
Aberrant B Cell Receptor Signaling in Naïve B Cells from Patients with Idiopathic Pulmonary Fibrosis

 , ,
, , 

Abstract
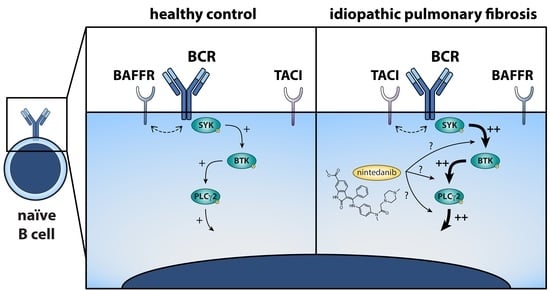
:
1. Introduction
2. Materials and Methods
2.1. Patient Characteristics and Study Design
2.2. Flow Cytometric Analysis
2.2.1. Flow Cytometry for B Cell Surface Markers and Intracellular BTK
2.2.2. BCR Signaling Measurement by Phosphoflow Cytometry
2.3. Cytokine Detection by Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Statistical and Computational Analysis
3. Results
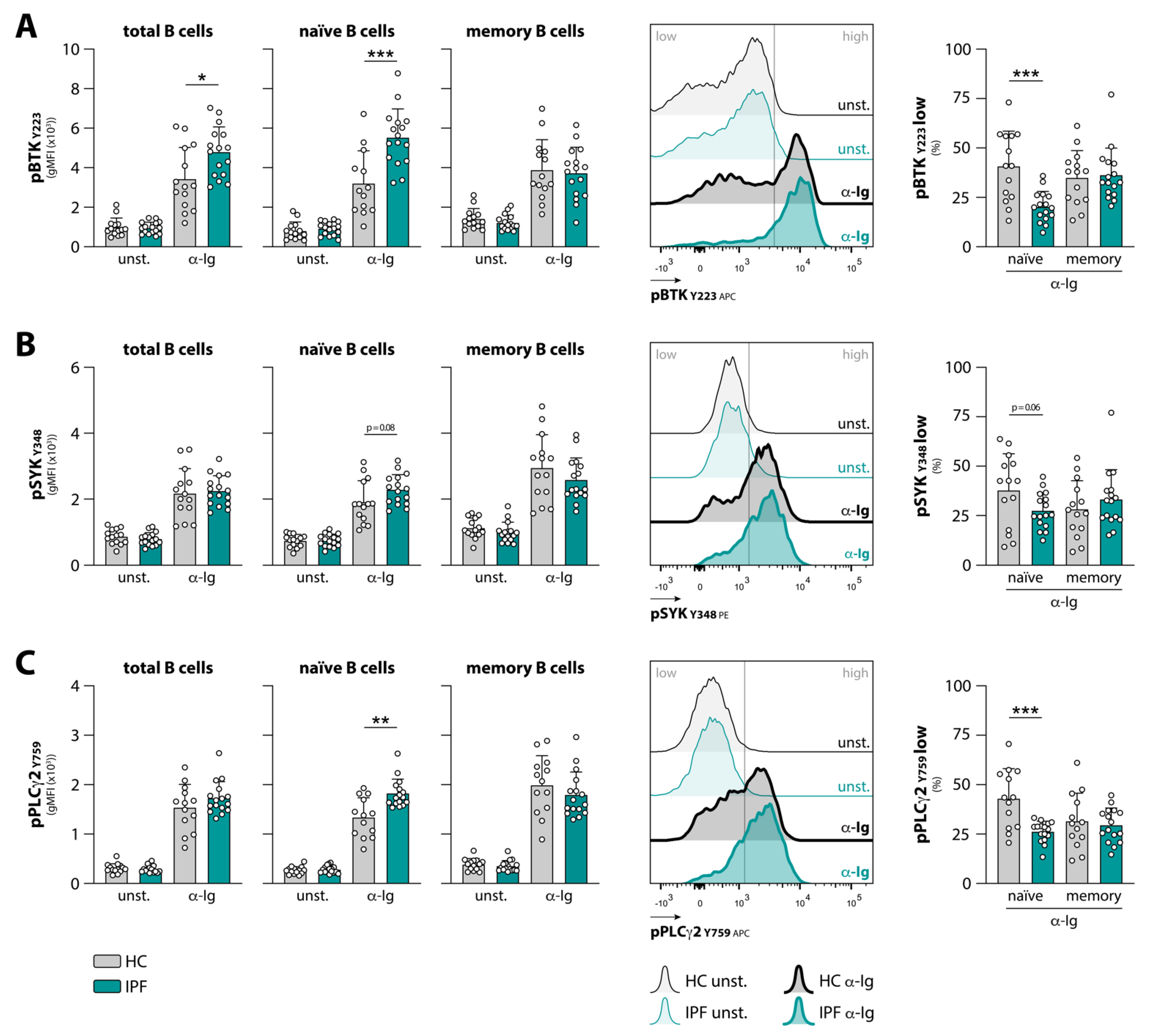
3.1. Naïve B Cells from IPF Patients Show Aberrant BCR Signaling upon Stimulation
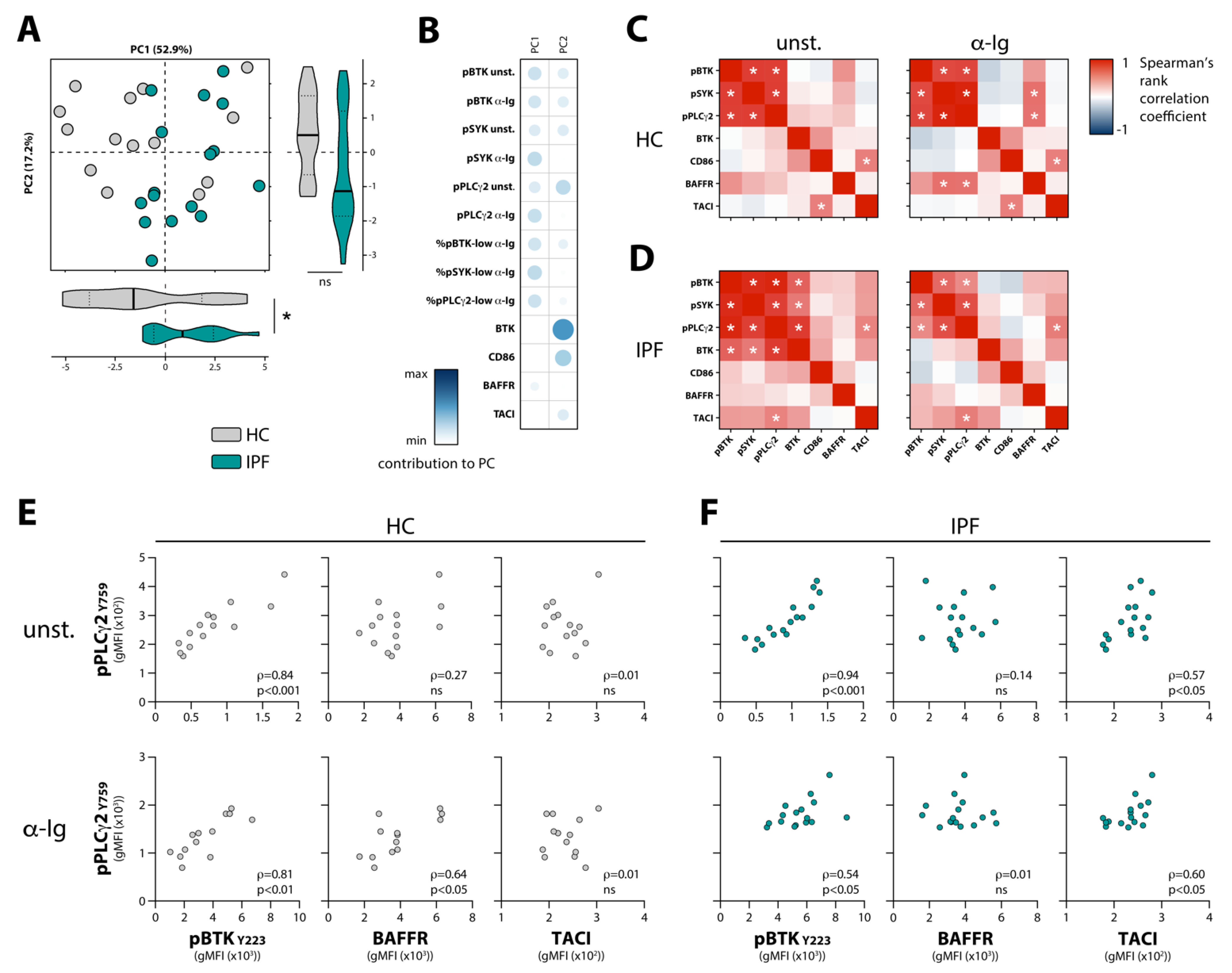
3.2. Enhanced BCR Signaling in Naïve B Cells from IPF Patients Correlates with TACI Expression
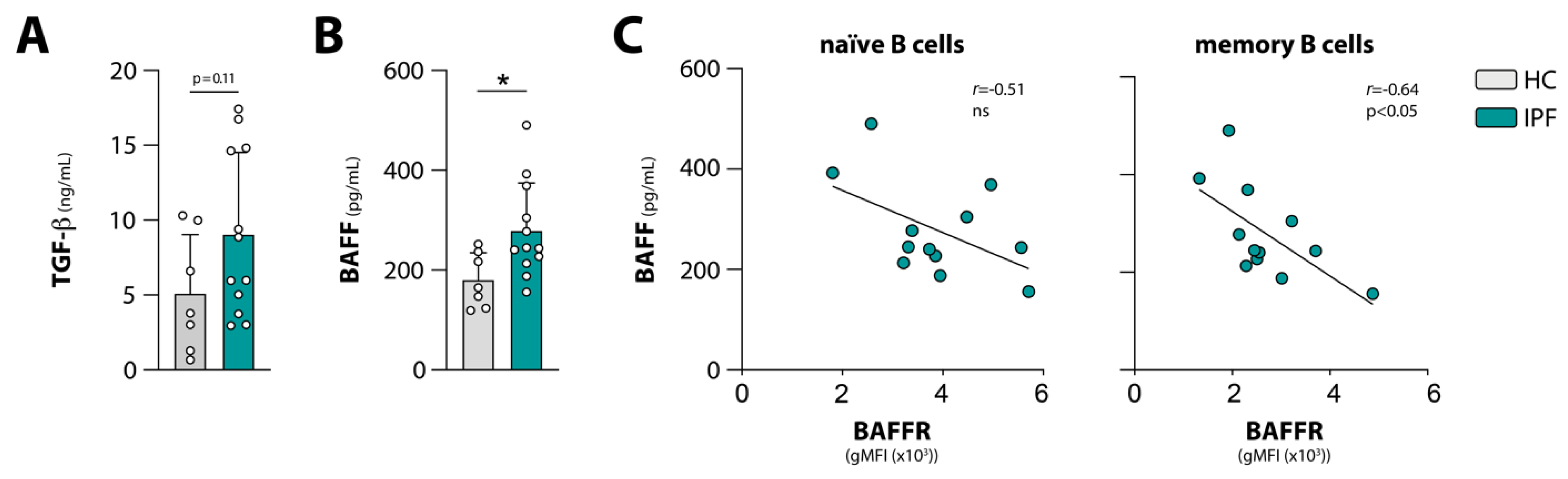
3.3. Negative Correlations between Circulating BAFF and BAFFR Expression, and between Circulating TGF-β and Phosphorylation of BCR Signalosome Molecules Following Stimulation
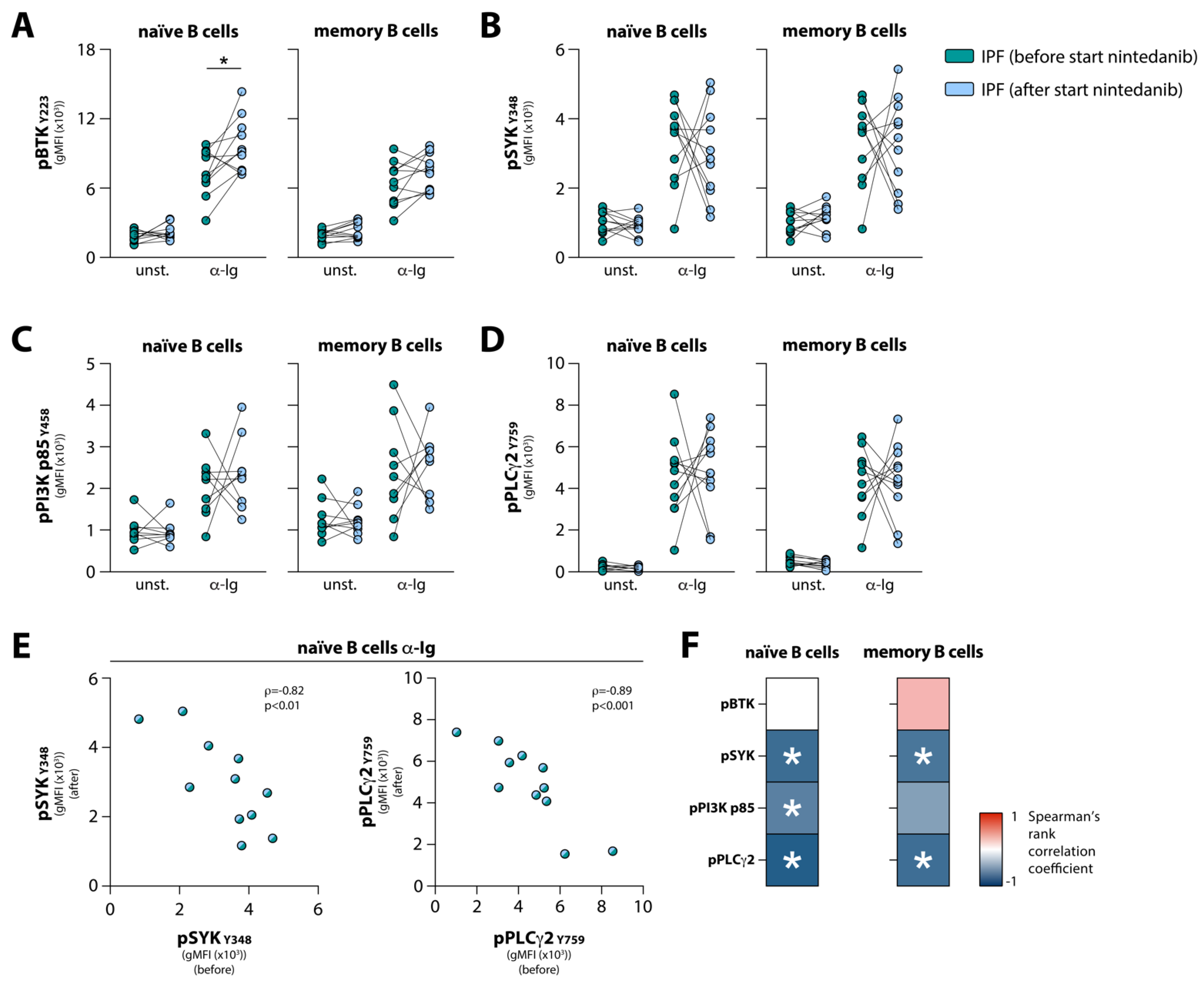
3.4. Nintedanib Treatment Induces Substantial Changes in BCR Signaling in Naïve and Memory B Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-de la Mora, D.A.; Sanchez-Roque, C.; Montoya-Buelna, M.; Sanchez-Enriquez, S.; Lucano-Landeros, S.; Macias-Barragan, J.; Armendariz-Borunda, J. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int. J. Med. Sci. 2015, 12, 840–847. [Google Scholar] [CrossRef] [Green Version]
- Heukels, P.; Moor, C.C.; von der Thüsen, J.H.; Wijsenbeek, M.S.; Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Marchal-Sommé, J.; Uzunhan, Y.; Marchand-Adam, S.; Valeyre, D.; Soumelis, V.; Crestani, B.; Soler, P. Cutting edge: Nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J. Immunol. 2006, 176, 5735–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuovo, G.J.; Hagood, J.S.; Magro, C.M.; Chin, N.; Kapil, R.; Davis, L.; Marsh, C.B.; Folcik, V.A. The distribution of immunomodulatory cells in the lungs of patients with idiopathic pulmonary fibrosis. Mod. Pathol. 