
Inflammation, Aging and Hematopoiesis: A Complex Relationship

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammatory Signaling as a Key Regulator of HSC Homeostasis
3. Inflammation Affects HSC Homeostasis via the BM Niche
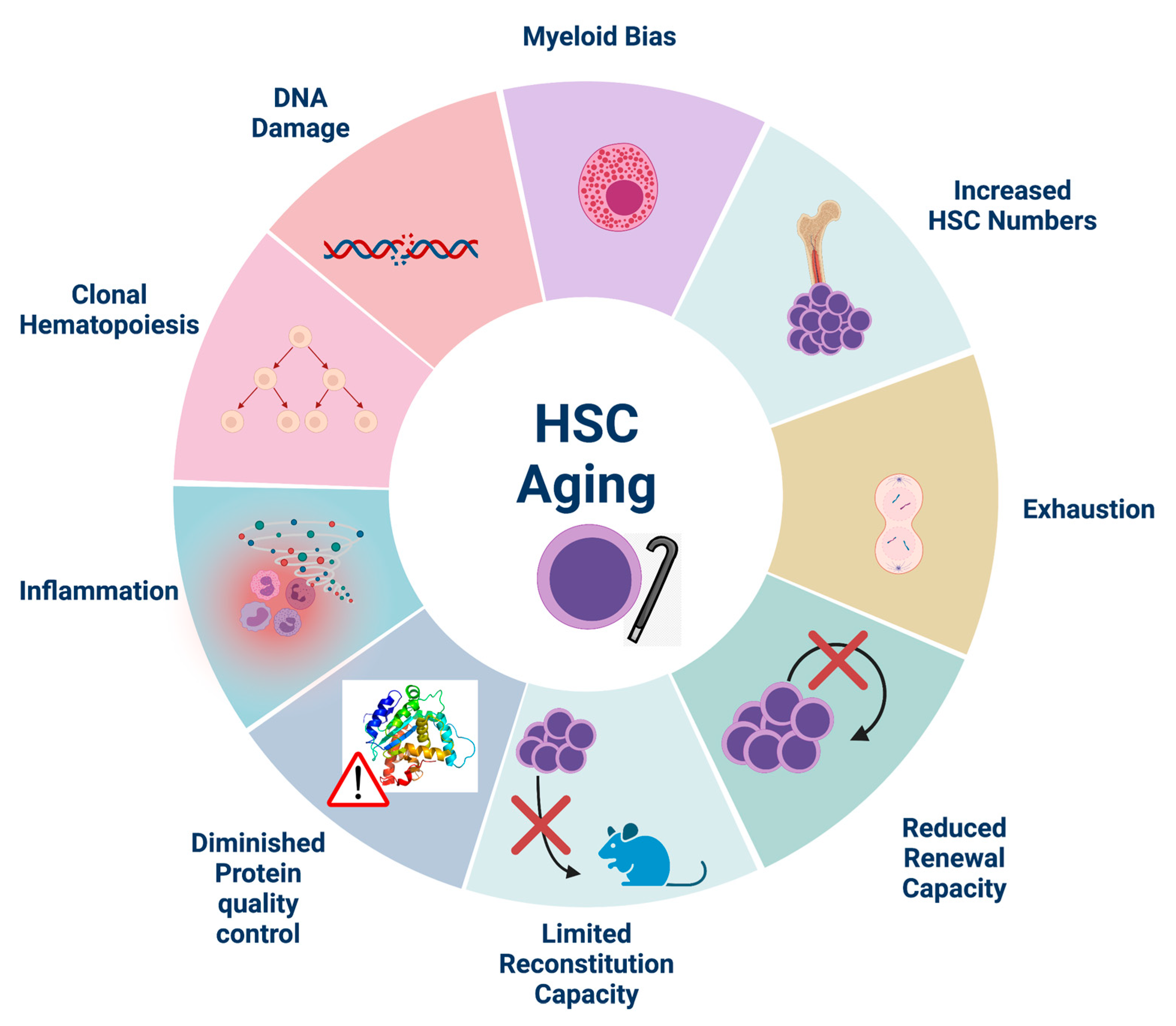
4. Inflammation and HSC Aging
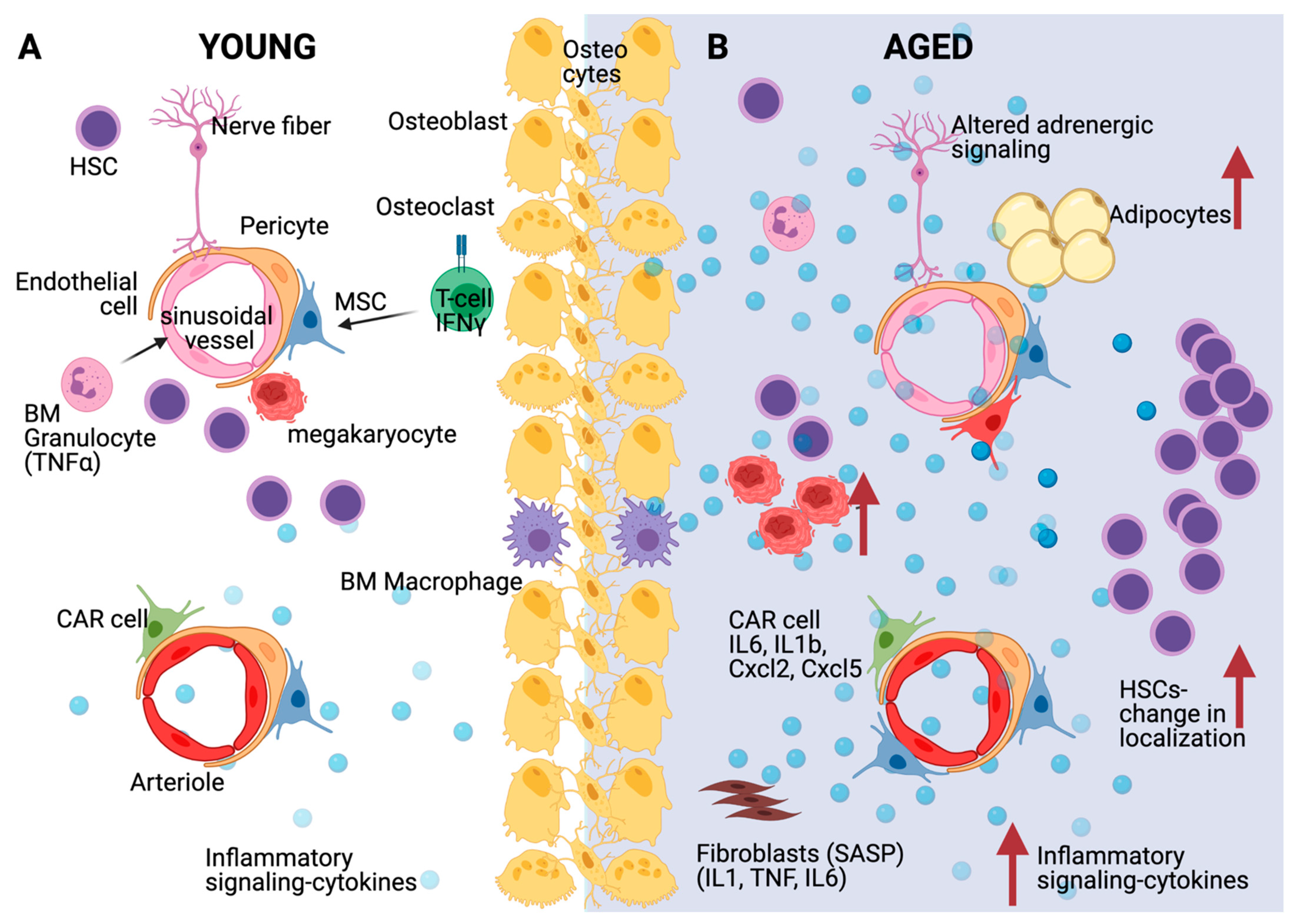
5. Inflammation Alters the Aged HSC Niche
6. Inflammation, Aging and Hematological Disorders
7. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGM | Aorta-Gonad-Mesonephros |
| AML | Acute Myeloid Leukemia |
| BM | Bone Marrow |
| CH | Clonal Hematopoiesis |
| CHIP | Clonal Hematopoiesis of Indeterminate Potential |
| CMA | Chaperone-Mediated Autophagy |
| CML | Chronic Myeloid Leukemia |
| FA | Fanconi Anemia |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| HSC | Hematopoietic Stem Cell |
| HSPC | Hematopoietic Stem and Progenitor Cell |
| IFN | Interferon |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| LT-HSC | Long Term Hematopoietic Stem Cell |
| MDS | Myelodysplastic Syndromes |
| MPN | Myeloproliferative Neoplasms |
| MPP | Multi-Potent Progenitor |
| MSC | Mesenchymal Stem Cell |
| SASP | Senescence-Associated Secretory Phenotype |
| ST-HSC | Short Term Hematopoietic Stem Cell |
| TLR | Toll-like Receptor |
| TNFα | Tumor Necrosis Factor α |
| VEGF | Vascular endothelial growth factor |
References
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, A.J.; Zon, L.I. The ‘definitive’ (and ‘primitive’) guide to zebrafish hematopoiesis. Oncogene 2004, 23, 7233–7246. [Google Scholar] [CrossRef] [Green Version]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Dzierzak, E.; Speck, N. Of lineage and legacy: The development of mammalian hematopoietic stem cells. Nat. Immunol. 2008, 9, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Golub, R.; Cumano, A. Embryonic hematopoiesis. Blood Cells Mol. Dis. 2013, 51, 226–231. [Google Scholar] [CrossRef]
- Yamane, T. Mouse Yolk Sac Hematopoiesis. Front. Cell Dev. Biol. 2018, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yoon, S.R.; Choi, I.; Jung, H. Causes and Mechanisms of Hematopoietic Stem Cell Aging. Int. J. Mol. Sci. 2019, 20, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M.; Warr, M.R.; Passegue, E. Cell cycle regulation in hematopoietic stem cells. J. Cell Biol. 2011, 195, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Clapes, T.; Lefkopoulos, S.; Trompouki, E. Stress and Non-Stress Roles of Inflammatory Signals during HSC Emergence and Maintenance. Front. Immunol. 2016, 7, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Xu, C.; Asada, N.; Frenette, P.S. The hematopoietic stem cell niche: From embryo to adult. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Sacma, M.; Pospiech, J.; Bogeska, R.; de Back, W.; Mallm, J.P.; Sakk, V.; Soller, K.; Marka, G.; Vollmer, A.; Karns, R.; et al. Haematopoietic stem cells in perisinusoidal niches are protected from ageing. Nat. Cell Biol. 2019, 21, 1309–1320. [Google Scholar] [CrossRef]
