
Negative Regulation of FGFR (Fibroblast Growth Factor Receptor) Signaling

 , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
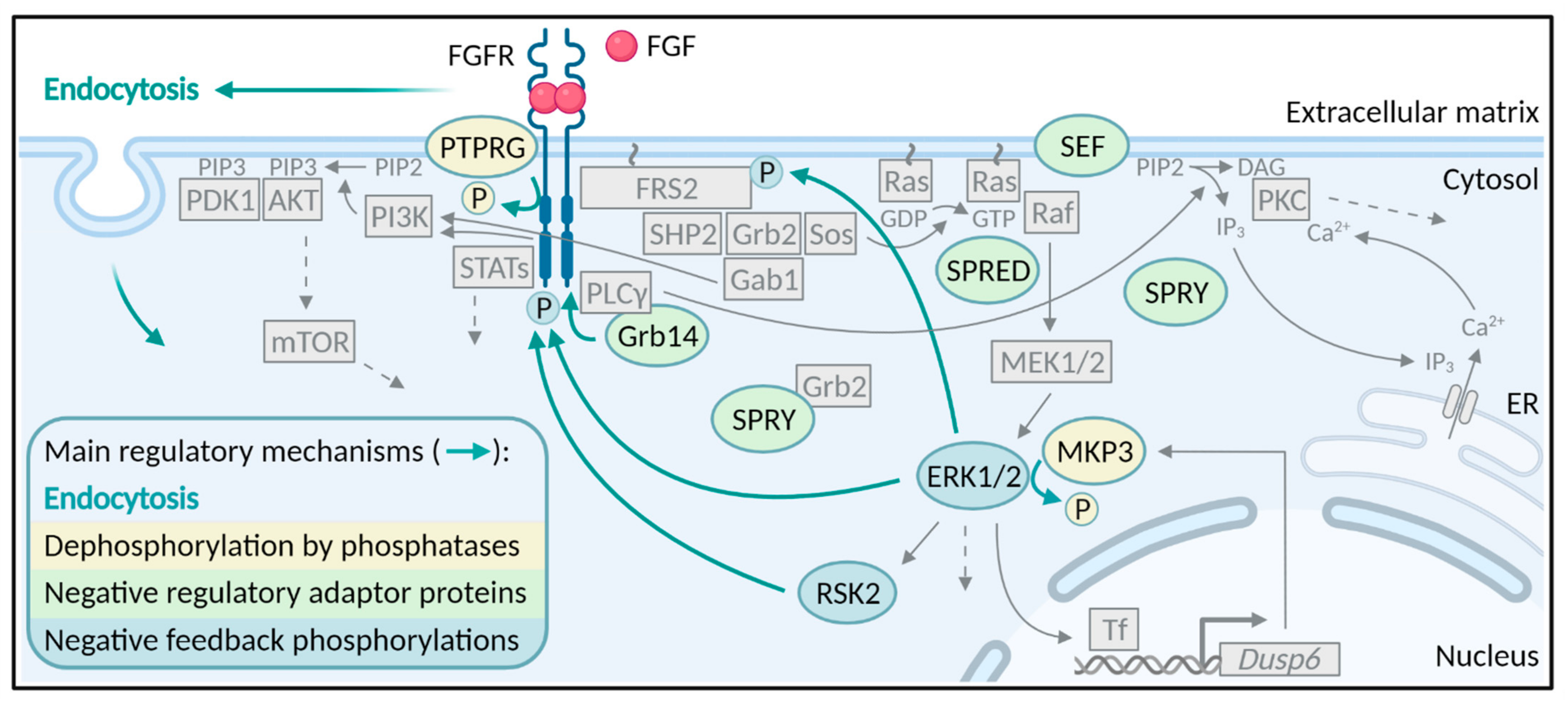
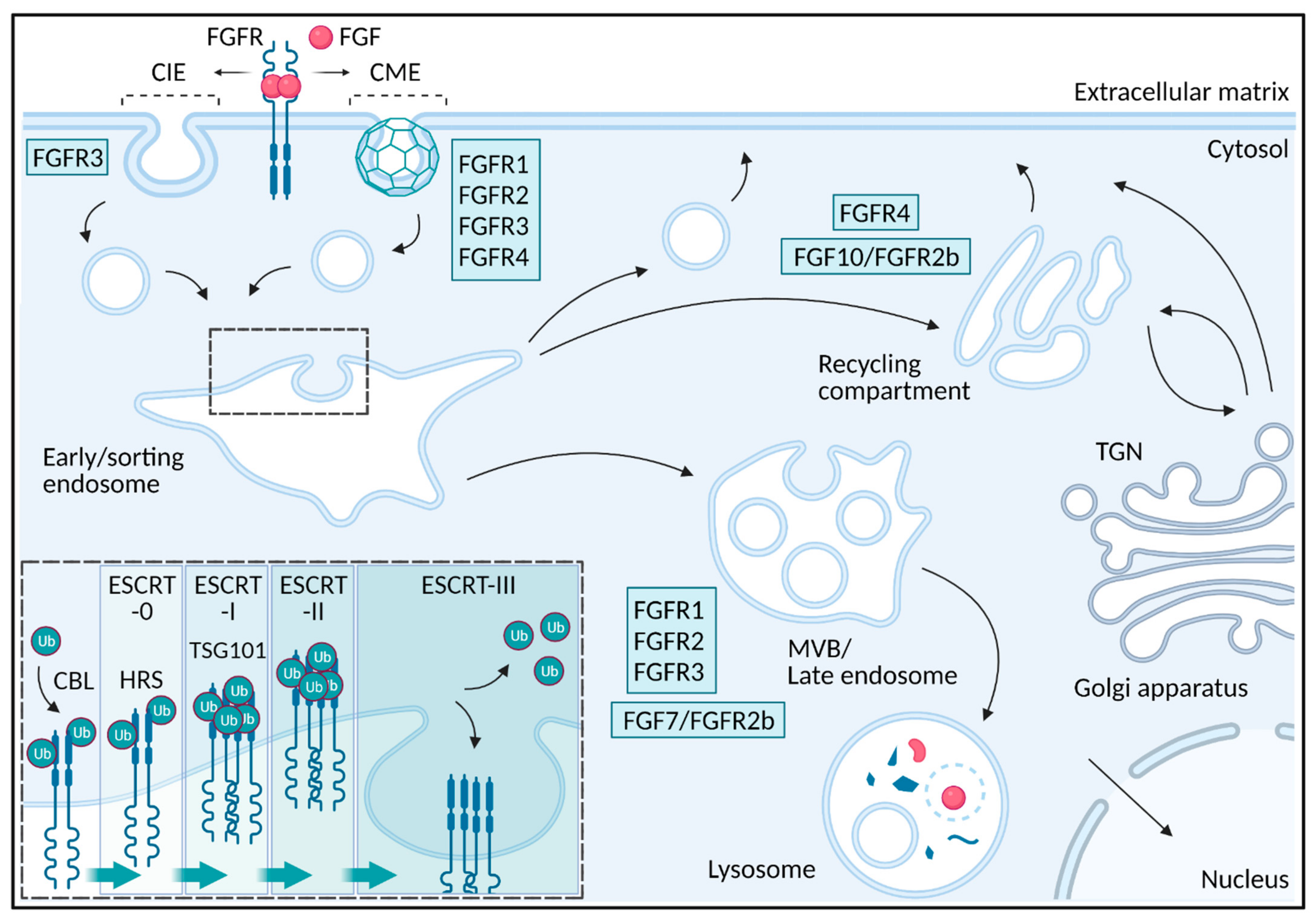
2. Endocytosis
3. Phosphatases
4. Negative Regulatory Proteins
5. Negative Feedback Phosphorylations
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP2 | Adaptor protein 2 |
| CBL | Casitas B-lineage lymphoma |
| CHIP | Carboxyl terminus of HSP70-interacting protein |
| CIE | Clathrin-independent endocytosis |
| CK1 | Casein kinase 1 |
| CLIC/GEEC | Clathrin-independent carriers/GPI-enriched early endosomal compartments |
| CME | Clathrin-mediated endocytosis |
| DUSP6 | Dual-specificity phosphatase 6 |
| DYRK1A | Dual-specificity tyrosine phosphorylation-regulated kinase 1A |
| Eps | Epidermal growth factor receptor kinase substrate |
| ERK 1/2 | Extracellular signal-regulated kinase 1/2 |
| ESCRT | Endosomal sorting complex required for transport |
| Esyt2 | Extended synaptotagmin-2 |
| FEME | Fast endophilin-mediated endocytosis |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FRS2 | FGFR substrate 2 |
| Grb2/14 | Growth factor receptor-bound 2/14 |
| HIF | Hypoxia-inducible factor |
| HRS | Hepatocyte growth factor-regulated tyrosine kinase substrate |
| Hsp90 | Heat shock protein 90 |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen-activated protein kinase kinase |
| MKP3 | MAPK phosphatase 3 |
| NBR1 | Neighbor of BRCA1 gene 1 protein |
| NCAM | Neural cell adhesion molecules |
| NEDD4 | Neural precursor cell expressed developmentally down-regulated protein 4 |
| NEGR1 | Neuronal growth regulator 1NF-1 Neurofibromin |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PKC | Protein kinase C |
| PLCγ | Phospholipase Cγ |
| PP2A | Protein phosphatase 2A |
| PTB | Phosphotyrosine binding |
| PTP | Protein tyrosine phosphatase |
| PTPN1/2/11 | Protein tyrosine phosphatase non-receptor type 1/2/11 |
| PTPRG | Protein tyrosine phosphatase receptor type G |
| RasGAP | Ras GTPase activating protein |
| RSK2 | Ribosomal s6 kinase 2 |
| RTK | Receptor tyrosine kinase |
| SEF | Similar expression to Fgf |
| SH3BP4 | SH3-binding protein 4 |
| SHIP2 | Phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase |
| SHP1/2 | Src homology region 2 domain-containing phosphatase 1/2 |
| Sos1 | Son of sevenless homolog 1 |
| SPRED | Sprouty related with EVH1 (Ena/VASP homology 1) |
| SPRY | Sprouty |
| STAM 1 | Signal transducing adaptor molecule 1 |
| STAT | Signal transducer and activator of transcription |
| TESK1 | Testicular protein kinase 1 |
| TGN | Trans Golgi network |
| TSG101 | Tumor susceptibility gene 101 protein |
| VHL | von Hippel-Lindau protein |
| VPS4 | Vacuolar protein sorting-associated protein 4 |
References
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murzin, A.G.; Lesk, A.M.; Chothia, C. beta-Trefoil fold. Patterns of structure and sequence in the Kunitz inhibitors interleukins-1 beta and 1 alpha and fibroblast growth factors. J. Mol. Biol. 1992, 223, 531–543. [Google Scholar] [CrossRef]
- Blaber, M.; DiSalvo, J.; Thomas, K.A. X-ray crystal structure of human acidic fibroblast growth factor. Biochemistry 1996, 35, 2086–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orengo, C.A.; Jones, D.T.; Thornton, J.M. Protein superfamilies and domain superfolds. Nature 1994, 372, 631–634. [Google Scholar] [CrossRef]
- Bertrand, S.; Iwema, T.; Escriva, H. FGF signaling emerged concomitantly with the origin of Eumetazoans. Mol. Biol. Evol. 2014, 31, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Philippe, H.; Derelle, R.; Lopez, P.; Pick, K.; Borchiellini, C.; Boury-Esnault, N.; Vacelet, J.; Renard, E.; Houliston, E.; Quéinnec, E.; et al. Phylogenomics revives traditional views on deep animal relationships. Curr. Biol. 2009, 19, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Duan, D.S.; de Vries, C.; Peters, K.G.; Johnson, D.E.; Williams, L.T. Differential splicing in the extracellular region of fibroblast growth factor receptor 1 generates receptor variants with different ligand-binding specificities. Mol. Cell. Biol. 1992, 12, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 1993, 60, 1–41. [Google Scholar]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Thisse, B.; Thisse, C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev. Biol. 2005, 287, 390–402. [Google Scholar] [CrossRef]