2012, 25, 416–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, N.W.; Scheraga, R.G.; Galvin, J.R.; Iacono, A.T.; Britt, E.J.; Luzina, I.G.; Burke, A.P.; Atamas, S.P. Lymphocyte aggregates persist and accumulate in the lungs of patients with idiopathic pulmonary fibrosis. J. Inflamm. Res. 2013, 6, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuga, L.J.; Tedrow, J.R.; Pandit, K.V.; Tan, J.; Kass, D.J.; Xue, J.; Chandra, D.; Leader, J.K.; Gibson, K.F.; Kaminski, N.; et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 966–974. [Google Scholar] [CrossRef] [PubMed]
- DePianto, D.J.; Chandriani, S.; Abbas, A.R.; Jia, G.; N’Diaye, E.N.; Caplazi, P.; Kauder, S.E.; Biswas, S.; Karnik, S.K.; Ha, C.; et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 2015, 70, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Kass, D.J.; Bon, J.; Vuga, L.; Tan, J.; Csizmadia, E.; Otterbein, L.; Soejima, M.; Levesque, M.C.; Gibson, K.F.; et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J. Immunol. 2013, 191, 2089–2095. [Google Scholar] [CrossRef]
- Ten Klooster, L.; van Moorsel, C.H.M.; Kwakkel-van Erp, J.M.; van Velzen-Blad, H.; Grutters, J.C. Immunoglobulin A in serum: An old acquaintance as a new prognostic biomarker in idiopathic pulmonary fibrosis. Clin. Exp. Immunol. 2015, 181, 357–361. [Google Scholar] [CrossRef] [Green Version]
- Hoyne, G.F.; Elliott, H.; Mutsaers, S.E.; Prêle, C.M. Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol. Cell Biol. 2017, 95, 577–583. [Google Scholar] [CrossRef]
- Donahoe, M.; Valentine, V.G.; Chien, N.; Gibson, K.F.; Raval, J.S.; Saul, M.; Xue, J.; Zhang, Y.; Duncan, S.R. Autoantibody-Targeted Treatments for Acute Exacerbations of Idiopathic Pulmonary Fibrosis. PLoS ONE 2015, 10, e0127771. [Google Scholar] [CrossRef]
- Enomoto, N.; Chida, K.; Suda, T.; Kaida, Y.; Taniguchi, M.; Azuma, A.; Hayashi, H.; Ogura, T.; Kitamura, H.; Yamaguchi, O.; et al. An exploratory trial of intravenous immunoglobulin therapy for idiopathic pulmonary fibrosis: A preliminary multicenter report. Clin. Respir. J. 2016, 10, 746–755. [Google Scholar] [CrossRef]
