
Silencing of Poly(ADP-Ribose) Polymerase-2 Induces Mitochondrial Reactive Species Production and Mitochondrial Fragmentation


 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Transient Transfection
2.4. In Vitro Cell Proliferation Assay (SRB Assay)
2.5. Detection of Cell Death
2.6. Determination of Cellular ATP Level
2.7. MitoTracker Red Staining
2.8. Immunofluorescence
2.9. Total RNA Preparation and Reverse Transcription–Quantitative PCR (RT-qPCR)
2.10. SDS-PAGE and Western Blotting
2.11. Determination of Lipid Peroxidation (TBARS Assay)
2.12. Determination of the Oxygen Consumption Rate (OCR)
2.13. Measurement of Superoxide Production (DHE Staining)
2.14. Electron Microscopy
2.15. Statistical Analysis
3. Results
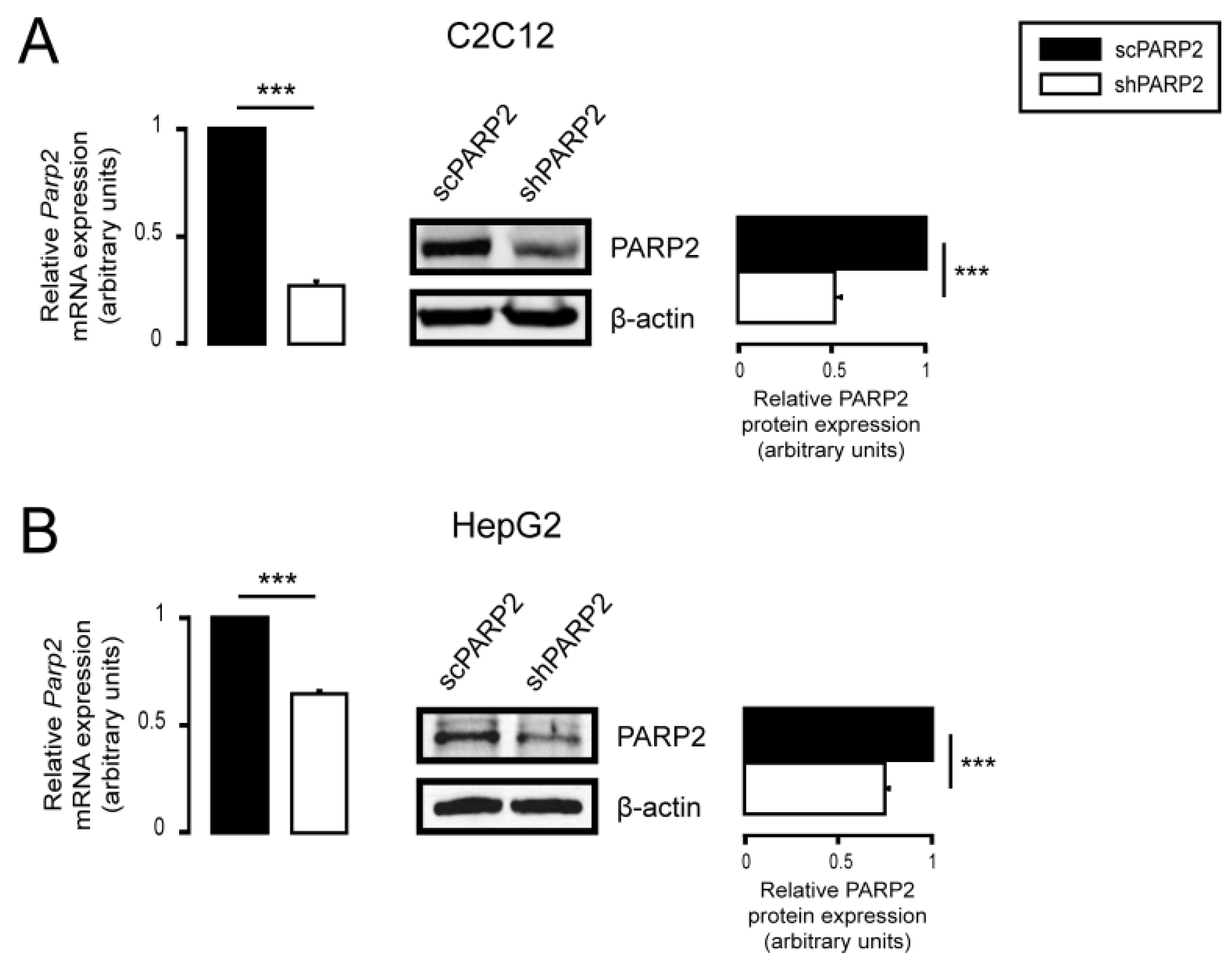
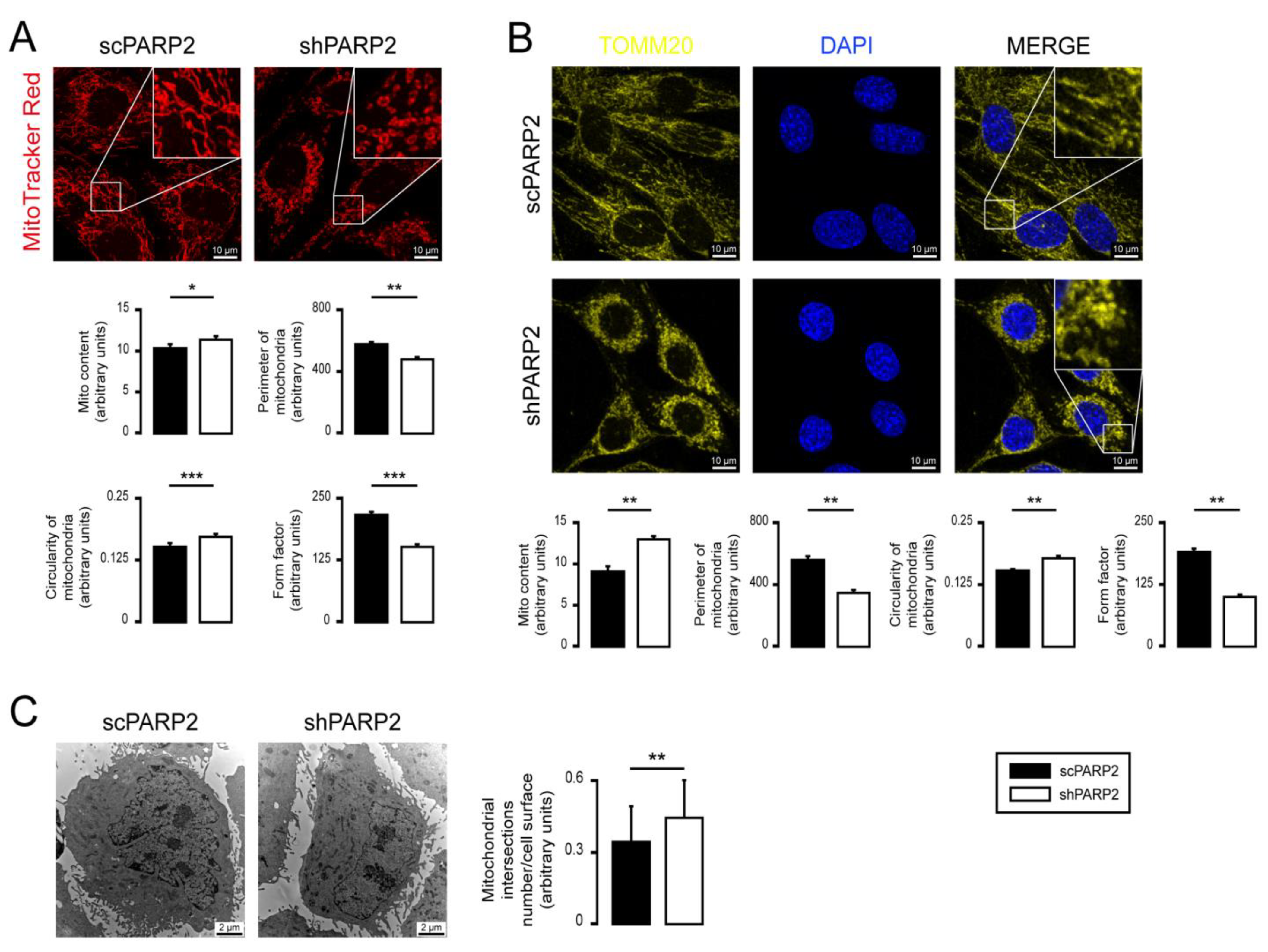
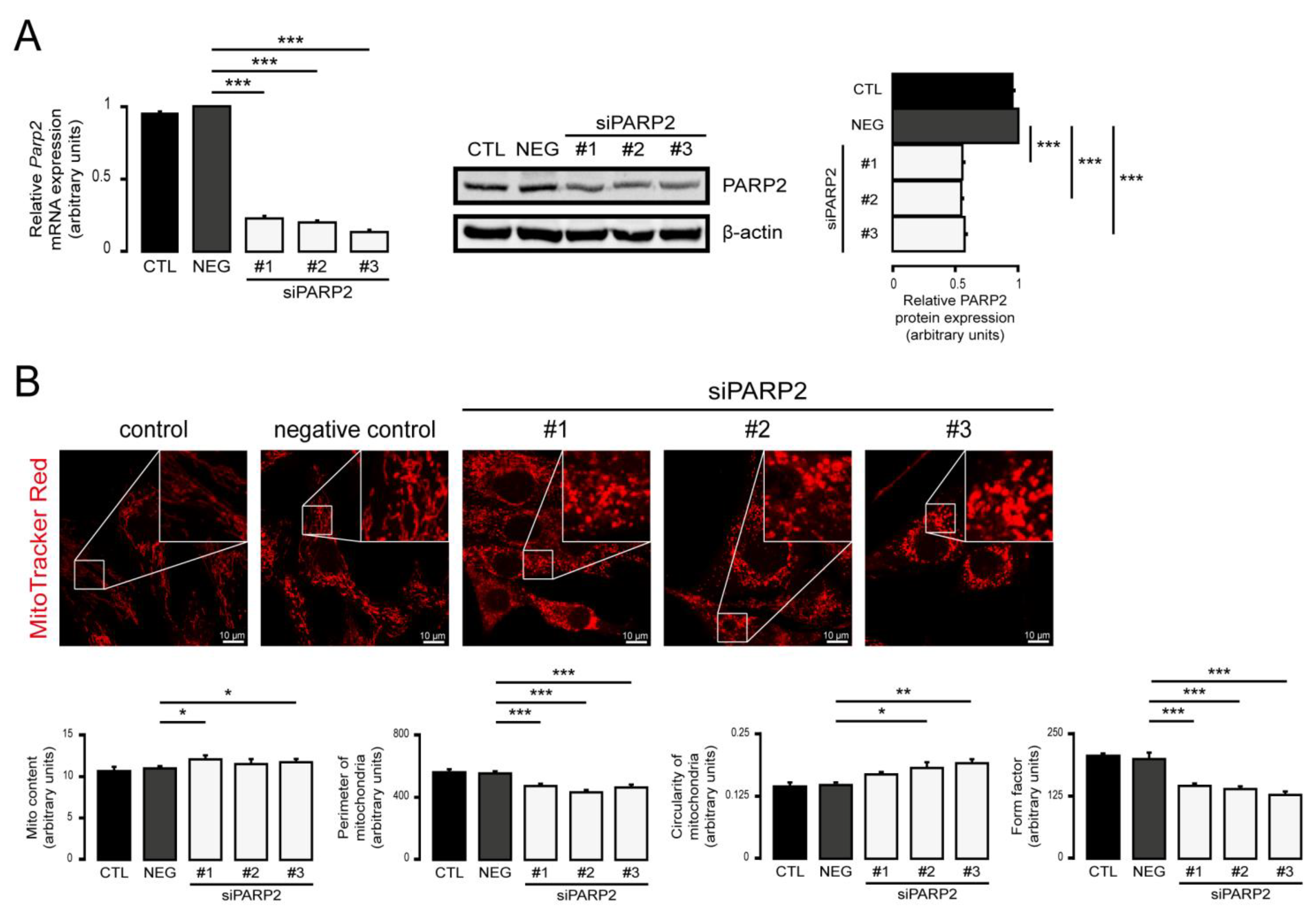
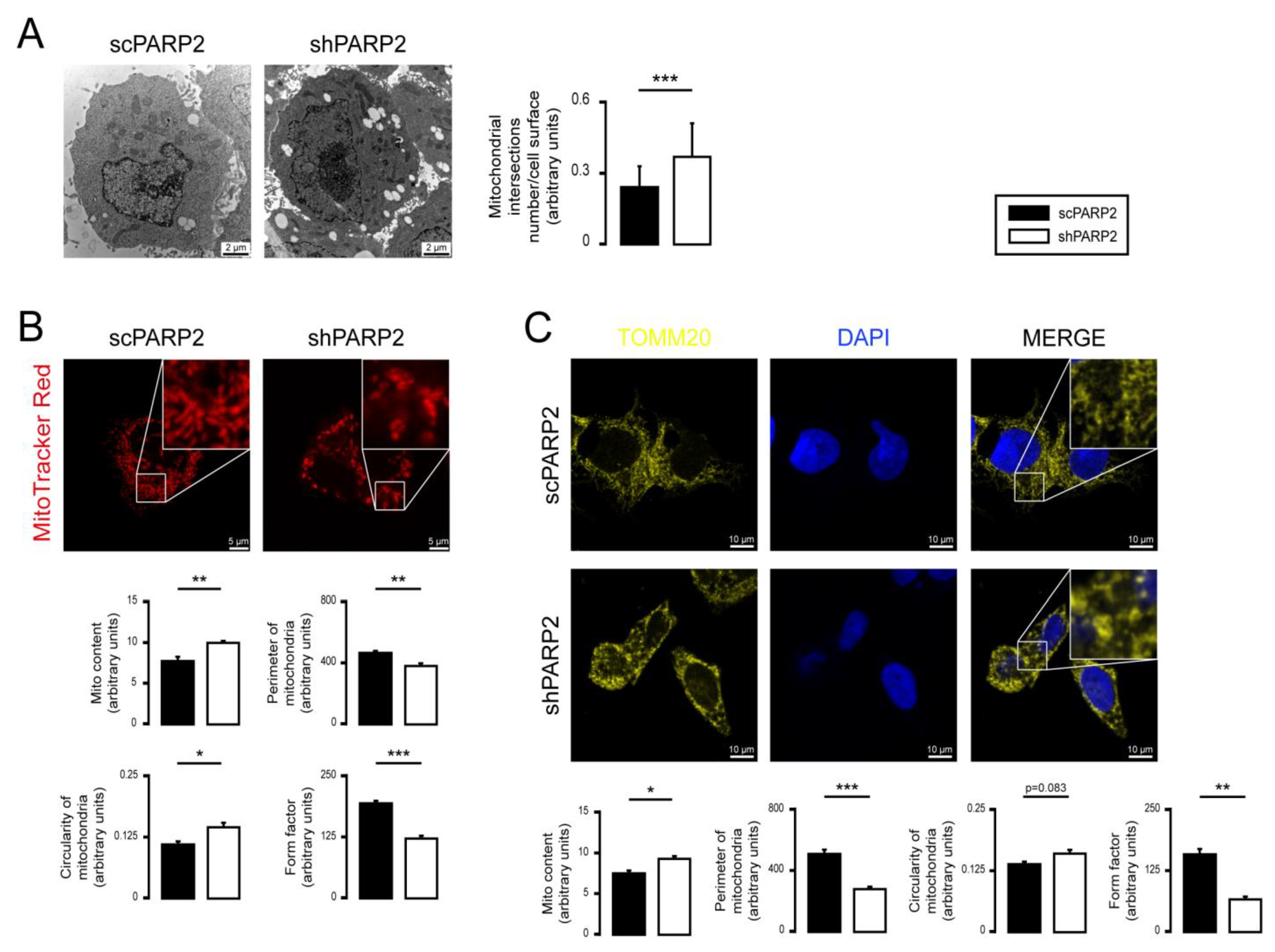
3.1. Silencing of PARP2 Leads to Fragmented Mitochondria
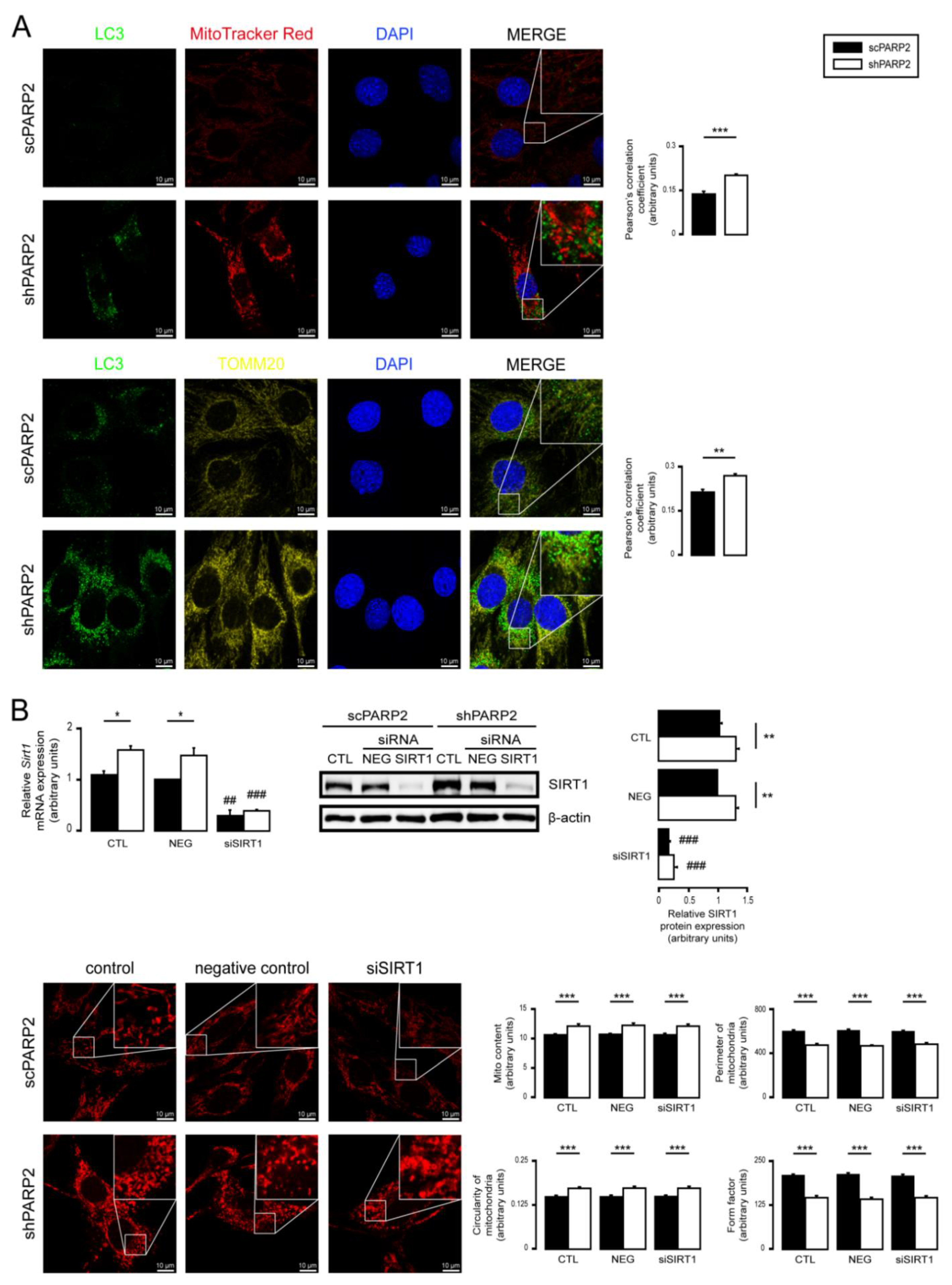
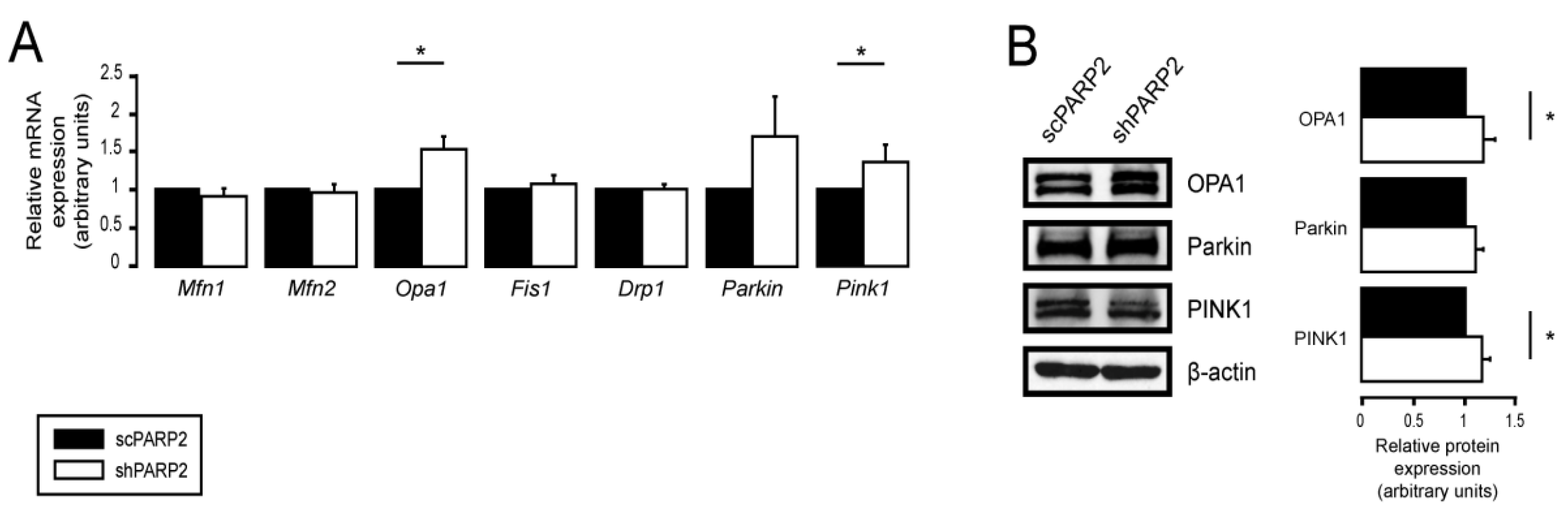
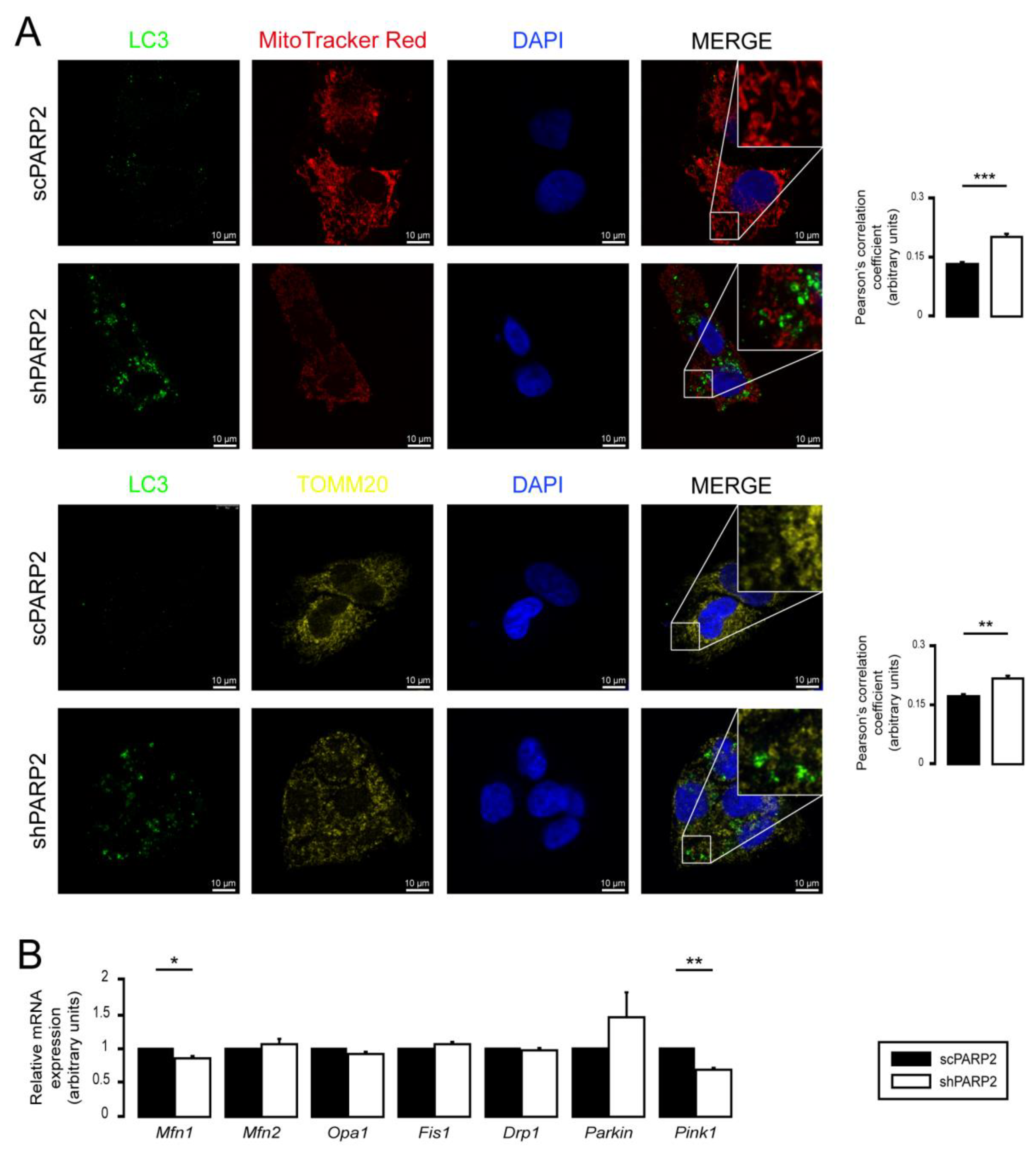
3.2. SIRT1 Activation, Mitophagy, mtUPR, and the Deregulation of the Mitochondrial Fusion–Fission System Are Not Involved in Mitochondrial Fragmentation in PARP2-Silenced Cells
3.3. Mitochondrial Fragmentation Is Not Specific for C2C12 Myoblasts
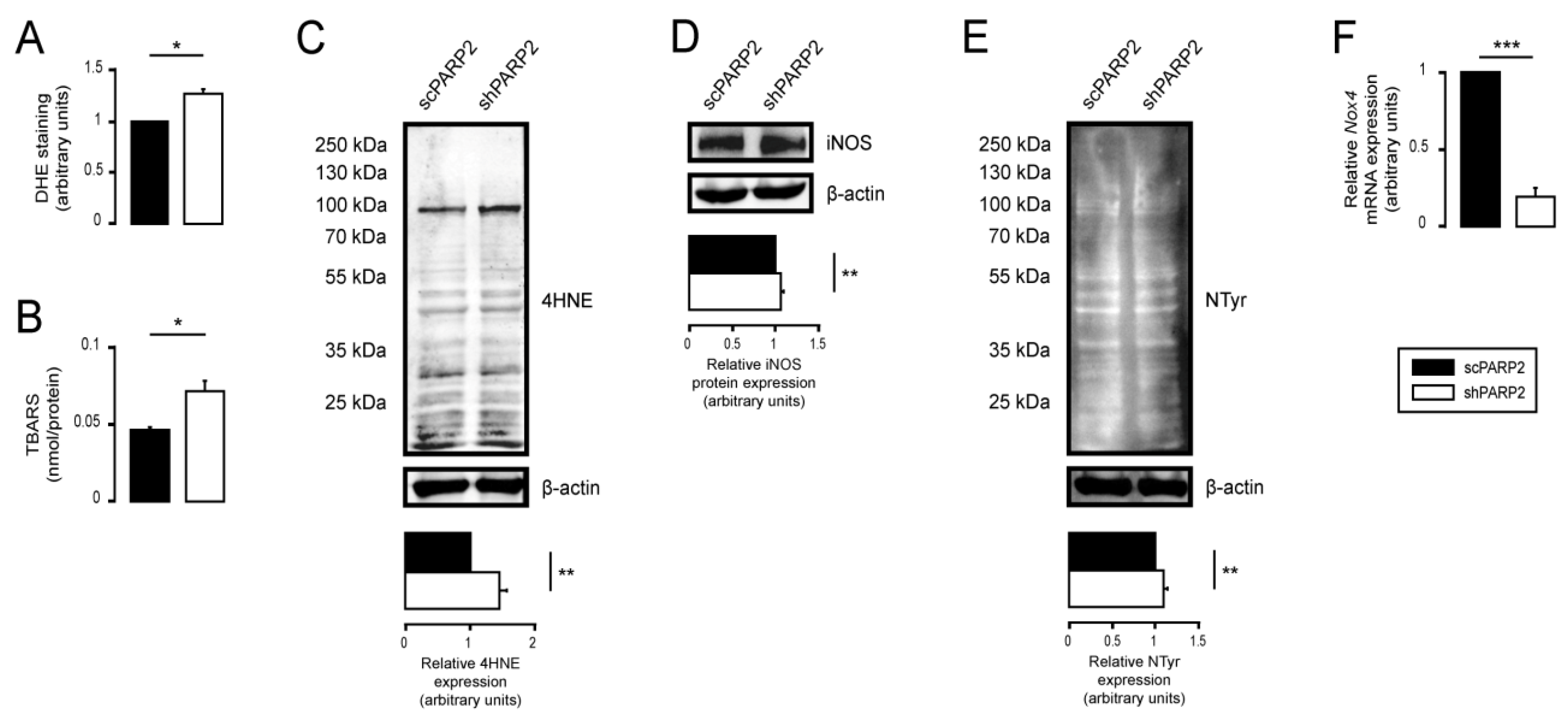
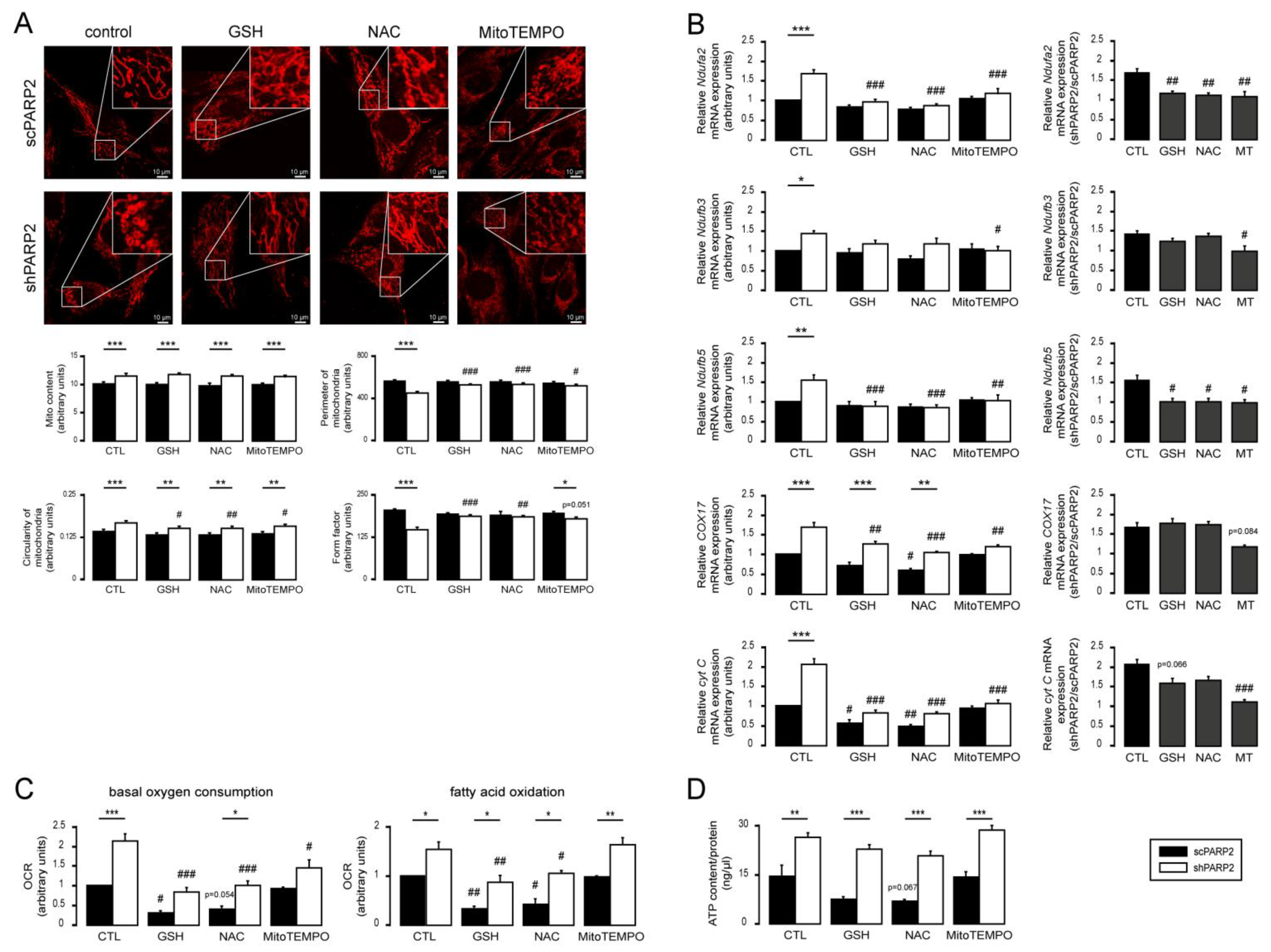
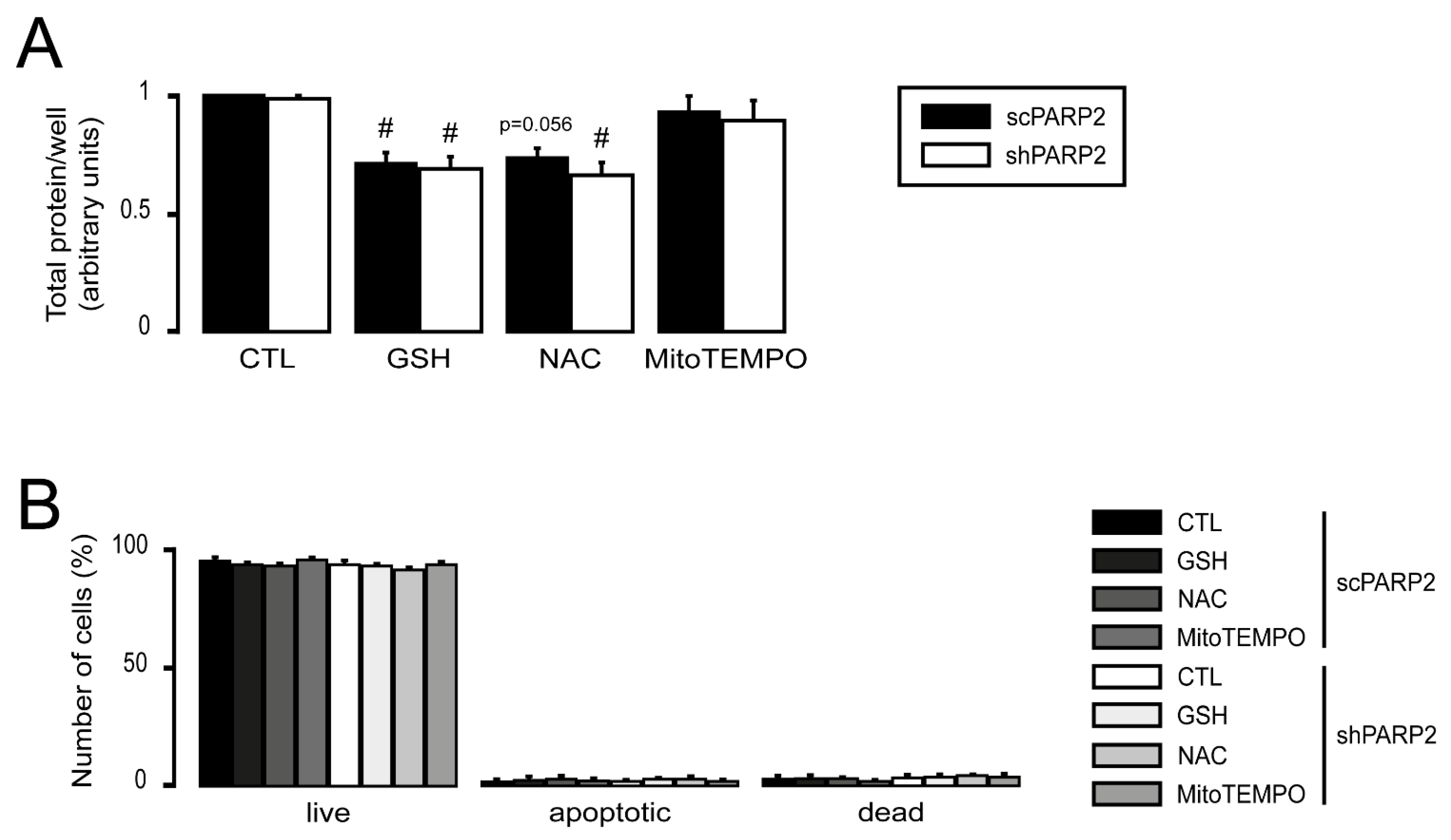
3.4. Silencing of PARP2 Leads to Oxidative Stress, Originating Partly from the Mitochondria through Mitochondrial Protein Imbalance and Fragmentation
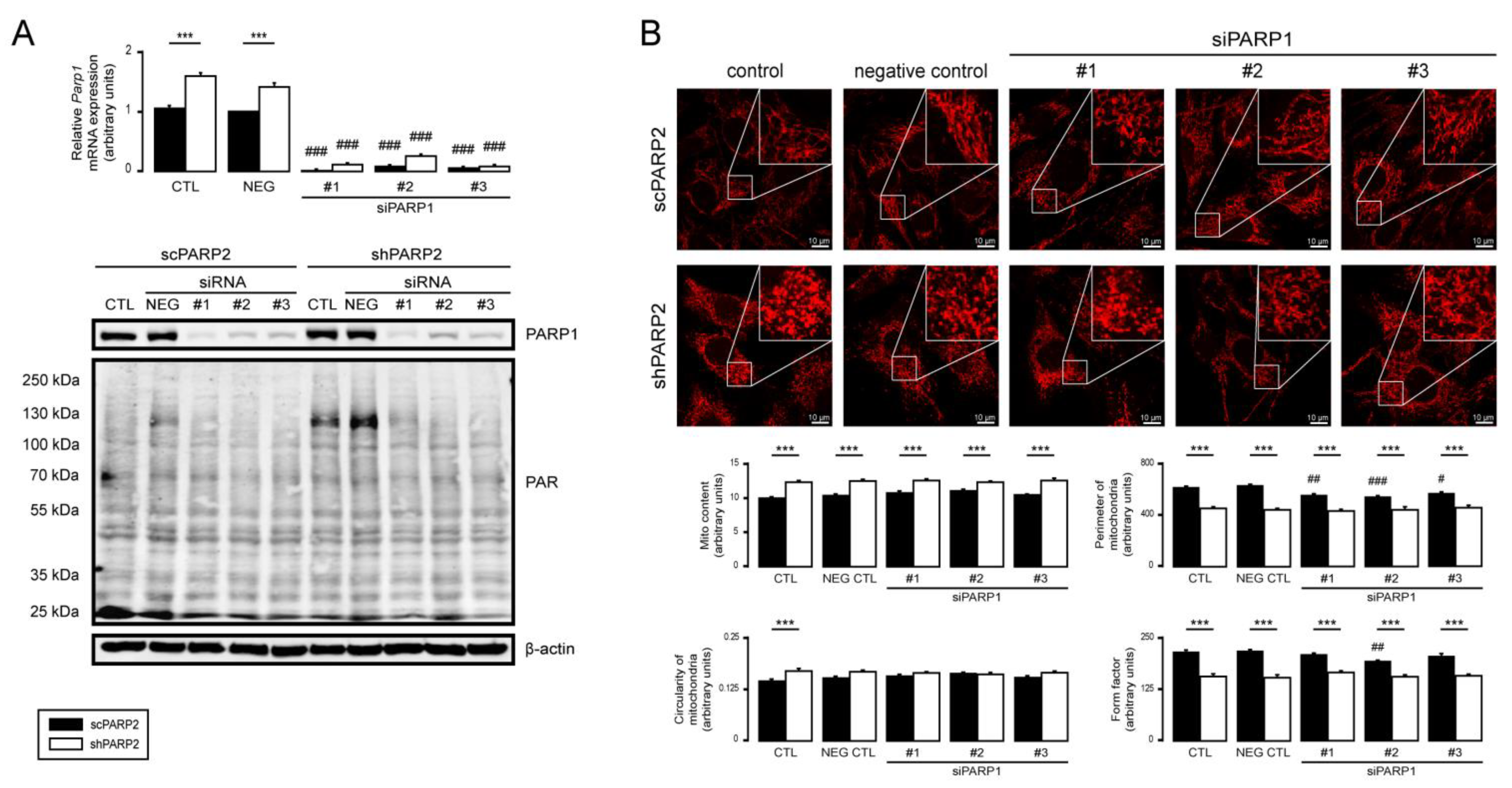
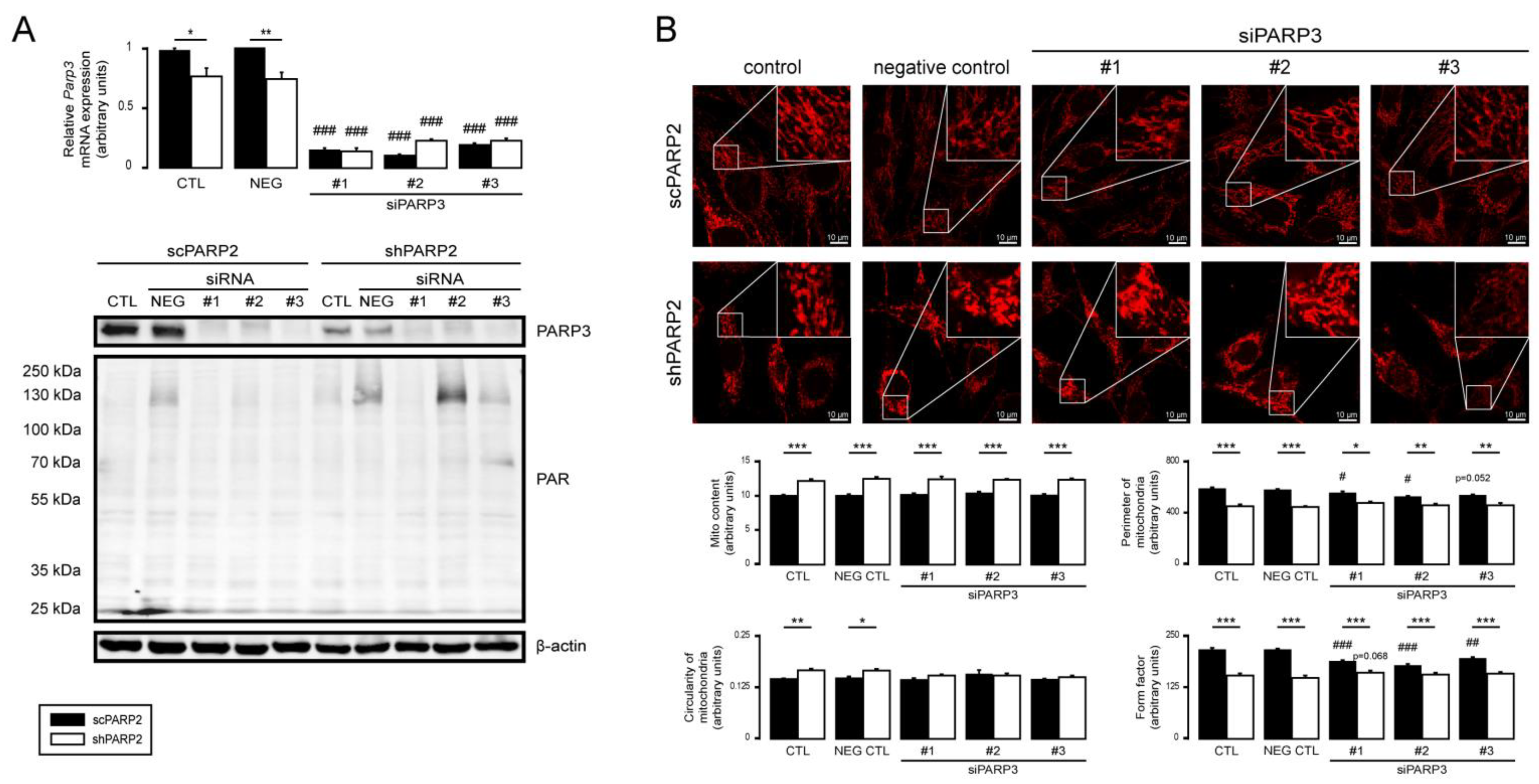
3.5. PARP1 and PARP3 Are Not Involved in Mitochondrial Fragmentation Elicited by the Silencing of PARP2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular communication: Shaping health and disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P.; Nagy, L.; Fodor, T.; Liaudet, L.; Pacher, P. Poly(ADP-ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol. Metab. 2015, 26, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Mottis, A.; Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response in mammalian physiology. Mamm. Genome 2014, 25, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell. Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Hill, S.; Van Remmen, H. Mitochondrial stress signaling in longevity: A new role for mitochondrial function in aging. Redox Biol. 2014, 2, 936–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; Sengupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD(+)/SIRT1 Reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on metabolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Ame, J.C.; Dolle, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Menissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef] [Green Version]
