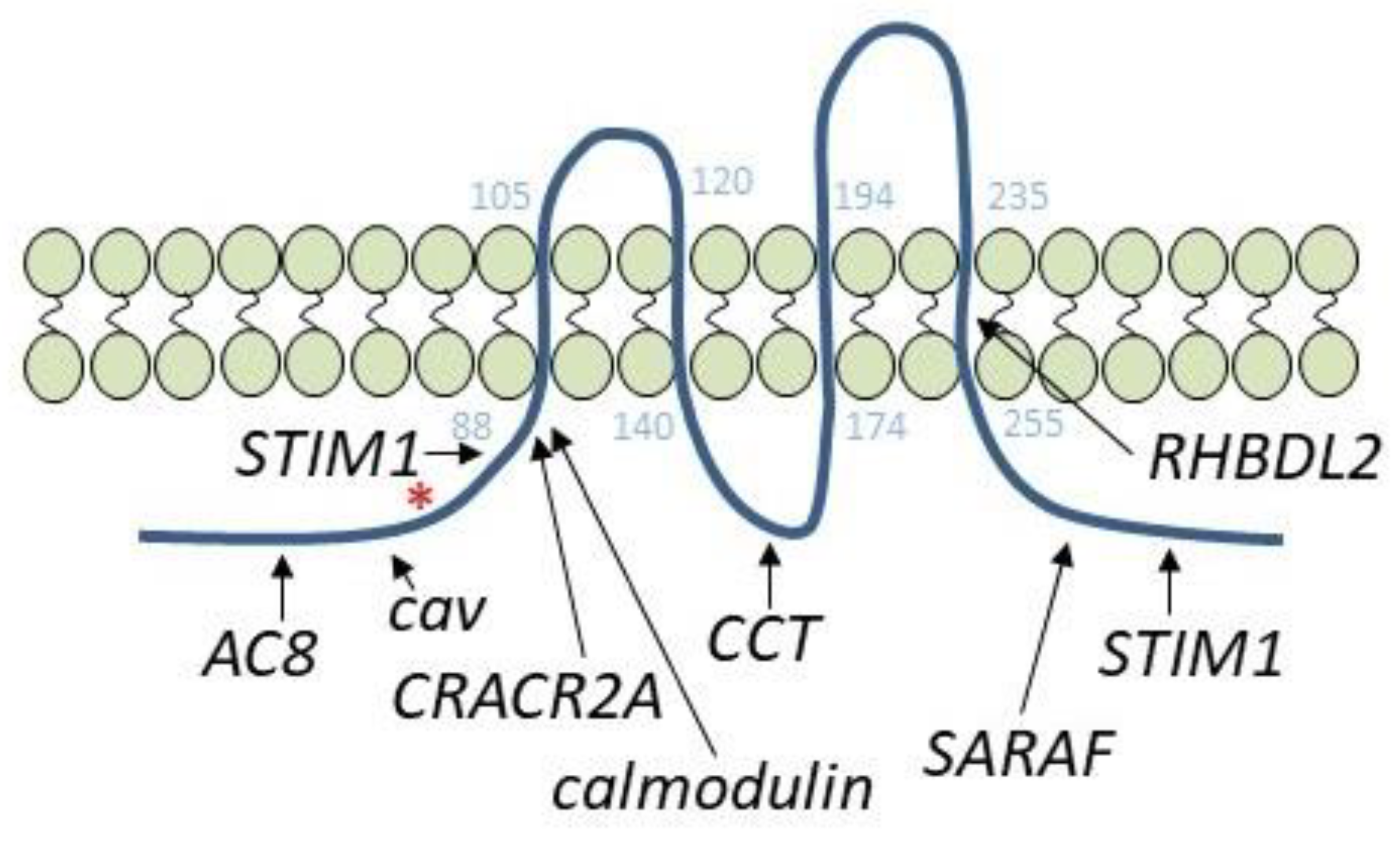
3. Orai1 Variants
4. Orai1-Interacting Proteins: Adenylyl Cyclase 8
5. Orai1-Adenylyl Cyclase 8 in Cancer Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Revs. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Parekh, A.B. Distinct spatial Ca2+ signatures selectively activate different NFAT transcription factor isoforms. Mol. Cell 2015, 58, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, J.I.; Straub, S.V.; Yule, D.I. Crosstalk between cAMP and Ca2+ signaling in non-excitable cells. Cell Calcium 2003, 34, 431–444. [Google Scholar] [CrossRef]
- Bruce, J.I.; Shuttleworth, T.J.; Giovannucci, D.R.; Yule, D.I. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J. Biol. Chem. 2002, 277, 1340–1348. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Lange, I.; Feske, S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 2009, 385, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.S.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Vig, M.; Beck, A.; Billingsley, J.M.; Lis, A.; Parvez, S.; Peinelt, C.; Koomoa, D.L.; Soboloff, J.; Gill, D.L.; Fleig, A.; et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006, 16, 2073–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinelt, C.; Vig, M.; Koomoa, D.L.; Beck, A.; Nadler, M.J.; Koblan-Huberson, M.; Lis, A.; Fleig, A.; Penner, R.; Kinet, J.P. Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell. Biol. 2006, 8, 771–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soboloff, J.; Spassova, M.A.; Tang, X.D.; Hewavitharana, T.; Xu, W.; Gill, D.L. Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 2006, 281, 20661–20665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [Green Version]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [Green Version]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca(2+) signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 2015, 8, ra74. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 2008, 283, 25296–25304. [Google Scholar] [CrossRef] [Green Version]
- Ong, H.L.; Subedi, K.P.; Son, G.Y.; Liu, X.; Ambudkar, I.S. Tuning store-operated calcium entry to modulate Ca2+-dependent physiological processes. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Hayashi, K.; Fujii, Y.; Mizushima, A.; Watarai, H.; Wakamori, M.; Numaga, T.; Mori, Y.; Iino, M.; Hikida, M.; et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2006, 103, 16704–16709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, X.; Loktionova, N.A.; Cai, X.; Nwokonko, R.M.; Vrana, E.; Wang, Y.; Rothberg, B.S.; Gill, D.L. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 2015, 6, 8395. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Nwokonko, R.M.; Loktionova, N.A.; Abdulqadir, R.; Baraniak, J.H., Jr.; Wang, Y.; Trebak, M.; Zhou, Y.; Gill, D.L. Pore properties of Orai1 calcium channel dimers and their activation by the STIM1 ER calcium sensor. J. Biol. Chem. 2018, 293, 12962–12974. [Google Scholar] [CrossRef] [Green Version]