- Tulin, S.; Stathopoulos, A. Extending the family table: Insights from beyond vertebrates into the regulation of embryonic development by FGFs. Birth Defects Res. Part C Embryo Today Rev. 2010, 90, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Popovici, C.; Roubin, R.; Coulier, F.; Birnbaum, D. An evolutionary history of the FGF superfamily. Bioessays 2005, 27, 849–857. [Google Scholar] [CrossRef]
- Brewer, J.R.; Mazot, P.; Soriano, P. Genetic insights into the mechanisms of Fgf signaling. Genes Dev. 2016, 30, 751–771. [Google Scholar] [CrossRef] [Green Version]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef]
- Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Church, D.M.; Fielder, T.J.; Bocian, M.; Winokur, S.T.; Wasmuth, J.J. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 1994, 78, 335–342. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Dikic, I.; Giordano, S. Negative receptor signalling. Curr. Opin. Cell Biol. 2003, 15, 128–135. [Google Scholar] [CrossRef]
- Jeger, J.L. Endosomes, lysosomes, and the role of endosomal and lysosomal biogenesis in cancer development. Mol. Biol. Rep. 2020, 47, 9801–9810. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Renard, H.F.; Boucrot, E. Unconventional endocytic mechanisms. Curr. Opin. Cell Biol. 2021, 71, 120–129. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Malecki, J.; Bjorklund, S.M.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef] [Green Version]
- Haugsten, E.M.; Zakrzewska, M.; Brech, A.; Pust, S.; Olsnes, S.; Sandvig, K.; Wesche, J. Clathrin- and dynamin-independent endocytosis of FGFR3—Implications for signalling. PLoS ONE 2011, 6, e21708. [Google Scholar] [CrossRef] [Green Version]
- Haugsten, E.M.; Sorensen, V.; Kunova Bosakova, M.; de Souza, G.A.; Krejci, P.; Wiedlocha, A.; Wesche, J. Proximity Labeling Reveals Molecular Determinants of FGFR4 Endosomal Transport. J. Proteome Res. 2016, 15, 3841–3855. [Google Scholar] [CrossRef]
- Belleudi, F.; Leone, L.; Nobili, V.; Raffa, S.; Francescangeli, F.; Maggio, M.; Morrone, S.; Marchese, C.; Torrisi, M.R. Keratinocyte growth factor receptor ligands target the receptor to different intracellular pathways. Traffic 2007, 8, 1854–1872. [Google Scholar] [CrossRef]
- Marchese, C.; Mancini, P.; Belleudi, F.; Felici, A.; Gradini, R.; Sansolini, T.; Frati, L.; Torrisi, M.R. Receptor-mediated endocytosis of keratinocyte growth factor. J. Cell Sci. 1998, 111 Pt 23, 3517–3527. [Google Scholar] [CrossRef]
- Auciello, G.; Cunningham, D.L.; Tatar, T.; Heath, J.K.; Rappoport, J.Z. Regulation of fibroblast growth factor receptor signalling and trafficking by Src and Eps8. J. Cell Sci. 2013, 126, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieber, S.; Gigout, A. Sprifermin (recombinant human FGF18) is internalized through clathrin- and dynamin-independent pathways and degraded in primary chondrocytes. Exp. Cell Res. 2020, 395, 112236. [Google Scholar] [CrossRef] [PubMed]
- Citores, L.; Khnykin, D.; Sørensen, V.; Wesche, J.; Klingenberg, O.; Wiedłocha, A.; Olsnes, S. Modulation of intracellular transport of acidic fibroblast growth factor by mutations in the cytoplasmic receptor domain. J. Cell Sci. 2001, 114, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Liao, W.X.; Luo, Q.; Zhang, H.H.; Wang, W.; Zheng, J.; Chen, D.B. Caveolin-1 orchestrates fibroblast growth factor 2 signaling control of angiogenesis in placental artery endothelial cell caveolae. J. Cell. Physiol. 2012, 227, 2480–2491. [Google Scholar] [CrossRef] [Green Version]
- Boucrot, E.; Ferreira, A.P.; Almeida-Souza, L.; Debard, S.; Vallis, Y.; Howard, G.; Bertot, L.; Sauvonnet, N.; McMahon, H.T. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 2015, 517, 460–465. [Google Scholar] [CrossRef]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Giangreco, G.; Malabarba, M.G.; Sigismund, S. Specialised endocytic proteins regulate diverse internalisation mechanisms and signalling outputs in physiology and cancer. Biol. Cell. 2021, 113, 165–182. [Google Scholar] [CrossRef]