- Szanto, M.; Rutkai, I.; Hegedus, C.; Czikora, A.; Rozsahegyi, M.; Kiss, B.; Virag, L.; Gergely, P.; Toth, A.; Bai, P. Poly(ADP-ribose) polymerase-2 depletion reduces doxorubicin-induced damage through SIRT1 induction. Cardiovasc. Res. 2011, 92, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Shieh, W.M.; Ame, J.C.; Wilson, M.V.; Wang, Z.Q.; Koh, D.W.; Jacobson, M.K.; Jacobson, E.L. Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J. Biol. Chem. 1998, 273, 30069–30072. [Google Scholar] [CrossRef] [Green Version]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [Green Version]
- Kutuzov, M.M.; Khodyreva, S.N.; Ame, J.C.; Ilina, E.S.; Sukhanova, M.V.; Schreiber, V.; Lavrik, O.I. Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie 2013, 95, 1208–1215. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Gatie, M.I.; Kelly, G.M. Serum-dependent and -independent regulation of PARP2. Biochem. Cell Biol. 2019, 97, 600–611. [Google Scholar] [CrossRef]
- Marton, J.; Peter, M.; Balogh, G.; Bodi, B.; Vida, A.; Szanto, M.; Bojcsuk, D.; Janko, L.; Bhattoa, H.P.; Gombos, I.; et al. Poly(ADP-ribose) polymerase-2 is a lipid-modulated modulator of muscular lipid homeostasis. Biochim. Biophys. Acta 2018, 1863, 1399–1412. [Google Scholar] [CrossRef]
- Schreiber, V.; Ricoul, M.; Amé, J.C.; Dantzer, F.; Meder, V.S.; Spenlehauer, C.; Stiegler, P.; Niedergang, C.; Sabatier, L.; Favaudon, V.; et al. PARP-2: Structure-function relationship. In Poly(ADP-ribosyl)ation; Springer: Boston, MA, USA, 2004; pp. 13–31. [Google Scholar]
- Yelamos, J.; Schreiber, V.; Dantzer, F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol. Med. 2008, 14, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Szanto, M.; Brunyánszki, A.; Kiss, B.; Nagy, L.; Gergely, P.; Virag, L.; Bai, P. Poly(ADP-ribose) polymerase-2: Emerging transcriptional roles of a DNA repair protein. Cell Mol. Life Sci. 2012, 69, 4079–4092. [Google Scholar] [CrossRef] [Green Version]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Menissier-de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Ame, J.C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, F.; Giraud-Panis, M.J.; Jaco, I.; Ame, J.C.; Schultz, I.; Blasco, M.; Koering, C.E.; Gilson, E.; Menissier-de Murcia, J.; de Murcia, G.; et al. Functional interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol. Cell Biol. 2004, 24, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, F.; Mark, M.; Quenet, D.; Scherthan, H.; Huber, A.; Liebe, B.; Monaco, L.; Chicheportiche, A.; Sassone-Corsi, P.; de Murcia, G.; et al. Poly(ADP-ribose) polymerase-2 contributes to the fidelity of male meiosis I and spermiogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 14854–14859. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, L.; Martinez, C.; Baro, C.; Rodriguez, M.; Baroja-Mazo, A.; Sole, F.; Flores, J.M.; Ampurdanes, C.; Dantzer, F.; Martin-Caballero, J.; et al. Loss of poly(ADP-ribose) polymerase-2 leads to rapid development of spontaneous T-cell lymphomas in p53-deficient mice. Oncogene 2010, 29, 2877–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yelamos, J.; Farres, J.; Llacuna, L.; Ampurdanes, C.; Martin-Caballero, J. PARP-1 and PARP-2: New players in tumour development. Am. J. Cancer Res. 2011, 1, 328–346. [Google Scholar]
- Mateu-Jimenez, M.; Cucarull-Martinez, B.; Yelamos, J.; Barreiro, E. Reduced tumor burden through increased oxidative stress in lung adenocarcinoma cells of PARP-1 and PARP-2 knockout mice. Biochimie 2016, 121, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, J.; Gozalbo-Lopez, B.; Mendez, A.C.; Dantzer, F.; Schreiber, V.; Martinez, C.; Arana, D.M.; Farres, J.; Revilla-Nuin, B.; Bueno, M.F.; et al. PARP-1/PARP-2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci. Rep. 2017, 7, 41962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Quenet, D.; Mark, M.; Govin, J.; van Dorsselear, A.; Schreiber, V.; Khochbin, S.; Dantzer, F. Parp2 is required for the differentiation of post-meiotic germ cells: Identification of a spermatid-specific complex containing Parp1, Parp2, TP2 and HSPA2. Exp. Cell Res. 2009, 315, 2824–2834. [Google Scholar] [CrossRef] [PubMed]
- Popoff, I.; Jijon, H.; Monia, B.; Tavernini, M.; Ma, M.; McKay, R.; Madsen, K. Antisense oligonucleotides to poly(ADP-ribose) polymerase-2 ameliorate colitis in interleukin-10-deficient mice. J. Pharmacol. Exp. Ther. 2002, 303, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Yelamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; de Murcia, G.; et al. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 2006, 25, 4350–4360. [Google Scholar] [CrossRef]
- Farres, J.; Martin-Caballero, J.; Martinez, C.; Lozano, J.J.; Llacuna, L.; Ampurdanes, C.; Ruiz-Herguido, C.; Dantzer, F.; Schreiber, V.; Villunger, A.; et al. PARP-2 is required to maintain hematopoiesis following sublethal gamma-irradiation in mice. Blood 2013, 122, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farres, J.; Llacuna, L.; Martin-Caballero, J.; Martinez, C.; Lozano, J.J.; Ampurdanes, C.; Lopez-Contreras, A.J.; Florensa, L.; Navarro, J.; Ottina, E.; et al. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 2015, 22, 1144–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Lama, L.; Galindo-Campos, M.A.; Martínez, C.; Comerma, L.; Vazquez, I.; Vernet-Tomas, M.; Ampurdanés, C.; Lutfi, N.; Martin-Caballero, J.; Dantzer, F.; et al. Coordinated signals from PARP-1 and PARP-2 are required to establish a proper T cell immune response to breast tumors in mice. Oncogene 2020, 39, 2835–2843. [Google Scholar] [CrossRef]
- Kofler, J.; Otsuka, T.; Zhang, Z.; Noppens, R.; Grafe, M.R.; Koh, D.W.; Dawson, V.L.; Menisser-de Murcia, J.; Hurn, P.D.; Traystman, R.J. Differential effect of PARP-2 deletion on brain injury after focal and global cerebral ischemia. J. Cereb. Blood Flow Metab. 2006, 26, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Klaus, J.A.; Zhang, J.; Xu, Z.; Kibler, K.K.; Andrabi, S.A.; Rao, K.; Yang, Z.J.; Dawson, T.M.; Dawson, V.L.; et al. Contributions of poly(ADP-ribose) polymerase-1 and -2 to nuclear translocation of apoptosis-inducing factor and injury from focal cerebral ischemia. J. Neurochem. 2010, 113, 1012–1022. [Google Scholar] [CrossRef] [Green Version]
- Szanto, M.; Brunyanszki, A.; Marton, J.; Vamosi, G.; Nagy, L.; Fodor, T.; Kiss, B.; Virag, L.; Gergely, P.; Bai, P. Deletion of PARP-2 induces hepatic cholesterol accumulation and decrease in HDL levels. Biochim. Biophys. Acta 2014, 1842, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P.; Canto, C.; Brunyánszki, A.; Huber, A.; Szántó, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Houten, S.M.; Huber, A.; Schreiber, V.; Watanabe, M.; Kiss, B.; de Murcia, G.; Auwerx, J.; Menissier-de Murcia, J. Poly(ADP-ribose) polymerase-2 controls adipocyte differentiation and adipose tissue function through the regulation of the activity of the retinoid X receptor/peroxisome proliferator-activated receptor-gamma heterodimer. J. Biol. Chem. 2007, 282, 37738–37746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, J.S.; Hajira, A.; Pardo, P.S.; Boriek, A.M. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1alpha network in skeletal muscle. Diabetes 2014, 63, 1546–1559. [Google Scholar] [CrossRef] [Green Version]
- Janko, L.; Sari, Z.; Kovacs, T.; Kis, G.; Szanto, M.; Antal, M.; Juhasz, G.; Bai, P. Silencing of PARP2 Blocks Autophagic Degradation. Cells 2020, 9, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacon-Cabrera, A.; Fermoselle, C.; Salmela, I.; Yelamos, J.; Barreiro, E. MicroRNA expression and protein acetylation pattern in respiratory and limb muscles of Parp-1−/− and Parp-2−/− mice with lung cancer cachexia. Biochim. Biophys. Acta 2015, 1850, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacon-Cabrera, A.; Mateu-Jimenez, M.; Langohr, K.; Fermoselle, C.; Garcia-Arumi, E.; Andreu, A.L.; Yelamos, J.; Barreiro, E. Role of PARP activity in lung cancer-induced cachexia: Effects on muscle oxidative stress, proteolysis, anabolic markers, and phenotype. J. Cell Physiol. 