- Asai, Y.; Chiba, H.; Nishikiori, H.; Kamekura, R.; Yabe, H.; Kondo, S.; Miyajima, S.; Shigehara, K.; Ichimiya, S.; Takahashi, H. Aberrant populations of circulating T follicular helper cells and regulatory B cells underlying idiopathic pulmonary fibrosis. Respir. Res. 2019, 20, 244. [Google Scholar] [CrossRef]
- Heukels, P.; van Hulst, J.A.C.; van Nimwegen, M.; Boorsma, C.E.; Melgert, B.N.; von der Thusen, J.H.; van den Blink, B.; Hoek, R.A.S.; Miedema, J.R.; Neys, S.F.H.; et al. Enhanced Bruton’s tyrosine kinase in B-cells and autoreactive IgA in patients with idiopathic pulmonary fibrosis. Respir. Res. 2019, 20, 232. [Google Scholar] [CrossRef]
- Hendriks, R.W.; Yuvaraj, S.; Kil, L.P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14, 219–232. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; de Bruijn, M.J.W.; Rip, J.; Lukkes, M.; van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes from Patients with Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef]
- von Borstel, A.; Abdulahad, W.H.; Sanders, J.S.; Rip, J.; Neys, S.F.H.; Hendriks, R.W.; Stegeman, C.A.; Heeringa, P.; Rutgers, A.; Corneth, O.B.J. Evidence for enhanced Bruton’s tyrosine kinase activity in transitional and naïve B cells of patients with granulomatosis with polyangiitis. Rheumatology 2019, 58, 2230–2239. [Google Scholar] [CrossRef] [Green Version]
- Kil, L.P.; de Bruijn, M.J.W.; van Nimwegen, M.; Corneth, O.B.J.; van Hamburg, J.P.; Dingjan, G.M.; Thaiss, F.; Rimmelzwaan, G.F.; Elewaut, D.; Delsing, D.; et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 2012, 119, 3744–3756. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; de Bruijn, M.J.W.; Rip, J.; Asmawidjaja, P.S.; Kil, L.P.; Hendriks, R.W. Enhanced Expression of Bruton’s Tyrosine Kinase in B Cells Drives Systemic Autoimmunity by Disrupting T Cell Homeostasis. J. Immunol. 2016, 1600208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, D.N.; Norman, K.C.; Xia, M.; Huang, Y.; Gurczynski, S.J.; Ashley, S.L.; White, E.S.; Flaherty, K.R.; Martinez, F.J.; Murray, S.; et al. The peripheral blood proteome signature of idiopathic pulmonary fibrosis is distinct from normal and is associated with novel immunological processes. Sci. Rep. 2017, 7, 46560. [Google Scholar] [CrossRef] [PubMed]