- Mettlen, M.; Chen, P.H.; Srinivasan, S.; Danuser, G.; Schmid, S.L. Regulation of Clathrin-Mediated Endocytosis. Annu. Rev. Biochem. 2018, 87, 871–896. [Google Scholar] [CrossRef]
- Muñoz, R.; Klingenberg, O.; Wiedłocha, A.; Rapak, A.; Falnes, P.O.; Olsnes, S. Effect of mutation of cytoplasmic receptor domain and of genistein on transport of acidic fibroblast growth factor into cells. Oncogene 1997, 15, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, A.; Mohammadi, M.; Huang, J.; Schlessinger, J. Internalization of fibroblast growth factor receptor is inhibited by a point mutation at tyrosine 766. J. Biol. Chem. 1994, 269, 17056–17061. [Google Scholar] [CrossRef]
- Ceridono, M.; Belleudi, F.; Ceccarelli, S.; Torrisi, M.R. Tyrosine 769 of the keratinocyte growth factor receptor is required for receptor signaling but not endocytosis. Biochem. Biophys. Res. Commun. 2005, 327, 523–532. [Google Scholar] [CrossRef]
- Nadratowska-Wesolowska, B.; Haugsten, E.M.; Zakrzewska, M.; Jakimowicz, P.; Zhen, Y.; Pajdzik, D.; Wesche, J.; Wiedlocha, A. RSK2 regulates endocytosis of FGF receptor 1 by phosphorylation on serine 789. Oncogene 2014, 33, 4823–4836. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.; Lamothe, B.; Lee, A.; Schlessinger, J.; Lax, I. FRS2 alpha attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc. Natl. Acad. Sci. USA 2002, 99, 6684–6689. [Google Scholar] [CrossRef] [Green Version]
- Persaud, A.; Alberts, P.; Hayes, M.; Guettler, S.; Clarke, I.; Sicheri, F.; Dirks, P.; Ciruna, B.; Rotin, D. Nedd4-1 binds and ubiquitylates activated FGFR1 to control its endocytosis and function. EMBO J. 2011, 30, 3259–3273. [Google Scholar] [CrossRef] [Green Version]
- Persaud, A.; Alberts, P.; Mari, S.; Tong, J.; Murchie, R.; Maspero, E.; Safi, F.; Moran, M.F.; Polo, S.; Rotin, D. Tyrosine phosphorylation of NEDD4 activates its ubiquitin ligase activity. Sci. Signal. 2014, 7, ra95. [Google Scholar] [CrossRef]
- Belleudi, F.; Visco, V.; Ceridono, M.; Leone, L.; Muraro, R.; Frati, L.; Torrisi, M.R. Ligand-induced clathrin-mediated endocytosis of the keratinocyte growth factor receptor occurs independently of either phosphorylation or recruitment of eps15. FEBS Lett. 2003, 553, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Jean, S.; Mikryukov, A.; Tremblay, M.G.; Baril, J.; Guillou, F.; Bellenfant, S.; Moss, T. Extended-synaptotagmin-2 mediates FGF receptor endocytosis and ERK activation in vivo. Dev. Cell 2010, 19, 426–439. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.; Adereth, Y.; Kose, N.; Dammai, V. Endocytic function of von Hippel-Lindau tumor suppressor protein regulates surface localization of fibroblast growth factor receptor 1 and cell motility. J. Biol. Chem. 2006, 281, 12069–12080. [Google Scholar] [CrossRef] [Green Version]
- Champion, K.J.; Guinea, M.; Dammai, V.; Hsu, T. Endothelial function of von Hippel-Lindau tumor suppressor gene: Control of fibroblast growth factor receptor signaling. Cancer Res. 2008, 68, 4649–4657. [Google Scholar] [CrossRef] [Green Version]
- Suyama, K.; Shapiro, I.; Guttman, M.; Hazan, R.B. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2002, 2, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Bryant, D.M.; Wylie, F.G.; Stow, J.L. Regulation of endocytosis, nuclear translocation, and signaling of fibroblast growth factor receptor 1 by E-cadherin. Mol. Biol. Cell 2005, 16, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Elfenbein, A.; Lanahan, A.; Zhou, T.X.; Yamasaki, A.; Tkachenko, E.; Matsuda, M.; Simons, M. Syndecan 4 regulates FGFR1 signaling in endothelial cells by directing macropinocytosis. Sci. Signal. 2012, 5, ra36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opaliński, Ł.; Sokołowska-Wędzina, A.; Szczepara, M.; Zakrzewska, M.; Otlewski, J. Antibody-induced dimerization of FGFR1 promotes receptor endocytosis independently of its kinase activity. Sci. Rep. 2017, 7, 7121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miaczynska, M. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a009035. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Brech, A.; Liestøl, K.; Norman, J.C.; Wesche, J. Photoactivation approaches reveal a role for Rab11 in FGFR4 recycling and signalling. Traffic 2014, 15, 665–683. [Google Scholar] [CrossRef] [Green Version]