- Baraniak, J.H., Jr.; Zhou, Y.; Nwokonko, R.M.; Gill, D.L. The Intricate Coupling Between STIM Proteins and Orai Channels. Curr. Opin. Physiol. 2020, 17, 106–114. [Google Scholar] [CrossRef]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential redox regulation of ORAI ion channels: A mechanism to tune cellular calcium signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [Green Version]
- Alansary, D.; Bogeski, I.; Niemeyer, B.A. Facilitation of Orai3 targeting and store-operated function by Orai1. Biochim. Biophys. Acta 2015, 1853, 1541–1550. [Google Scholar] [CrossRef] [Green Version]
- Saul, S.; Gibhardt, C.S.; Schmidt, B.; Lis, A.; Pasieka, B.; Conrad, D.; Jung, P.; Gaupp, R.; Wonnenberg, B.; Diler, E.; et al. A calcium-redox feedback loop controls human monocyte immune responses: The role of ORAI Ca2+ channels. Sci. Signal. 2016, 9, ra26. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.; Johnson, R.; Pashos, M.; Cummings, C.; Kang, C.; Sampedro, G.R.; Tycksen, E.; McBride, H.J.; Sah, R.; Lowell, C.A.; et al. ORAI1 and ORAI2 modulate murine neutrophil calcium signaling, cellular activation, and host defense. Proc. Natl. Acad. Sci. USA 2020, 117, 24403–24414. [Google Scholar] [CrossRef] [PubMed]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta 2017, 1864, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Yuan, J.P.; Zeng, W.; So, I.; Worley, P.F.; Muallem, S. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc. Natl. Acad. Sci. USA 2009, 106, 14687–14692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; Putney, J.W., Jr. ORAI Calcium Channels. Physiology 2017, 32, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.; Everett, K.L.; Halls, M.L.; Pacheco, J.; Skroblin, P.; Vaca, L.; Klussmann, E.; Cooper, D.M. Direct binding between Orai1 and AC8 mediates dynamic interplay between Ca2+ and cAMP signaling. Sci. Signal. 2012, 5, ra29. [Google Scholar] [CrossRef]
- Kawasaki, T.; Ueyama, T.; Lange, I.; Feske, S.; Saito, N. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J. Biol. Chem. 2010, 285, 25720–25730. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Z.C.; Zhang, P.; Poon, E.; Kong, C.W.; Boheler, K.R.; Huang, Y.; Li, R.A.; Yao, X. Nitric Oxide-cGMP-PKG Pathway Acts on Orai1 to Inhibit the Hypertrophy of Human Embryonic Stem Cell-Derived Cardiomyocytes. Stem Cells 2015, 33, 2973–2984. [Google Scholar] [CrossRef]