2017, 232, 3744–3761. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Hunter, T.C.; Loudy, D.E.; Dave, V.; Schreiber, V.; Whitsett, J.A. PARP-2 interacts with TTF-1 and regulates expression of surfactant protein-B. J. Biol. Chem 2006, 281, 9600–9606. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Puigserver, P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. USA 2007, 104, 12861–12866. [Google Scholar] [CrossRef] [Green Version]
- Fodor, T.; Szanto, M.; Abdul-Rahman, O.; Nagy, L.; Der, A.; Kiss, B.; Bai, P. Combined Treatment of MCF-7 Cells with AICAR and Methotrexate, Arrests Cell Cycle and Reverses Warburg Metabolism through AMP-Activated Protein Kinase (AMPK) and FOXO1. PLoS ONE 2016, 11, e0150232. [Google Scholar] [CrossRef] [Green Version]
- Hegedűs, C.; Juhász, T.; Fidrus, E.; Janka, E.A.; Juhász, G.; Boros, G.; Paragh, G.; Uray, K.; Emri, G.; Remenyik, É.; et al. Cyclobutane pyrimidine dimers from UVB exposure induce a hypermetabolic state in keratinocytes via mitochondrial oxidative stress. Redox Biol. 2021, 38, 101808. [Google Scholar] [CrossRef]
- Dagda, R.K. Mito-Morphology Macro. Available online: https://imagejdocu.tudor.lu/plugin/morphology/mitochondrial_morphology_macro_plug-in/start (accessed on 24 June 2020).
- Russ, J.C.; Neal, B.F. The Image Processing Handbook; CRC Publications: Boca Raton, FL, USA, 2016. [Google Scholar] [CrossRef]
- Stauffer, W.; Sheng, H.; Lim, H.N. EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep. 2018, 8, 15764. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Sári, Z.; Mikó, E.; Kovács, T.; Jankó, L.; Csonka, T.; Sebő, E.; Toth, J.; Tóth, D.; Árkosy, P.; Boratkó, A.; et al. Indolepropionic acid, a metabolite of the microbiome, has cytostatic properties in breast cancer by activating AHR and PXR receptors and inducing oxidative stress. Cancers 2020, 12, 2411. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Long, A.; Kristian, T. Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3-dependent mechanism in male mice. J. Neurosci. Res. 2019, 97, 975–990. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Long, A.; Scafidi, S.; Kristian, T. Interplay between NAD+ and acetyl-CoA metabolism in ischemia-induced mitochondrial pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2060–2067. [Google Scholar] [CrossRef]