- Porębska, N.; Latko, M.; Kucińska, M.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment. J. Clin. Med. 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Coleman, S.J.; Bruce, C.; Chioni, A.M.; Kocher, H.M.; Grose, R.P. The ins and outs of fibroblast growth factor receptor signalling. Clin. Sci. 2014, 127, 217–231. [Google Scholar] [CrossRef]
- Chioni, A.M.; Grose, R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J. Cell Biol. 2012, 197, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef]
- Lee, J.E.; Shin, S.-H.; Shin, H.-W.; Chun, Y.-S.; Park, J.-W. Nuclear FGFR2 negatively regulates hypoxia-induced cell invasion in prostate cancer by interacting with HIF-1 and HIF-2. Sci. Rep. 2019, 9, 3480. [Google Scholar] [CrossRef] [Green Version]
- Goh, L.K.; Sorkin, A. Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017459. [Google Scholar] [CrossRef] [Green Version]
- Haugsten, E.M.; Sorensen, V.; Brech, A.; Olsnes, S.; Wesche, J. Different intracellular trafficking of FGF1 endocytosed by the four homologous FGF receptors. J. Cell Sci. 2005, 118, 3869–3881. [Google Scholar] [CrossRef] [Green Version]
- Francavilla, C.; Rigbolt, K.T.; Emdal, K.B.; Carraro, G.; Vernet, E.; Bekker-Jensen, D.B.; Streicher, W.; Wikström, M.; Sundström, M.; Bellusci, S.; et al. Functional proteomics defines the molecular switch underlying FGF receptor trafficking and cellular outputs. Mol. Cell 2013, 51, 707–722. [Google Scholar] [CrossRef] [Green Version]
- Francavilla, C.; Cattaneo, P.; Berezin, V.; Bock, E.; Ami, D.; de Marco, A.; Christofori, G.; Cavallaro, U. The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J. Cell Biol. 2009, 187, 1101–1116. [Google Scholar] [CrossRef]
- Szczurkowska, J.; Pischedda, F.; Pinto, B.; Managò, F.; Haas, C.A.; Summa, M.; Bertorelli, R.; Papaleo, F.; Schäfer, M.K.; Piccoli, G.; et al. NEGR1 and FGFR2 cooperatively regulate cortical development and core behaviours related to autism disorders in mice. Brain 2018, 141, 2772–2794. [Google Scholar] [CrossRef]
- Bryant, D.M.; Stow, J.L. Nuclear translocation of cell-surface receptors: Lessons from fibroblast growth factor. Traffic 2005, 6, 947–954. [Google Scholar] [CrossRef]
- Olsnes, S.; Klingenberg, O.; Wiedłocha, A. Transport of exogenous growth factors and cytokines to the cytosol and to the nucleus. Physiol. Rev. 2003, 83, 163–182. [Google Scholar] [CrossRef] [Green Version]
- Polo, S. Signaling-mediated control of ubiquitin ligases in endocytosis. BMC Biol. 2012, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- McCullough, J.; Frost, A.; Sundquist, W.I. Structures, Functions, and Dynamics of ESCRT-III/Vps4 Membrane Remodeling and Fission Complexes. Annu. Rev. Cell Dev. Biol. 2018, 34, 85–109. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Kon, E.; Calvo-Jiménez, E.; Cossard, A.; Na, Y.; Cooper, J.A.; Jossin, Y. N-cadherin-regulated FGFR ubiquitination and degradation control mammalian neocortical projection neuron migration. eLife 2019, 8. [Google Scholar] [CrossRef]
- Belleudi, F.; Leone, L.; Maggio, M.; Torrisi, M.R. Hrs regulates the endocytic sorting of the fibroblast growth factor receptor 2b. Exp. Cell Res. 2009, 315, 2181–2191. [Google Scholar] [CrossRef]
- Cho, J.Y.; Guo, C.; Torello, M.; Lunstrum, G.P.; Iwata, T.; Deng, C.; Horton, W.A. Defective lysosomal targeting of activated fibroblast growth factor receptor 3 in achondroplasia. Proc. Natl. Acad. Sci. USA 2004, 101, 609–614. [Google Scholar] [CrossRef] [Green Version]
- Bonaventure, J.; Horne, W.C.; Baron, R. The localization of FGFR3 mutations causing thanatophoric dysplasia type I differentially affects phosphorylation, processing and ubiquitylation of the receptor. FEBS J. 2007, 274, 3078–3093. [Google Scholar] [CrossRef]