- Zhang, X.; Pathak, T.; Yoast, R.; Emrich, S.; Xin, P.; Nwokonko, R.M.; Johnson, M.; Wu, S.; Delierneux, C.; Gueguinou, M.; et al. A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction. Nat. Commun. 2019, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Parekh, A.B. Distinct structural domains of caveolin-1 independently regulate Ca2+ release-activated Ca2+ channels and Ca2+ microdomain-dependent gene expression. Mol. Cell Biol. 2015, 35, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J. Physiol. 2008, 586, 185–195. [Google Scholar] [CrossRef]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J. Physiol. 2007, 579, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, W.; Gonzalez-Cobos, J.C.; Jardin, I.; Romanin, C.; Matrougui, K.; Trebak, M. Complex role of STIM1 in the activation of store-independent Orai1/3 channels. J. Gen. Physiol. 2014, 143, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, P.; Lin, Y.P.; Bhardwaj, R.; Tucker, C.J.; Bird, G.S.; Hediger, M.A.; Monico, C.; Amin, N.; Parekh, A.B. The N terminus of Orai1 couples to the AKAP79 signaling complex to drive NFAT1 activation by local Ca2+ entry. Proc. Natl. Acad. Sci. USA 2021, 118, e2012908118. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Samanta, K.; Kramer, H.; Morris, O.; Bakowski, D.; Parekh, A.B. Dynamic assembly of a membrane signaling complex enables selective activation of NFAT by Orai1. Curr. Biol. 2014, 24, 1361–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanth, S.; Jung, H.J.; Kim, K.D.; Souda, P.; Whitelegge, J.; Gwack, Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat. Cell Biol. 2010, 12, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Hodeify, R.; Nandakumar, M.; Own, M.; Courjaret, R.J.; Graumann, J.; Hubrack, S.Z.; Machaca, K. The CCT chaperonin is a novel regulator of Ca2+ signaling through modulation of Orai1 trafficking. Sci. Adv. 2018, 4, eaau1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarran, L.; Lopez, J.J.; Amor, N.B.; Martin-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Albarran, L.; Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Berna-Erro, A.; Smani, T.; Camello, P.J.; Salido, G.M.; Rosado, J.A. EFHB is a Novel Cytosolic Ca2+ Sensor That Modulates STIM1-SARAF Interaction. Cell Physiol. Biochem. 2018, 51, 1164–1178. [Google Scholar] [CrossRef]
- Zhu, H.; Weisleder, N.; Wu, P.; Cai, C.; Chen, J.W. Caveolae/caveolin-1 are important modulators of store-operated calcium entry in Hs578/T breast cancer cells. J. Pharmacol. Sci. 2008, 106, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Prakash, Y.S.; Thompson, M.A.; Vaa, B.; Matabdin, I.; Peterson, T.E.; He, T.; Pabelick, C.M. Caveolins and intracellular calcium regulation in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1118–L1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Sun, L.; Machaca, K. Constitutive recycling of the store-operated Ca2+ channel Orai1 and its internalization during meiosis. J. Cell Biol. 2010, 191, 523–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Sun, L.; Machaca, K. Orai1 internalization and STIM1 clustering inhibition modulate SOCE inactivation during meiosis. Proc. Natl. Acad. Sci. USA 2009, 106, 17401–17406. [Google Scholar] [CrossRef] [Green Version]