- Schweighoffer, E.; Vanes, L.; Nys, J.; Cantrell, D.; McCleary, S.; Smithers, N.; Tybulewicz, V.L.J. The BAFF Receptor Transduces Survival Signals by Co-opting the B Cell Receptor Signaling Pathway. Immunity 2013, 38, 475–488. [Google Scholar] [CrossRef] [Green Version]
- Shinners, N.P.; Carlesso, G.; Castro, I.; Hoek, K.L.; Corn, R.A.; Woodland, R.T.; Scott, M.L.; Wang, D.; Khan, W.N. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J. Immunol. 2007, 179, 3872–3880. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Rip, J.; de Bruijn, M.J.W.; Kaptein, A.; Hendriks, R.W.; Corneth, O.B.J. Phosphoflow Protocol for Signaling Studies in Human and Murine B Cell Subpopulations. J. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rip, J.; Hendriks, R.W.; Corneth, O.B.J. A Versatile Protocol to Quantify BCR-mediated Phosphorylation in Human and Murine B Cell Subpopulations. Bio-Protocol 2021, 11, e3902. [Google Scholar] [CrossRef] [PubMed]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Distler, J.H.W.; Györfi, A.-H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Metzler, G.; Wray-Dutra, M.; Jackson, S.W. Altered B cell signalling in autoimmunity. Nat. Rev. Immunol. 2017, 17, 421–436. [Google Scholar] [CrossRef] [Green Version]
- Khan, W.N. B Cell Receptor and BAFF Receptor Signaling Regulation of B Cell Homeostasis. J. Immunol. 2009, 183, 3561–3567. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [Green Version]
- Hilberg, F.; Tontsch-Grunt, U.; Baum, A.; Le, A.T.; Doebele, R.C.; Lieb, S.; Gianni, D.; Voss, T.; Garin-Chesa, P.; Haslinger, C.; et al. Triple Angiokinase Inhibitor Nintedanib Directly Inhibits Tumor Cell Growth and Induces Tumor Shrinkage via Blocking Oncogenic Receptor Tyrosine Kinases. J. Pharmacol. Exp. Ther. 2018, 364, 494–503. [Google Scholar] [CrossRef]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant autoantibody production by early human B cell precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.J.; Ford, B.R.; Rihanek, M.; Coleman, B.M.; Getahun, A.; Sarapura, V.D.; Gottlieb, P.A.; Cambier, J.C. Elevated PTEN expression maintains anergy in human B cells and reveals unexpectedly high repertoire autoreactivity. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef]
- Thien, M.; Phan, T.G.; Gardam, S.; Amesbury, M.; Basten, A.; Mackay, F.; Brink, R. Excess BAFF Rescues Self-Reactive B Cells from Peripheral Deletion and Allows Them to Enter Forbidden Follicular and Marginal Zone Niches. Immunity 2004, 20, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesley, R.; Xu, Y.; Kalled, S.L.; Hess, D.M.; Schwab, S.R.; Shu, H.-B.; Cyster, J.G. Reduced Competitiveness of Autoantigen-Engaged B Cells due to Increased Dependence on BAFF. Immunity 2004, 20, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Fanny, M.; Gombault, A.; Francois, F.; Villeret, B.; Ryffel, B.; Wachhsmann, D.; Gottenberg, J.-E.; Couillin, I. B-cell activating factor regulates IL-1β- and IL-17A-mediated pulmonary fibrosis in mice. Rev. Mal. Respir. 2015, 32, 311. [Google Scholar] [CrossRef]
- François, A.; Gombault, A.; Villeret, B.; Alsaleh, G.; Fanny, M.; Gasse, P.; Adam, S.M.; Crestani, B.; Sibilia, J.; Schneider, P.; et al. B cell activating factor is central to bleomycin- and IL-17-mediated experimental pulmonary fibrosis. J. Autoimmun. 2015, 56, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figgett, W.A.; Deliyanti, D.; Fairfax, K.A.; Quah, P.S.; Wilkinson-Berka, J.L.; Mackay, F. Deleting the BAFF receptor TACI protects against systemic lupus erythematosus without extensive reduction of B cell numbers. J. Autoimmun. 2015, 61, 9–16. [Google Scholar] [CrossRef]
- Jacobs, H.M.; Thouvenel, C.D.; Leach, S.; Arkatkar, T.; Metzler, G.; Scharping, N.E.; Kolhatkar, N.S.; Rawlings, D.J.; Jackson, S.W. Cutting Edge: BAFF Promotes Autoantibody Production via TACI-Dependent Activation of Transitional B Cells. J. Immunol. 2016, 196, 3525–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, J.; Zhang, Y.-M.; Zhang, X.-M.; Tao, J. Effect of TACI Signaling on Humoral Immunity and Autoimmune Diseases. J. Immunol. Res. 2015, 2015, 247426. [Google Scholar] [CrossRef]