- Monsonego-Ornan, E.; Adar, R.; Rom, E.; Yayon, A. FGF receptors ubiquitylation: Dependence on tyrosine kinase activity and role in downregulation. FEBS Lett. 2002, 528, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Dufour, C.; Guenou, H.; Kaabeche, K.; Bouvard, D.; Sanjay, A.; Marie, P.J. FGFR2-Cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone 2008, 42, 1032–1039. [Google Scholar] [CrossRef]
- Kaabeche, K.; Lemonnier, J.; Le Mee, S.; Caverzasio, J.; Marie, P.J. Cbl-mediated degradation of Lyn and Fyn induced by constitutive fibroblast growth factor receptor-2 activation supports osteoblast differentiation. J. Biol. Chem. 2004, 279, 36259–36267. [Google Scholar] [CrossRef] [Green Version]
- Sévère, N.; Miraoui, H.; Marie, P.J. The Casitas B lineage lymphoma (Cbl) mutant G306E enhances osteogenic differentiation in human mesenchymal stromal cells in part by decreased Cbl-mediated platelet-derived growth factor receptor alpha and fibroblast growth factor receptor 2 ubiquitination. J. Biol. Chem. 2011, 286, 24443–24450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, M.; Machate, A.; Yu, S.R.; Gupta, M.; Brand, M. Interpretation of the FGF8 morphogen gradient is regulated by endocytic trafficking. Nat. Cell Biol. 2011, 13, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Laederich, M.B.; Degnin, C.R.; Lunstrum, G.P.; Holden, P.; Horton, W.A. Fibroblast growth factor receptor 3 (FGFR3) is a strong heat shock protein 90 (Hsp90) client: Implications for therapeutic manipulation. J. Biol. Chem. 2011, 286, 19597–19604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degnin, C.R.; Laederich, M.B.; Horton, W.A. Ligand activation leads to regulated intramembrane proteolysis of fibroblast growth factor receptor 3. Mol. Biol. Cell 2011, 22, 3861–3873. [Google Scholar] [CrossRef] [PubMed]
- Hausott, B.; Förste, A.; Zach, F.; Mangger, S.; Haugsten, E.M.; Klimaschewski, L. Endocytosis and Transport of Growth Factor Receptors in Peripheral Axon Regeneration: Novel Lessons from Neurons Expressing Lysine-Deficient FGF Receptor Type 1 in vitro. Anat. Rec. 2019, 302, 1268–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausott, B.; Rietzler, A.; Vallant, N.; Auer, M.; Haller, I.; Perkhofer, S.; Klimaschewski, L. Inhibition of fibroblast growth factor receptor 1 endocytosis promotes axonal branching of adult sensory neurons. Neuroscience 2011, 188, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Scholpp, S.; Brand, M. Endocytosis controls spreading and effective signaling range of Fgf8 protein. Curr. Biol. 2004, 14, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Tonks, N.K. Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostas, M.; Haugsten, E.M.; Zhen, Y.; Sorensen, V.; Szybowska, P.; Fiorito, E.; Lorenz, S.; Jones, N.; de Souza, G.A.; Wiedlocha, A.; et al. Protein Tyrosine Phosphatase Receptor Type G (PTPRG) Controls Fibroblast Growth Factor Receptor (FGFR) 1 Activity and Influences Sensitivity to FGFR Kinase Inhibitors. Mol. Cell. Proteom. 2018, 17, 850–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Germain, J.R.; Taylor, P.; Zhang, W.; Li, Z.; Ketela, T.; Moffat, J.; Neel, B.G.; Trudel, S.; Moran, M.F. Differential regulation of FGFR3 by PTPN1 and PTPN2. Proteomics 2015, 15, 419–433. [Google Scholar] [CrossRef]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Matsumoto, K.; Nishida, E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J. Biol. Chem. 2004, 279, 22992–22995. [Google Scholar] [CrossRef] [Green Version]
- Hadari, Y.R.; Kouhara, H.; Lax, I.; Schlessinger, J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol. Cell. Biol. 1998, 18, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Fafilek, B.; Balek, L.; Bosakova, M.K.; Varecha, M.; Nita, A.; Gregor, T.; Gudernova, I.; Krenova, J.; Ghosh, S.; Piskacek, M.; et al. The inositol phosphatase SHIP2 enables sustained ERK activation downstream of FGF receptors by recruiting Src kinases. Sci. Signal. 2018, 11, eaap8608. [Google Scholar] [CrossRef] [Green Version]