- Lovy, A.; Ahumada-Castro, U.; Bustos, G.; Farias, P.; Gonzalez-Billault, C.; Molgó, J.; Cardenas, C. Concerted Action of AMPK and Sirtuin-1 Induces Mitochondrial Fragmentation Upon Inhibition of Ca2+ Transfer to Mitochondria. Front. Cell Dev. Biol. 2020, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD+ precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef]
- Song, S.B.; Park, J.S.; Jang, S.Y.; Hwang, E.S. Nicotinamide Treatment Facilitates Mitochondrial Fission through Drp1 Activation Mediated by SIRT1-Induced Changes in Cellular Levels of cAMP and Ca2+. Cells 2021, 10, 612. [Google Scholar] [CrossRef]
- Liu, G.; Park, S.H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2017, 26, 849–863. [Google Scholar] [CrossRef]
- Pirinen, E.; Cantó, E.; Jo, S.K.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef]
- Rodriguez-Vargas, J.M.; Martin-Hernandez, K.; Wang, W.; Kunath, N.; Suganthan, R.; Amé, J.C.; Oliver, F.J.; Ye, J.; Bjørås, M.; Dantzer, F. Parp3 promotes astrocytic differentiation through a tight regulation of Nox4-induced ROS and mTorc2 activation. Cell Death Dis. 2020, 11, 954. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [Green Version]
- White, A.T.; Schenk, S. NAD+/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E308–E321. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.T.; Huang, D.Y.; Hu, C.J.; Wu, D.; Lin, W.W. Energy adaptive response during parthanatos is enhanced by PD98059 and involves mitochondrial function but not autophagy induction. Biochim. Biophys. Acta 2013, 1843, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, P.; Horvath, B.; Rajesh, M.; Varga, Z.V.; Gariani, K.; Ryu, D.; Cao, Z.; Holovac, E.; Park, O.; Zhou, Z.; et al. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J. Hepatol. 2017, 66, 589–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakouri, N.B.; Durhuus, J.A.; Regnell, C.E.; Angleys, M.; Desler, C.; Hasan-Olive, M.M.; Martin-Pardillos, A.; Tsaalbi-Shtylik, A.; Thomsen, K.; Lauritzen, M.; et al. Rev1 contributes to proper mitochondrial function via the PARP-NAD+-SIRT1-PGC1alpha axis. Sci. Rep. 2017, 7, 12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mázala, D.A.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.; Rauch, B.; Balla, N.; Ujlaki, G.; Kis, G.; Abdul-Rahman, O.; Kristof, E.; Sipos, A.; Antal, M.; Toth, A.; et al. Olaparib induces browning of in vitro cultures of human primary white adipocytes. Biochem. Pharmacol. 2019, 25, 30250–30253. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Bernstein, D. Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium 2016, 60, 88–101. [Google Scholar] [CrossRef] [Green Version]