- Cheng, H.; Zheng, Z.; Cheng, T. New paradigms on hematopoietic stem cell differentiation. Protein Cell 2020, 11, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, S.; Xia, J.; Liu, F. Hematopoietic Hierarchy—An Updated Roadmap. Trends Cell Biol. 2018, 28, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Trumpp, A. An Intrinsic Interferon Program Protects Stem Cells from Viral Infection. Dev. Cell 2018, 44, 279–280. [Google Scholar] [CrossRef] [Green Version]
- Demerdash, Y.; Kain, B.; Essers, M.A.G.; King, K.Y. Yin and Yang: The dual effects of interferons on hematopoiesis. Exp. Hematol. 2021, 96, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Frame, J.M.; Kubaczka, C.; Long, T.L.; Esain, V.; Soto, R.A.; Hachimi, M.; Jing, R.; Shwartz, A.; Goessling, W.; Daley, G.Q.; et al. Metabolic Regulation of Inflammasome Activity Controls Embryonic Hematopoietic Stem and Progenitor Cell Production. Dev. Cell 2020, 55, 133–149.e136. [Google Scholar] [CrossRef] [PubMed]
- Lefkopoulos, S.; Polyzou, A.; Derecka, M.; Bergo, V.; Clapes, T.; Cauchy, P.; Jerez-Longres, C.; Onishi-Seebacher, M.; Yin, N.; Martagon-Calderon, N.A.; et al. Repetitive Elements Trigger RIG-I-like Receptor Signaling that Regulates the Emergence of Hematopoietic Stem and Progenitor Cells. Immunity 2020, 53, 934–951.e939. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, J.T.; Ghazale, N.; Pradhan, K.; Gupta, V.; Potts, K.S.; Tricomi, B.; Daniels, N.J.; Padgett, R.A.; De Oliveira, S.; Verma, A.; et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev. Cell 2021, 56, 627–640.e625. [Google Scholar] [CrossRef] [PubMed]
- Espin-Palazon, R.; Stachura, D.L.; Campbell, C.A.; Garcia-Moreno, D.; Del Cid, N.; Kim, A.D.; Candel, S.; Meseguer, J.; Mulero, V.; Traver, D. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell 2014, 159, 1070–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamiphak, S.; Kontarakis, Z.; Stainier, D.Y. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev. Cell 2014, 31, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Esain, V.; Teng, L.; Xu, J.; Kwan, W.; Frost, I.M.; Yzaguirre, A.D.; Cai, X.; Cortes, M.; Maijenburg, M.W.; et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 2014, 28, 2597–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Zhang, C.; Wang, L.; Zhang, P.; Ma, D.; Lv, J.; Liu, F. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 2015, 125, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef]
- Pietras, E.M.; Lakshminarasimhan, R.; Techner, J.M.; Fong, S.; Flach, J.; Binnewies, M.; Passegue, E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med. 2014, 211, 245–262. [Google Scholar] [CrossRef]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Florez, M.A.; Matatall, K.A.; Jeong, Y.; Ortinau, L.; Shafer, P.W.; Lynch, A.M.; Jaksik, R.; Kimmel, M.; Park, D.; King, K.Y. Interferon Gamma Mediates Hematopoietic Stem Cell Activation and Niche Relocalization through BST2. Cell Rep. 2020, 33, 108530. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Signer, R.A.J.; Morrison, S.J. Mechanisms that Regulate Stem Cell Aging and Life Span. Cell Stem Cell 2013, 12, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Verovskaya, E.V.; Dellorusso, P.V.; Passegue, E. Losing Sense of Self and Surroundings: Hematopoietic Stem Cell Aging and Leukemic Transformation. Trends Mol. Med. 2019, 25, 494–515. [Google Scholar] [CrossRef]