- Hodeify, R.; Dib, M.; Alcantara-Adap, E.; Courjaret, R.; Nader, N.; Reyes, C.Z.; Hammad, A.S.; Hubrack, S.; Yu, F.; Machaca, K. The carboxy terminal coiled-coil modulates Orai1 internalization during meiosis. Sci. Rep. 2021, 11, 2290. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Chen, G.L.; Garcia-Vaz, E.; Bhandari, S.; Daskoulidou, N.; Berglund, L.M.; Jiang, H.; Hallett, T.; Zhou, L.P.; Huang, L.; et al. ORAI channels are critical for receptor-mediated endocytosis of albumin. Nat. Commun. 2017, 8, 1920. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Jeon, I.S.; Han, N.E.; Song, H.J.; Kim, E.G.; Choi, J.W.; Song, K.D.; Lee, H.K.; Choi, J.K. Ubiquilin 1 interacts with Orai1 to regulate calcium mobilization. Mol. Cells 2013, 35, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Grieve, A.G.; Yeh, Y.C.; Zarcone, L.; Breuning, J.; Johnson, N.; Stříšovský, K.; Brown, M.H.; Parekh, A.B.; Freeman, M. Conformational surveillance of Orai1 by a rhomboid intramembrane protease prevents inappropriate CRAC channel activation. BioRxiv 2020. [CrossRef] [Green Version]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Cross, B.M.; Hack, A.; Reinhardt, T.A.; Rao, R. SPCA2 regulates Orai1 trafficking and store independent Ca2+ entry in a model of lactation. PLoS ONE 2013, 8, e67348. [Google Scholar] [CrossRef] [PubMed]
- Cantonero, C.; Sanchez-Collado, J.; Gonzalez-Nunez, M.A.; Salido, G.M.; Lopez, J.J.; Jardin, I.; Rosado, J.A. Store-independent Orai1-mediated Ca2+ entry and cancer. Cell Calcium 2019, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Collado, J.; Lopez, J.J.; Jardin, I.; Camello, P.J.; Falcon, D.; Regodon, S.; Salido, G.M.; Smani, T.; Rosado, J.A. Adenylyl Cyclase Type 8 Overexpression Impairs Phosphorylation-Dependent Orai1 Inactivation and Promotes Migration in MDA-MB-231 Breast Cancer Cells. Cancers 2019, 11, 1624. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.C.; Willoughby, D.; Ciruela, A.; Ayling, L.J.; Pagano, M.; Wachten, S.; Tengholm, A.; Cooper, D.M. Capacitative Ca2+ entry via Orai1 and stromal interacting molecule 1 (STIM1) regulates adenylyl cyclase type 8. Mol. Pharmacol. 2009, 75, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Cooper, D.M. Calmodulin-binding sites on adenylyl cyclase type VIII. J. Biol. Chem. 1999, 274, 8012–8021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, R.E.; Ciruela, A.; Cooper, D.M.F. The role of calmodulin recruitment in Ca2+ stimulation of adenylyl cyclase type 8. J. Biol. Chem. 2006, 281, 17379–17389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagan, K.A.; Mahey, R.; Cooper, D.M. Functional co-localization of transfected Ca2+-stimulable adenylyl cyclases with capacitative Ca2+ entry sites. J. Biol. Chem. 1996, 271, 12438–12444. [Google Scholar] [CrossRef] [Green Version]
- Fagan, K.A.; Graf, R.A.; Tolman, S.; Schaack, J.; Cooper, D.M. Regulation of a Ca2+-sensitive adenylyl cyclase in an excitable cell. Role of voltage-gated versus capacitative Ca2+ entry. J. Biol. Chem. 2000, 275, 40187–40194. [Google Scholar] [CrossRef] [Green Version]