- Camps, M.; Nichols, A.; Gillieron, C.; Antonsson, B.; Muda, M.; Chabert, C.; Boschert, U.; Arkinstall, S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 1998, 280, 1262–1265. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Scott, D.A.; Hatch, E.; Tian, X.; Mansour, S.L. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development 2007, 134, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Eblaghie, M.C.; Lunn, J.S.; Dickinson, R.J.; Münsterberg, A.E.; Sanz-Ezquerro, J.J.; Farrell, E.R.; Mathers, J.; Keyse, S.M.; Storey, K.; Tickle, C. Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr. Biol. 2003, 13, 1009–1018. [Google Scholar] [CrossRef]
- Kawakami, Y.; Rodríguez-León, J.; Koth, C.M.; Büscher, D.; Itoh, T.; Raya, A.; Ng, J.K.; Esteban, C.R.; Takahashi, S.; Henrique, D.; et al. MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat. Cell Biol. 2003, 5, 513–519. [Google Scholar] [CrossRef]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J. 2008, 412, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.M.; Morrison, D.J.; Bassit, B.; Dimri, M.; Band, H.; Licht, J.D.; Gross, I. Tyrosine phosphorylation of Sprouty proteins regulates their ability to inhibit growth factor signaling: A dual feedback loop. Mol. Biol. Cell 2004, 15, 2176–2188. [Google Scholar] [CrossRef] [Green Version]
- Lake, D.; Correa, S.A.; Muller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Nishida, E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 2002, 4, 850–858. [Google Scholar] [CrossRef]
- Yusoff, P.; Lao, D.H.; Ong, S.H.; Wong, E.S.; Lim, J.; Lo, T.L.; Leong, H.F.; Fong, C.W.; Guy, G.R. Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. J. Biol. Chem. 2002, 277, 3195–3201. [Google Scholar] [CrossRef] [Green Version]
- Lao, D.H.; Yusoff, P.; Chandramouli, S.; Philp, R.J.; Fong, C.W.; Jackson, R.A.; Saw, T.Y.; Yu, C.Y.; Guy, G.R. Direct binding of PP2A to Sprouty2 and phosphorylation changes are a prerequisite for ERK inhibition downstream of fibroblast growth factor receptor stimulation. J. Biol. Chem. 2007, 282, 9117–9126. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Degnin, C.R.; Laederich, M.B.; Lunstrum, G.P.; Holden, P.; Bihlmaier, J.; Krakow, D.; Cho, Y.J.; Horton, W.A. Sprouty 2 disturbs FGFR3 degradation in thanatophoric dysplasia type II: A severe form of human achondroplasia. Cell. Signal. 2008, 20, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Harkins, L.K.; Zubanova, O.; Harrington, A.; Kovalenko, D.; Nadeau, R.J.; Chen, P.Y.; Toher, J.L.; Lindner, V.; Liaw, L.; et al. Overexpression of Spry1 in chondrocytes causes attenuated FGFR ubiquitination and sustained ERK activation resulting in chondrodysplasia. Dev. Biol. 2008, 321, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Yim, D.G.; Ghosh, S.; Guy, G.R.; Virshup, D.M. Casein kinase 1 regulates Sprouty2 in FGF-ERK signaling. Oncogene 2015, 34, 474–484. [Google Scholar] [CrossRef]
- Friedmacher, F.; Gosemann, J.H.; Fujiwara, N.; Alvarez, L.A.; Corcionivoschi, N.; Puri, P. Spatiotemporal alterations in Sprouty-2 expression and tyrosine phosphorylation in nitrofen-induced pulmonary hypoplasia. J. Pediatr. Surg. 2013, 48, 2219–2225. [Google Scholar] [CrossRef]
- Aranda, S.; Alvarez, M.; Turro, S.; Laguna, A.; de la Luna, S. Sprouty2-mediated inhibition of fibroblast growth factor signaling is modulated by the protein kinase DYRK1A. Mol. Cell. Biol. 2008, 28, 5899–5911. [Google Scholar] [CrossRef] [Green Version]
- Chandramouli, S.; Yu, C.Y.; Yusoff, P.; Lao, D.H.; Leong, H.F.; Mizuno, K.; Guy, G.R. Tesk1 interacts with Spry2 to abrogate its inhibition of ERK phosphorylation downstream of receptor tyrosine kinase signaling. J. Biol. Chem. 2008, 283, 1679–1691. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Miyazaki, S.; Tanimura, S.; Kohno, M. Efficient suppression of FGF-2-induced ERK activation by the cooperative interaction among mammalian Sprouty isoforms. J. Cell Sci. 2005, 118, 5861–5871. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, C.; McCormick, F. SPRED proteins and their roles in signal transduction, development, and malignancy. Genes Dev. 2020, 34, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- McClatchey, A.I.; Cichowski, K. SPRED proteins provide a NF-ty link to Ras suppression. Genes Dev. 2012, 26, 1515–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowe, I.B.; Mercado, E.L.; Stowe, T.R.; Bell, E.L.; Oses-Prieto, J.A.; Hernández, H.; Burlingame, A.L.; McCormick, F. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012, 26, 1421–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakioka, T.; Sasaki, A.; Kato, R.; Shouda, T.; Matsumoto, A.; Miyoshi, K.; Tsuneoka, M.; Komiya, S.; Baron, R.; Yoshimura, A. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001, 412, 647–651. [Google Scholar] [CrossRef]