- Tsuchida, Y.; Sumitomo, S.; Ishigaki, K.; Suzuki, A.; Kochi, Y.; Tsuchiya, H.; Ota, M.; Komai, T.; Inoue, M.; Morita, K.; et al. TGF-β3 Inhibits Antibody Production by Human B Cells. PLoS ONE 2017, 12, e0169646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roes, J.; Choi, B.K.; Cazac, B.B. Redirection of B cell responsiveness by transforming growth factor beta receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 7241–7246. [Google Scholar] [CrossRef] [Green Version]
- Yong, S.J.; Adlakha, A.; Limper, A.H. Circulating transforming growth factor-beta(1): A potential marker of disease activity during idiopathic pulmonary fibrosis. Chest 2001, 120, 68S–70S. [Google Scholar] [CrossRef]
- Bergeron, A.; Soler, P.; Kambouchner, M.; Loiseau, P.; Milleron, B.; Valeyre, D.; Hance, A.J.; Tazi, A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-β and IL-10. Eur. Respir. J. 2003, 22, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhamad, E.H.; Cal, J.G.; Shakoor, Z.; Almogren, A.; AlBoukai, A.A. Cytokine gene polymorphisms and serum cytokine levels in patients with idiopathic pulmonary fibrosis. BMC Med. Genet. 2013, 14, 66. [Google Scholar] [CrossRef] [Green Version]
- Guiot, J.; Bondue, B.; Henket, M.; Corhay, J.L.; Louis, R. Raised serum levels of IGFBP-1 and IGFBP-2 in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2016, 16, 86. [Google Scholar] [CrossRef]
- Rangarajan, S.; Locy, M.L.; Luckhardt, T.R.; Thannickal, V.J. Targeted Therapy for Idiopathic Pulmonary Fibrosis: Where To Now? Drugs 2016, 76, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef]