- Smith, J.N.P.; Zhang, Y.; Li, J.J.; McCabe, A.; Jo, H.J.; Maloney, J.; MacNamara, K.C. Type I IFNs drive hematopoietic stem and progenitor cell collapse via impaired proliferation and increased RIPK1-dependent cell death during shock-like ehrlichial infection. PLoS Pathog. 2018, 14, e1007234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.M.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejia-Ramirez, E.; Florian, M.C. Understanding intrinsic hematopoietic stem cell aging. Haematologica 2020, 105, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef]
- Leimkuhler, N.B.; Schneider, R.K. Inflammatory bone marrow microenvironment. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 294–302. [Google Scholar] [CrossRef]
- Ehninger, A.; Boch, T.; Uckelmann, H.; Essers, M.A.; Mudder, K.; Sleckman, B.P.; Trumpp, A. Posttranscriptional regulation of c-Myc expression in adult murine HSCs during homeostasis and interferon-alpha-induced stress response. Blood 2014, 123, 3909–3913. [Google Scholar] [CrossRef] [Green Version]
- King, K.Y.; Baldridge, M.T.; Weksberg, D.C.; Chambers, S.M.; Lukov, G.L.; Wu, S.; Boles, N.C.; Jung, S.Y.; Qin, J.; Liu, D.; et al. Irgm1 protects hematopoietic stem cells by negative regulation of IFN signaling. Blood 2011, 118, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, A.M.; Voermans, C.; Nolte, M.A. Impact of interferon-gamma on hematopoiesis. Blood 2014, 124, 2479–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Zhang, C. The Regulatory Role of IFN-gamma on the Proliferation and Differentiation of Hematopoietic Stem and Progenitor Cells. Stem Cell Rev. Rep. 2017, 13, 705–712. [Google Scholar] [CrossRef] [PubMed]
- MacNamara, K.C.; Oduro, K.; Martin, O.; Jones, D.D.; McLaughlin, M.; Choi, K.; Borjesson, D.L.; Winslow, G.M. Infection-Induced Myelopoiesis during Intracellular Bacterial Infection Is Critically Dependent upon IFN-gamma Signaling. J. Immunol. 2011, 186, 1032–1043. [Google Scholar] [CrossRef]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, A.M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naude, P.J.; den Boer, J.A.; Luiten, P.G.; Eisel, U.L. Tumor necrosis factor receptor cross-talk. FEBS J. 2011, 278, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Sherry, B.; Jue, D.M.; Zentella, A.; Cerami, A. Characterization of high molecular weight glycosylated forms of murine tumor necrosis factor. Biochem. Biophys. Res. Commun. 1990, 173, 1072–1078. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuettpelz, L.G.; Link, D.C. Regulation of hematopoietic stem cell activity by inflammation. Front. Immunol. 2013, 4, 204. [Google Scholar] [CrossRef] [Green Version]