- Bundschu, K.; Knobeloch, K.P.; Ullrich, M.; Schinke, T.; Amling, M.; Engelhardt, C.M.; Renne, T.; Walter, U.; Schuh, K. Gene disruption of Spred-2 causes dwarfism. J. Biol. Chem. 2005, 280, 28572–28580. [Google Scholar] [CrossRef] [Green Version]
- Mardakheh, F.K.; Yekezare, M.; Machesky, L.M.; Heath, J.K. Spred2 interaction with the late endosomal protein NBR1 down-regulates fibroblast growth factor receptor signaling. J. Cell Biol. 2009, 187, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Lock, P.; Stacey, T.T.; Straffon, A.F.; Schieb, H.; Hovens, C.M.; Stylli, S.S. Spred-2 steady-state levels are regulated by phosphorylation and Cbl-mediated ubiquitination. Biochem. Biophys. Res. Commun. 2006, 351, 1018–1023. [Google Scholar] [CrossRef]
- Meng, S.; Zhang, M.; Pan, W.; Li, Z.; Anderson, D.H.; Zhang, S.; Ge, B.; Wang, C. Tyrosines 303/343/353 within the Sprouty-related domain of Spred2 are essential for its interaction with p85 and inhibitory effect on Ras/ERK activation. Int. J. Biochem. Cell Biol. 2012, 44, 748–758. [Google Scholar] [CrossRef]
- Furthauer, M.; Lin, W.; Ang, S.L.; Thisse, B.; Thisse, C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 2002, 4, 170–174. [Google Scholar] [CrossRef]
- Torii, S.; Nakayama, K.; Yamamoto, T.; Nishida, E. Regulatory mechanisms and function of ERK MAP kinases. J. Biochem. 2004, 136, 557–561. [Google Scholar] [CrossRef]
- Darby, S.; Murphy, T.; Thomas, H.; Robson, C.N.; Leung, H.Y.; Mathers, M.E.; Gnanapragasam, V.J. Similar expression to FGF (Sef) inhibits fibroblast growth factor-induced tumourigenic behaviour in prostate cancer cells and is downregulated in aggressive clinical disease. Br. J. Cancer 2009, 101, 1891–1899. [Google Scholar] [CrossRef]
- Ren, Y.; Cheng, L.; Rong, Z.; Li, Z.; Li, Y.; Li, H.; Wang, Z.; Chang, Z. hSef co-localizes and interacts with Ras in the inhibition of Ras/MAPK signaling pathway. Biochem. Biophys. Res. Commun. 2006, 347, 988–993. [Google Scholar] [CrossRef]
- Kovalenko, D.; Yang, X.; Nadeau, R.J.; Harkins, L.K.; Friesel, R. Sef inhibits fibroblast growth factor signaling by inhibiting FGFR1 tyrosine phosphorylation and subsequent ERK activation. J. Biol. Chem. 2003, 278, 14087–14091. [Google Scholar] [CrossRef] [Green Version]
- Ziv, I.; Fuchs, Y.; Preger, E.; Shabtay, A.; Harduf, H.; Zilpa, T.; Dym, N.; Ron, D. The human sef-a isoform utilizes different mechanisms to regulate receptor tyrosine kinase signaling pathways and subsequent cell fate. J. Biol. Chem. 2006, 281, 39225–39235. [Google Scholar] [CrossRef] [Green Version]
- Newitt, P.; Boros, J.; Madakashira, B.P.; Robinson, M.L.; Reneker, L.W.; McAvoy, J.W.; Lovicu, F.J. Sef is a negative regulator of fiber cell differentiation in the ocular lens. Differentiation 2010, 80, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Kovalenko, D.; Yang, X.; Chen, P.Y.; Nadeau, R.J.; Zubanova, O.; Pigeon, K.; Friesel, R. A role for extracellular and transmembrane domains of Sef in Sef-mediated inhibition of FGF signaling. Cell. Signal. 2006, 18, 1958–1966. [Google Scholar] [CrossRef]
- Kilkenny, D.M.; Rocheleau, J.V. Chapter Two—The FGF21 Receptor Signaling Complex: Klothoβ, FGFR1c, and Other Regulatory Interactions. Vitam. Horm. 2016, 101, 17–58. [Google Scholar]
- Silva, P.N.; Altamentova, S.M.; Kilkenny, D.M.; Rocheleau, J.V. Fibroblast growth factor receptor like-1 (FGFRL1) interacts with SHP-1 phosphatase at insulin secretory granules and induces beta-cell ERK1/2 protein activation. J. Biol. Chem. 2013, 288, 17859–17870. [Google Scholar] [CrossRef] [Green Version]