- Onodera, Y.; Nam, J.M.; Horikawa, M.; Shirato, H.; Sabe, H. Arf6-driven cell invasion is intrinsically linked to TRAK1-mediated mitochondrial anterograde trafficking to avoid oxidative catastrophe. Nat. Commun. 2018, 9, 2682. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, F.M.; Torelli, N.Q.; Kowaltowski, A.J. Mitochondrial Retrograde Signaling: Triggers, Pathways, and Outcomes. Oxid. Med. Cell. Longev. 2015, 2015, 482582. [Google Scholar] [CrossRef] [Green Version]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Curtin, N.; Szabo, C. Therapeutic Applications of PARP Inhibitors: Anticancer Therapy and Beyond. Mol. Asp. Med. 2013, 6, 1043–1258. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Burkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.A.; Kang, H.C.; Haince, J.F.; Lee, Y.I.; Zhang, J.; Chi, Z.; West, A.B.; Koehler, R.C.; Poirier, G.G.; Dawson, T.M.; et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat. Med. 2011, 17, 692–699. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Catalog Number | Dilution |
|---|---|---|---|
| TOMM20 | Abcam (Cambridge, MA, USA) | ab56783 | 1:200 |
| LC3A/B Alexa Fluor 488 conjugate | Cell Signaling Technology (Danvers, MA, USA) | 13082 | 1:50 |
| Goat-anti mouse IgG Alexa Fluor 546 | Thermo Fisher Scientific (Walthan, MA, USA) | A-11003 | 1:1000 |
| Gene Name | Primers |
|---|---|
| COX17 | 5′-CGTGATGCGTGCATCATTGA-3′ 5′-CATTCACAAAGTAGGCCACC-3′ |
| Cyclophilin | 5′-TGGAGAGCACCAAGACAGACA-3′ 5′-TGCCGGAGTCGACAATGAT-3′ |
| Cyt C | 5′-TCCATCAGGGTATCCTCTCC-3′ 5′-GGAGGCAAGCATAAGACTGG-3′ |
| Drp1 | 5′-TCGAGTCCCCATTCATTGCAGT-3′ 5′-GAAGAAGGTCCCTGCCCACTAG-3′ |
| Fis1 | 5′-CTGGACTCATTGGACTGGCTGT-3′ 5′-AGAGGTAGACTACAGGGGTGCA-3′ |
| Mfn1 | 5′-GGTCCTGCAATCACTCTGTCCT-3′ 5′-CCCATTTCACCCCTTCAGACCT-3′ |
| Mfn2 | 5′-GTGATCAGGTTCAGCGTCCTCT-3′ 5′-CCACTCCTCCGACCACAAGAAT-3′ |
| Ndufa2 | 5′-GCACACATTTCCCCACACTG-3′ 5′-CCCAACCTGCCCATTCTGAT-3′ |
| Ndufb3 | 5′-TACCACAAACGCAGCAAACC-3′ 5′-AAGGGACGCCATTAGAAACG-3′ |
| Ndufb5 | 5′-CTTCGAACTTCCTGCTCCTT-3′ 5′-GGCCCTGAAAAGAACTACG-3′ |
| Nox4 | 5′-GCAGATTTACTCTGTGTGTTGCAT-3′ 5′-TCCCATCTGTTTGACTGAGGT-3′ |
| Opa1 | 5′-ATTGTGTGCTCTCAGTCAGGCT-3′ 5′-ACCTTTCCCTGACGCCTAGTTC-3′ |
| Parkin | 5′-AAATGCATCTGGAGGGGACGAA-3′ 5′-TAACTGGACCTCTGGCTGCTTC-3′ |
| Parp1 | 5′-GGAGCTGCTCATCTTCAACC-3′ 5′-GCAGTGACATCCCCAGTACA-3′ |
| Parp2 | 5′-GGAAGGCGAGTGCTAAATGAA-3′ 5′-AAGGTCTTCACAGAGTCTCGATTG-3′ |
| Parp3 | 5′-CCTGCTGATAATCGGGTCAT-3′ 5′-TTGTTGTTGTTGCCGATGTT-3′ |
| Pink1 | 5′-GGGAAGAACAGCCTTGAACAGC-3′ 5′-GCAAAGTTCAGTGTTGGCCTCA-3′ |
| Sirt1 | 5′-TGTGAAGTTACTGCAGGAGTGTAAA-3′ 5′-GCATAGATACCGTCTCTTGATCTGAA-3′ |