- Dybedal, I.; Bryder, D.; Fossum, A.; Rusten, L.S.; Jacobsen, S.E. Tumor necrosis factor (TNF)-mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood 2001, 98, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Selleri, C.; Sato, T.; Anderson, S.; Young, N.S.; Maciejewski, J.P. Interferon-gamma and tumor necrosis factor-alpha suppress both early and late stages of hematopoiesis and induce programmed cell death. J. Cell Physiol. 1995, 165, 538–546. [Google Scholar] [CrossRef]
- Rezzoug, F.; Huang, Y.; Tanner, M.K.; Wysoczynski, M.; Schanie, C.L.; Chilton, P.M.; Ratajczak, M.Z.; Fugier-Vivier, I.J.; Ildstad, S.T. TNF-alpha is critical to facilitate hemopoietic stem cell engraftment and function. J. Immunol. 2008, 180, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Pronk, C.J.; Veiby, O.P.; Bryder, D.; Jacobsen, S.E. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: Involvement of two distinct receptors. J. Exp. Med. 2011, 208, 1563–1570. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Passegue, E. TNF-alpha Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 2019, 25, 357–372.e357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzrodt, M.; Ahmed, N.; Hoppe, P.S.; Loeffler, D.; Skylaki, S.; Hilsenbeck, O.; Kokkaliaris, K.D.; Kaltenbach, H.M.; Stelling, J.; Nerlov, C.; et al. Inflammatory signals directly instruct PU.1 in HSCs via TNF. Blood 2019, 133, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.S.; Rabe, J.L.; Loeffler, D.; Higa, K.C.; Hernandez, G.; Mills, T.S.; Ahmed, N.; Gessner, R.L.; Ke, Z.; Idler, B.M.; et al. PU.1 enforces quiescence and limits hematopoietic stem cell expansion during inflammatory stress. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, J.; Mizoguchi, I.; Chiba, Y.; Hisada, M.; Kobayashi, F.; Yoshida, H.; Nakae, S.; Tsuchida, A.; Matsumoto, T.; Ema, H.; et al. Promotion of Expansion and Differentiation of Hematopoietic Stem Cells by Interleukin-27 into Myeloid Progenitors to Control Infection in Emergency Myelopoiesis. PLoS Pathog. 2016, 12, e1005507. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Li, X.; Meng, Y.; Yuan, B.; Liu, T.; Jiao, M.; Wang, X.; Liu, Y.; Yin, H. Interleukin-33 regulates hematopoietic stem cell regeneration after radiation injury. Stem Cell Res. Ther. 2019, 10, 123. [Google Scholar] [CrossRef]
- Zhang, S.; Morita, M.; Wang, Z.; Ooehara, J.; Zhang, S.; Xie, M.; Bai, H.; Yu, W.; Wang, X.; Dong, F.; et al. Interleukin-12 supports in vitro self-renewal of long-term hematopoietic stem cells. Blood Sci. 2019, 1, 92–101. [Google Scholar] [CrossRef]
- Zhao, J.L.; Ma, C.; O’ Connell, R.M.; Mehta, A.; DiLoreto, R.; Heath, J.R.; Baltimore, D. Conversion of Danger Signals into Cytokine Signals by Hematopoietic Stem and Progenitor Cells for Regulation of Stress-Induced Hematopoiesis. Cell Stem Cell 2014, 14, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yao, J.C.; Li, J.T.; Schmidt, A.P.; Link, D.C. TLR7/8 agonist treatment induces an increase in bone marrow resident dendritic cells and hematopoietic progenitor expansion and mobilization. Exp. Hematol. 2021, 96, 35–43.e37. [Google Scholar] [CrossRef] [PubMed]