- Tomasovic, A.; Traub, S.; Tikkanen, R. Molecular networks in FGF signaling: Flotillin-1 and cbl-associated protein compete for the binding to fibroblast growth factor receptor substrate 2. PLoS ONE 2012, 7, e29739. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Melo, F.A.; Ghosh, R.; Suen, K.M.; Stagg, L.J.; Kirkpatrick, J.; Arold, S.T.; Ahmed, Z.; Ladbury, J.E. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell 2012, 149, 1514–1524. [Google Scholar] [CrossRef] [Green Version]
- Reilly, J.F.; Mickey, G.; Maher, P.A. Association of fibroblast growth factor receptor 1 with the adaptor protein Grb14. Characterization of a new receptor binding partner. J. Biol. Chem. 2000, 275, 7771–7778. [Google Scholar] [CrossRef]
- Mohammadi, M.; Honegger, A.M.; Rotin, D.; Fischer, R.; Bellot, F.; Li, W.; Dionne, C.A.; Jaye, M.; Rubinstein, M.; Schlessinger, J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol. Cell. Biol. 1991, 11, 5068–5078. [Google Scholar] [CrossRef] [Green Version]
- Cailliau, K.; Perdereau, D.; Lescuyer, A.; Chen, H.; Garbay, C.; Vilain, J.P.; Burnol, A.F.; Browaeys-Poly, E. FGF receptor phosphotyrosine 766 is a target for Grb14 to inhibit MDA-MB-231 human breast cancer cell signaling. Anticancer Res. 2005, 25, 3877–3882. [Google Scholar]
- Browaeys-Poly, E.; Blanquart, C.; Perdereau, D.; Antoine, A.F.; Goenaga, D.; Luzy, J.P.; Chen, H.; Garbay, C.; Issad, T.; Cailliau, K.; et al. Grb14 inhibits FGF receptor signaling through the regulation of PLCγ recruitment and activation. FEBS Lett. 2010, 584, 4383–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lax, I.; Wong, A.; Lamothe, B.; Lee, A.; Frost, A.; Hawes, J.; Schlessinger, J. The docking protein FRS2alpha controls a MAP kinase-mediated negative feedback mechanism for signaling by FGF receptors. Mol. Cell 2002, 10, 709–719. [Google Scholar] [CrossRef]
- Zakrzewska, M.; Haugsten, E.M.; Nadratowska-Wesolowska, B.; Oppelt, A.; Hausott, B.; Jin, Y.; Otlewski, J.; Wesche, J.; Wiedlocha, A. ERK-mediated phosphorylation of fibroblast growth factor receptor 1 on Ser777 inhibits signaling. Sci. Signal. 2013, 6, ra11. [Google Scholar] [CrossRef] [PubMed]
- Szybowska, P.; Kostas, M.; Wesche, J.; Wiedlocha, A.; Haugsten, E.M. Cancer Mutations in FGFR2 Prevent a Negative Feedback Loop Mediated by the ERK1/2 Pathway. Cells 2019, 8, 518. [Google Scholar] [CrossRef] [Green Version]
- Lonic, A.; Powell, J.A.; Kong, Y.; Thomas, D.; Holien, J.K.; Truong, N.; Parker, M.W.; Guthridge, M.A. Phosphorylation of serine 779 in fibroblast growth factor receptor 1 and 2 by protein kinase C(epsilon) regulates Ras/mitogen-activated protein kinase signaling and neuronal differentiation. J. Biol. Chem. 2013, 288, 14874–14885. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.Y.; Maddileti, S.; Mitin, N.; Harden, T.K.; Der, C.J. Aberrant receptor internalization and enhanced FRS2-dependent signaling contribute to the transforming activity of the fibroblast growth factor receptor 2 IIIb C3 isoform. J. Biol. Chem. 2009, 284, 6227–6240. [Google Scholar] [CrossRef] [Green Version]
- Moffa, A.B.; Tannheimer, S.L.; Ethier, S.P. Transforming potential of alternatively spliced variants of fibroblast growth factor receptor 2 in human mammary epithelial cells. Mol. Cancer Res. 2004, 2, 643–652. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szybowska, P.; Kostas, M.; Wesche, J.; Haugsten, E.M.; Wiedlocha, A. Negative Regulation of FGFR (Fibroblast Growth Factor Receptor) Signaling. Cells 2021, 10, 1342. https://doi.org/10.3390/cells10061342
Szybowska P, Kostas M, Wesche J, Haugsten EM, Wiedlocha A. Negative Regulation of FGFR (Fibroblast Growth Factor Receptor) Signaling. Cells. 2021; 10(6):1342. https://doi.org/10.3390/cells10061342
Chicago/Turabian StyleSzybowska, Patrycja, Michal Kostas, Jørgen Wesche, Ellen Margrethe Haugsten, and Antoni Wiedlocha. 2021. "Negative Regulation of FGFR (Fibroblast Growth Factor Receptor) Signaling" Cells 10, no. 6: 1342. https://doi.org/10.3390/cells10061342
APA StyleSzybowska, P., Kostas, M., Wesche, J., Haugsten, E. M., & Wiedlocha, A. (2021). Negative Regulation of FGFR (Fibroblast Growth Factor Receptor) Signaling. Cells, 10(6), 1342. https://doi.org/10.3390/cells10061342