| 36B4 | 5′-AGATTCGGGATATGCTGTTGG-3′ 5′-AAAGCCTGGAAGAAGGAGGTC-3′ |
| Antibody | Company | Catalog Number | Dilution |
|---|---|---|---|
| GAPDH | Sigma-Aldrich | G9545 | 1:10,000 |
| HSP25 | Enzo Life Sciences | ADI-SPA-801 | 1:1500 |
| HSP70/HSP72 (C92F3A-5) | Enzo Life Sciences | ADI-SPA-810 | 1:8000 |
| HSC70/HSP70 (N27F3-4) | Enzo Life Sciences | ADI-SPA-820 | 1:1000 |
| HSP40 (C-20) | Santa Cruz Biotechnology | sc-1800 | 1:1000 |
| HSP90 (AC88) | Enzo Life Sciences | ADI-SPA-830 | 1:1000 |
| HSP110 | Becton Dickinson Biosciences | 610510 | 1:2500 |
| iNOS | Novus Biologicals | NB300-605 | 1:1000 |
| NTyr | Millipore | 06-284 | 1:1000 |
| OPA1 | Thermo Fisher Scientific | MA5-16149 | 1:1000 |
| Parkin | Thermo Fisher Scientific | PA5-13399 | 1:1000 |
| PARP1 | Cell Signaling Technology | 9532 | 1:2000 |
| PARP2 | Enzo Life Sciences | ALX-210-899 | 1:2000 |
| PARP3 | Novus Biologicals | NBP1-31415 | 1:1000 |
| PAR | Sigma-Aldrich | MABC547 | 1:1000 |
| PINK1 | Novus Biologicals | BC100-494 | 1:1000 |
| SIRT1 | Millipore | 07-131 | 1:1000 |
| 4HNE | Abcam | ab46545 | 1:1000 |
| Anti-mouse IgG, HRP-linked | Sigma-Aldrich | A9044 | 1:2000 |
| Anti-rabbit IgG, HRP-linked | Cell Signaling Technology | 7074 | 1:2000 |
| Anti-β-actin-peroxidase | Sigma-Aldrich | A3854 | 1:20,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jankó, L.; Kovács, T.; Laczik, M.; Sári, Z.; Ujlaki, G.; Kis, G.; Horváth, I.; Antal, M.; Vígh, L.; Bálint, B.L.; et al. Silencing of Poly(ADP-Ribose) Polymerase-2 Induces Mitochondrial Reactive Species Production and Mitochondrial Fragmentation. Cells 2021, 10, 1387. https://doi.org/10.3390/cells10061387
Jankó L, Kovács T, Laczik M, Sári Z, Ujlaki G, Kis G, Horváth I, Antal M, Vígh L, Bálint BL, et al. Silencing of Poly(ADP-Ribose) Polymerase-2 Induces Mitochondrial Reactive Species Production and Mitochondrial Fragmentation. Cells. 2021; 10(6):1387. https://doi.org/10.3390/cells10061387
Chicago/Turabian StyleJankó, Laura, Tünde Kovács, Miklós Laczik, Zsanett Sári, Gyula Ujlaki, Gréta Kis, Ibolya Horváth, Miklós Antal, László Vígh, Bálint L. Bálint, and et al. 2021. "Silencing of Poly(ADP-Ribose) Polymerase-2 Induces Mitochondrial Reactive Species Production and Mitochondrial Fragmentation" Cells 10, no. 6: 1387. https://doi.org/10.3390/cells10061387
APA StyleJankó, L., Kovács, T., Laczik, M., Sári, Z., Ujlaki, G., Kis, G., Horváth, I., Antal, M., Vígh, L., Bálint, B. L., Uray, K., & Bai, P. (2021). Silencing of Poly(ADP-Ribose) Polymerase-2 Induces Mitochondrial Reactive Species Production and Mitochondrial Fragmentation. Cells, 10(6), 1387. https://doi.org/10.3390/cells10061387