- Aluri, J.; Bach, A.; Kaviany, S.; Chiquetto Paracatu, L.; Kitcharoensakkul, M.; Walkiewicz, M.A.; Putnam, C.D.; Shinawi, M.; Saucier, N.; Rizzi, E.M.; et al. Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain-of-function. Blood 2021, 18, 2450–2462. [Google Scholar] [CrossRef] [PubMed]
- Schuettpelz, L.G.; Borgerding, J.N.; Christopher, M.J.; Gopalan, P.K.; Romine, M.P.; Herman, A.C.; Woloszynek, J.R.; Greenbaum, A.M.; Link, D.C. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia 2014, 28, 1851–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.D.; et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 2017, 21, 225–240.e225. [Google Scholar] [CrossRef]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Kalafati, L.; Bornhauser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol. 2020, 11, 1540. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, A.N.; Maloney, J.M.; MacNamara, K.C. Macrophages Orchestrate Hematopoietic Programs and Regulate HSC Function During Inflammatory Stress. Front. Immunol. 2020, 11, 1499. [Google Scholar] [CrossRef]
- Batsivari, A.; Haltalli, M.L.R.; Passaro, D.; Pospori, C.; Lo Celso, C.; Bonnet, D. Dynamic responses of the haematopoietic stem cell niche to diverse stresses. Nat. Cell Biol. 2020, 22, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, S.; Gerosa, R.C.; Radpour, R.; Bauer, J.; Ampenberger, F.; Heikenwalder, M.; Kopf, M.; Manz, M.G. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood 2014, 124, 1393–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbling, P.M.; Pineiro-Yanez, E.; Gerosa, R.; Boettcher, S.; Al-Shahrour, F.; Manz, M.G.; Nombela-Arrieta, C. Global Transcriptomic Profiling of the Bone Marrow Stromal Microenvironment during Postnatal Development, Aging, and Inflammation. Cell Rep. 2019, 29, 3313–3330.e3314. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, S.; Ziegler, P.; Schmid, M.A.; Takizawa, H.; van Rooijen, N.; Kopf, M.; Heikenwalder, M.; Manz, M.G. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J. Immunol. 2012, 188, 5824–5828. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.S.; Li, H.; Kang, Y.L.; Chen, W.C.; Cheng, W.C.; Lai, D.M. Loss of Cxcl12/Sdf-1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood 2011, 117, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Depond, M.; He, L.; Foudi, A.; Kwarteng, E.O.; Lauret, E.; Plo, I.; Desterke, C.; Dessen, P.; Fujii, N.; et al. CXCR4/CXCL12 axis counteracts hematopoietic stem cell exhaustion through selective protection against oxidative stress. Sci. Rep. 2016, 6, 37827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurch, C.M.; Riether, C.; Ochsenbein, A.F. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 2014, 14, 460–472. [Google Scholar] [CrossRef] [Green Version]