- Wollin, L.; Distler, J.H.W.; Redente, E.F.; Riches, D.W.H.; Stowasser, S.; Schlenker-Herceg, R.; Maher, T.M.; Kolb, M. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019, 54, 1900161. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Huang, T.-T.; Chu, P.-Y.; Huang, C.-T.; Lee, C.-H.; Wang, W.-L.; Lau, K.-Y.; Tsai, W.-C.; Chao, T.-I.; Su, J.-C.; et al. The tyrosine kinase inhibitor nintedanib activates SHP-1 and induces apoptosis in triple-negative breast cancer cells. Exp. Mol. Med. 2017, 49, e366. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Ostermann, A.; Williams, C. Nintedanib inhibits pro-fibrotic mediators from T cells with relevance to connective tissue disease-associated interstitial lung disease. Eur. Respir. J. 2017, 50, PA903. [Google Scholar] [CrossRef]
- Feghali-Bostwick, C.A.; Tsai, C.G.; Valentine, V.G.; Kantrow, S.; Stoner, M.W.; Pilewski, J.M.; Gadgil, A.; George, M.P.; Gibson, K.F.; Choi, A.M.K.; et al. Cellular and Humoral Autoreactivity in Idiopathic Pulmonary Fibrosis. J. Immunol. 2007, 179, 2592–2599. [Google Scholar] [CrossRef] [Green Version]
- Labirua, A.; Lundberg, I.E. Interstitial lung disease and idiopathic inflammatory myopathies: Progress and pitfalls. Curr. Opin. Rheumatol. 2010, 22, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Prednisone, Azathioprine, and N-Acetylcysteine for Pulmonary Fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [CrossRef]
- Saunders, P.; Tsipouri, V.; Keir, G.J.; Ashby, D.; Flather, M.D.; Parfrey, H.; Babalis, D.; Renzoni, E.A.; Denton, C.P.; Wells, A.U.; et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): Study protocol for a randomised controlled trial. Trials 2017, 18, 275. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Huang, B.; Yang, Y.; Qi, M.; Lu, G.; Xia, D.; Li, H. Ibrutinib Exacerbates Bleomycin-Induced Pulmonary Fibrosis via Promoting Inflammation. Inflammation 2018, 41, 904–913. [Google Scholar] [CrossRef]
- Sun, B.; Liu, X.; Zheng, X.; Wang, C.; Meng, Q.; Sun, H.; Shu, X.; Liu, K.; Sun, X.; Li, Y.; et al. Novel Pyrimidines as Multitarget Protein Tyrosine Kinase Inhibitors for the Treatment of Idiopathic Pulmonary Fibrosis (IPF). ChemMedChem 2020, 15, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Neys, S.F.H.; Hendriks, R.W.; Corneth, O.B.J. Targeting Bruton’s Tyrosine Kinase in Inflammatory and Autoimmune Pathologies. Front. Cell Dev. Biol. 2021, 9, 668131. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IPF Cohort 1 | IPF Cohort 2 | |