- Everett, K.L.; Cooper, D.M. An improved targeted cAMP sensor to study the regulation of adenylyl cyclase 8 by Ca2+ entry through voltage-gated channels. PLoS ONE 2013, 8, e75942. [Google Scholar] [CrossRef] [Green Version]
- Shuttleworth, T.J.; Thompson, J.L. Discriminating between capacitative and arachidonate-activated Ca2+ entry pathways in HEK293 cells. J. Biol. Chem. 1999, 274, 31174–31178. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.C.; Cooper, D.M. Capacitative and 1-oleyl-2-acetyl-sn-glycerol-activated Ca2+ entry distinguished using adenylyl cyclase type 8. Mol. Pharmacol. 2006, 70, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehbe, N.; Slika, H.; Mesmar, J.; Nasser, S.A.; Pintus, G.; Baydoun, S.; Badran, A.; Kobeissy, F.; Eid, A.H.; Baydoun, E. The Role of Epac in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 6489. [Google Scholar] [CrossRef]
- Roberts-Thomson, S.J.; Chalmers, S.B.; Monteith, G.R. The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb. Perspect. Biol. 2019, 11, a035204. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.G.; Kim, S.J.; Kim, C.; Jeong, J. Molecular Classification of Triple-Negative Breast Cancer. J. Breast Cancer 2016, 19, 223–230. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.Y.; Jiang, Z.; Zacksenhaus, E. Stratifying the stratifiers of triple negative breast cancer. Oncotarget 2020, 11, 306–308. [Google Scholar] [CrossRef]
- So, C.L.; Saunus, J.M.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium signalling and breast cancer. Sem. Cell Dev. Biol. 2019, 94, 74–83. [Google Scholar] [CrossRef]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [Green Version]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Gupta, S.; Dabral, S.; Singh, S.; Sehrawat, S. Role of exchange protein directly activated by cAMP (EPAC1) in breast cancer cell migration and apoptosis. Mol. Cell Biochem. 2017, 430, 115–125. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Collado, J.; López, J.J.; Rosado, J.A. The Orai1-AC8 Interplay: How Breast Cancer Cells Escape from Orai1 Channel Inactivation. Cells 2021, 10, 1308. https://doi.org/10.3390/cells10061308
Sánchez-Collado J, López JJ, Rosado JA. The Orai1-AC8 Interplay: How Breast Cancer Cells Escape from Orai1 Channel Inactivation. Cells. 2021; 10(6):1308. https://doi.org/10.3390/cells10061308
Chicago/Turabian StyleSánchez-Collado, José, José J. López, and Juan A. Rosado. 2021. "The Orai1-AC8 Interplay: How Breast Cancer Cells Escape from Orai1 Channel Inactivation" Cells 10, no. 6: 1308. https://doi.org/10.3390/cells10061308
APA StyleSánchez-Collado, J., López, J. J., & Rosado, J. A. (2021). The Orai1-AC8 Interplay: How Breast Cancer Cells Escape from Orai1 Channel Inactivation. Cells, 10(6), 1308. https://doi.org/10.3390/cells10061308