- Umemoto, T.; Matsuzaki, Y.; Shiratsuchi, Y.; Hashimoto, M.; Yoshimoto, T.; Nakamura-Ishizu, A.; Petrich, B.; Yamato, M.; Suda, T. Integrin alphavbeta3 enhances the suppressive effect of interferon-gamma on hematopoietic stem cells. EMBO J. 2017, 36, 2390–2403. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, A.M.; Kuck, A.; van Essen, M.; Haas, S.; Blaszkiewicz, S.; Essers, M.A. IFNalpha-mediated remodeling of endothelial cells in the bone marrow niche. Haematologica 2017, 102, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, E.; Slaughter, A.; Frenette, P.S.; Kuick, R.; Pello, O.M.; Lucas, D. Granulocyte-derived TNFalpha promotes vascular and hematopoietic regeneration in the bone marrow. Nat. Med. 2018, 24, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Xu, P.; Zhang, X.; Liao, M.; Dong, Q.; Cong, T.; Tang, B.; Yang, X.; Ye, M.; Chang, Y.; et al. Aging-induced IL27Ra signaling impairs hematopoietic stem cells. Blood 2020, 136, 183–198. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Amro, E.M.; Becker, F.; Holzer, M.; Rasa, S.M.M.; Njeru, S.N.; Han, B.; Di Sanzo, S.; Chen, Y.; Tang, D.; et al. Cohesin-mediated NF-kappaB signaling limits hematopoietic stem cell self-renewal in aging and inflammation. J. Exp. Med. 2019, 216, 152–175. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, Y.; Karigane, D.; Ootomo, Y.; Kobayashi, H.; Morikawa, T.; Otsu, K.; Kubota, Y.; Okamoto, S.; Goda, N.; Takubo, K. p38alpha plays differential roles in hematopoietic stem cell activity dependent on aging contexts. J. Biol. Chem. 2021, 296, 100563. [Google Scholar] [CrossRef]
- Mann, M.; Mehta, A.; de Boer, C.G.; Kowalczyk, M.S.; Lee, K.; Haldeman, P.; Rogel, N.; Knecht, A.R.; Farouq, D.; Regev, A.; et al. Heterogeneous Responses of Hematopoietic Stem Cells to Inflammatory Stimuli Are Altered with Age. Cell Rep. 2018, 25, 2992–3005.e2995. [Google Scholar] [CrossRef] [Green Version]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [Green Version]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Bogeska, R.; Kaschutnig, P.; Fawaz, M.; Mikecin, A.-M.; Büchler-Schäff, M.; Paffenholz, S.; Asada, N.; Frauhammer, F.; Buettner, F.; Ball, M.; et al. Hematopoietic stem cells fail to regenerate following inflammatory challenge. Biorxiv 2020. [Google Scholar] [CrossRef]
- Sera, Y.; Nakata, Y.; Ueda, T.; Yamasaki, N.; Koide, S.; Kobayashi, H.; Ikeda, K.I.; Kobatake, K.; Iwasaki, M.; Oda, H.; et al. UTX maintains the functional integrity of the murine hematopoietic system by globally regulating aging-associated genes. Blood 2021, 137, 908–922. [Google Scholar] [CrossRef]
- Kobatake, K.; Ikeda, K.I.; Nakata, Y.; Yamasaki, N.; Ueda, T.; Kanai, A.; Sentani, K.; Sera, Y.; Hayashi, T.; Koizumi, M.; et al. Kdm6a Deficiency Activates Inflammatory Pathways, Promotes M2 Macrophage Polarization, and Causes Bladder Cancer in Cooperation with p53 Dysfunction. Clin. Cancer Res. 2020, 26, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Verovskaya, E.V.; Mitchell, C.A.; Calero-Nieto, F.J.; Hérault, A.; Dellorusso, P.D.; Wang, X.; Zhang, S.Y.; Svendsen, A.F.; Pietras, E.M.; Bakker, S.T. Stromal inflammation is a targetable driver of hematopoietic aging. Biorxiv 2021. [Google Scholar] [CrossRef]