|---|---|---|
| subjects, n | 16 | 12 |
| age (years), mean (±SD) | 68.5 (±7.2) | 69.1 (±7.7) |
| male sex, n | 10 (63%) | 9 (75%) |
| time to diagnosis (years), mean (min–max) | 3.1 (0.1–12.0) | 1.8 (0.2–7.0) |
| CT diagnosis, definite UIP/probable UIP/inconsistent * | 8/6/2 | 5/6/1 |
| smoking, active/previous/never | 0/15/1 | 1/11/0 |
| PY (years), mean (min–max) * | 23.0 (0–80) | 24.8 (1–60) |
| FVC (L), mean (±SD) * | 2.8 (±0.8) | 2.5 (±1.0) |
| FVC % predicted, mean (±SD) | 80.1 (±14.1) | 75.3 (±22.9) |
| Tiffeneau index, mean (±SD) | 82.9 (±5.4) | 77.3 (±11.2) |
| prednisone, n | 0 | 0 |
| nintedanib, n | 0 | 0 |
| pirfenidone, n | 0 | 10 (83%) |
| time between last intake of medication and starting nintedanib (weeks), mean (±SD) | - | 2.3 (±2.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neys, S.F.H.; Heukels, P.; van Hulst, J.A.C.; Rip, J.; Wijsenbeek, M.S.; Hendriks, R.W.; Corneth, O.B.J. Aberrant B Cell Receptor Signaling in Naïve B Cells from Patients with Idiopathic Pulmonary Fibrosis. Cells 2021, 10, 1321. https://doi.org/10.3390/cells10061321
Neys SFH, Heukels P, van Hulst JAC, Rip J, Wijsenbeek MS, Hendriks RW, Corneth OBJ. Aberrant B Cell Receptor Signaling in Naïve B Cells from Patients with Idiopathic Pulmonary Fibrosis. Cells. 2021; 10(6):1321. https://doi.org/10.3390/cells10061321
Chicago/Turabian StyleNeys, Stefan F. H., Peter Heukels, Jennifer A. C. van Hulst, Jasper Rip, Marlies S. Wijsenbeek, Rudi W. Hendriks, and Odilia B. J. Corneth. 2021. "Aberrant B Cell Receptor Signaling in Naïve B Cells from Patients with Idiopathic Pulmonary Fibrosis" Cells 10, no. 6: 1321. https://doi.org/10.3390/cells10061321
APA StyleNeys, S. F. H., Heukels, P., van Hulst, J. A. C., Rip, J., Wijsenbeek, M. S., Hendriks, R. W., & Corneth, O. B. J. (2021). Aberrant B Cell Receptor Signaling in Naïve B Cells from Patients with Idiopathic Pulmonary Fibrosis. Cells, 10(6), 1321. https://doi.org/10.3390/cells10061321