- Ergen, A.V.; Boles, N.C.; Goodell, M.A. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 2012, 119, 2500–2509. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.H.; Mendez-Ferrer, S. Microenvironmental contributions to hematopoietic stem cell aging. Haematologica 2020, 105, 38–46. [Google Scholar] [CrossRef]
- Frisch, B.J.; Hoffman, C.M.; Latchney, S.E.; LaMere, M.W.; Myers, J.; Ashton, J.; Li, A.J.; Saunders, J., 2nd; Palis, J.; Perkins, A.S.; et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via Interleukin1B. JCI Insight 2019, 4, e124213. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef]
- Hart, E.C.; Charkoudian, N. Sympathetic neural regulation of blood pressure: Influences of sex and aging. Physiology 2014, 29, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.H.; Del Toro, R.; Rivera-Torres, J.; Rak, J.; Korn, C.; Garcia-Garcia, A.; Macias, D.; Gonzalez-Gomez, C.; Del Monte, A.; Wittner, M.; et al. Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 2019, 25, 407–418.e406. [Google Scholar] [CrossRef] [Green Version]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Ganan-Gomez, I.; Wei, Y.; Starczynowski, D.T.; Colla, S.; Yang, H.; Cabrero-Calvo, M.; Bohannan, Z.S.; Verma, A.; Steidl, U.; Garcia-Manero, G. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015, 29, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Qian, Y.; Eksioglu, E.; Epling-Burnette, P.K.; Wei, S. The inflammatory microenvironment in MDS. Cell Mol. Life Sci. 2015, 72, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef] [Green Version]
- Shetty, V.; Mundle, S.; Alvi, S.; Showel, M.; Broady-Robinson, L.; Dar, S.; Borok, R.; Showel, J.; Gregory, S.; Rifkin, S.; et al. Measurement of apoptosis, proliferation and three cytokines in 46 patients with myelodysplastic syndromes. Leuk. Res. 1996, 20, 891–900. [Google Scholar] [CrossRef]
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, D.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef] [Green Version]
- Kuninaka, N.; Kurata, M.; Yamamoto, K.; Suzuki, S.; Umeda, S.; Kirimura, S.; Arai, A.; Nakagawa, Y.; Suzuki, K.; Kitagawa, M. Expression of Toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia. Exp. Mol. Pathol. 2010, 88, 293–298. [Google Scholar] [CrossRef]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Menssen, A.J.; Walter, M.J. Genetics of progression from MDS to secondary leukemia. Blood 2020, 136, 50–60. [Google Scholar] [CrossRef]
- Caligaris-Cappio, F. Inflammation, the microenvironment and chronic lymphocytic leukemia. Haematologica 2011, 96, 353–355. [Google Scholar] [CrossRef] [Green Version]
- Giles, F.J.; Krawczyk, J.; O’Dwyer, M.; Swords, R.; Freeman, C. The role of inflammation in leukaemia. Adv. Exp. Med. Biol. 2014, 816, 335–360. [Google Scholar] [CrossRef]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef]
- Geissler, K.; Tricot, G.; Leemhuis, T.; Walker, E.; Broxmeyer, H.E. Differentiation-inducing effect of recombinant human tumor necrosis factor alpha and gamma-interferon in vitro on blast cells from patients with acute myeloid leukemia and myeloid blast crisis of chronic myeloid leukemia. Cancer Res. 1989, 49, 3057–3062. [Google Scholar] [PubMed]
- Habbel, J.; Arnold, L.; Chen, Y.; Mollmann, M.; Bruderek, K.; Brandau, S.; Duhrsen, U.; Hanoun, M. Inflammation-driven activation of JAK/STAT signaling reversibly accelerates acute myeloid leukemia in vitro. Blood Adv. 2020, 4, 3000–3010. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Nievergall, E.; Reynolds, J.; Kok, C.H.; Watkins, D.B.; Biondo, M.; Busfield, S.J.; Vairo, G.; Fuller, K.; Erber, W.N.; Sadras, T.; et al. TGF-alpha and IL-6 plasma levels selectively identify CML patients who fail to achieve an early molecular response or progress in the first year of therapy. Leukemia 2016, 30, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Challen, G.A.; Goodell, M.A. Clonal hematopoiesis: Mechanisms driving dominance of stem cell clones. Blood 2020, 136, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833–849.e835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hormaechea-Agulla, D.; Matatall, K.A.; Le, D.T.; Kain, B.; Long, X.; Kus, P.; Jaksik, R.; Challen, G.A.; Kimmel, M.; King, K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell 2021. [Google Scholar] [CrossRef]
- Hu, L.; Huang, W.; Hjort, E.; Eklund, E.A. Increased Fanconi C expression contributes to the emergency granulopoiesis response. J. Clin. Investig. 2013, 123, 3952–3966. [Google Scholar] [CrossRef]
- Dufour, C.; Corcione, A.; Svahn, J.; Haupt, R.; Poggi, V.; Beka’ssy, A.N.; Scime, R.; Pistorio, A.; Pistoia, V. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood 2003, 102, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sejas, D.P.; Zhang, X.; Qiu, Y.; Nattamai, K.J.; Rani, R.; Rathbun, K.R.; Geiger, H.; Williams, D.A.; Bagby, G.C.; et al. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J. Clin. Investig. 2007, 117, 3283–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bousounis, P.; Bergo, V.; Trompouki, E. Inflammation, Aging and Hematopoiesis: A Complex Relationship. Cells 2021, 10, 1386. https://doi.org/10.3390/cells10061386
Bousounis P, Bergo V, Trompouki E. Inflammation, Aging and Hematopoiesis: A Complex Relationship. Cells. 2021; 10(6):1386. https://doi.org/10.3390/cells10061386
Chicago/Turabian StyleBousounis, Pavlos, Veronica Bergo, and Eirini Trompouki. 2021. "Inflammation, Aging and Hematopoiesis: A Complex Relationship" Cells 10, no. 6: 1386. https://doi.org/10.3390/cells10061386
APA StyleBousounis, P., Bergo, V., & Trompouki, E. (2021). Inflammation, Aging and Hematopoiesis: A Complex Relationship. Cells, 10(6), 1386. https://doi.org/10.3390/cells10061386