
To Trap a Pathogen: Neutrophil Extracellular Traps and Their Role in Mucosal Epithelial and Skin Diseases

 , , , , , and
, , , , , and
Abstract
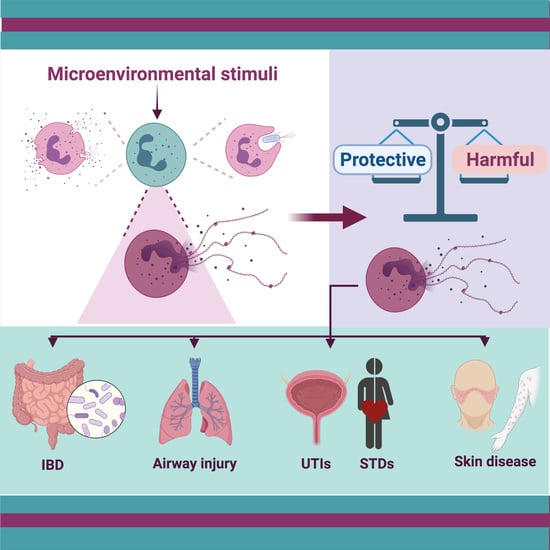
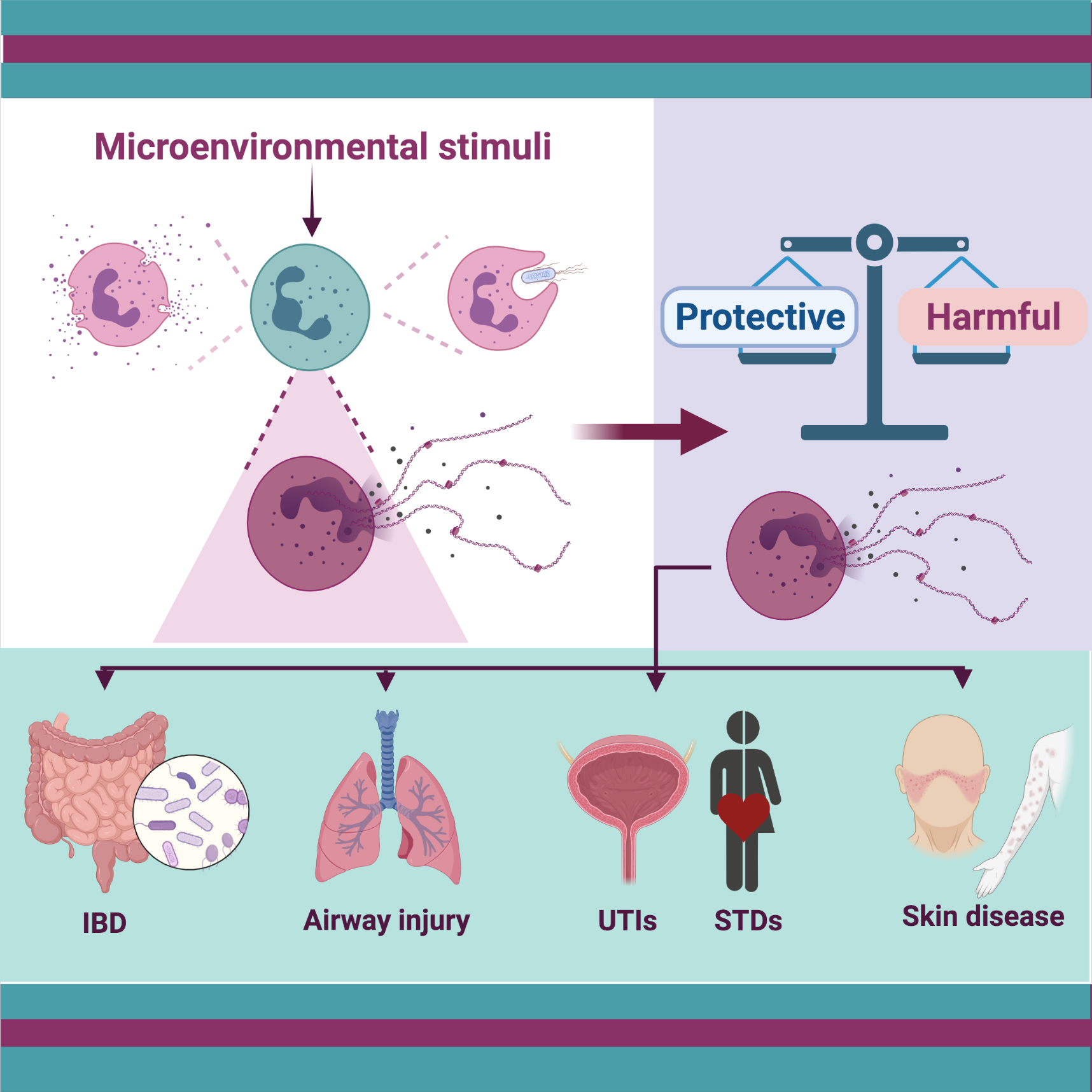
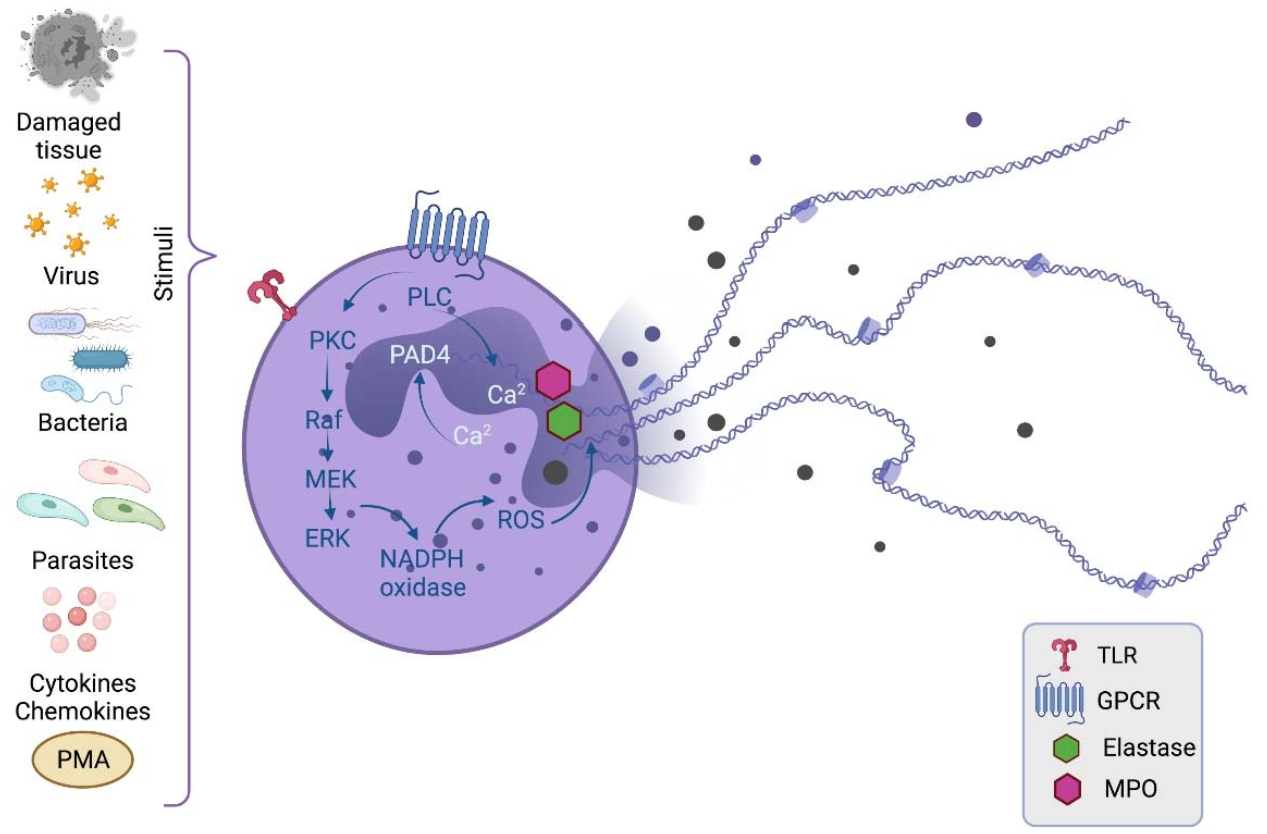
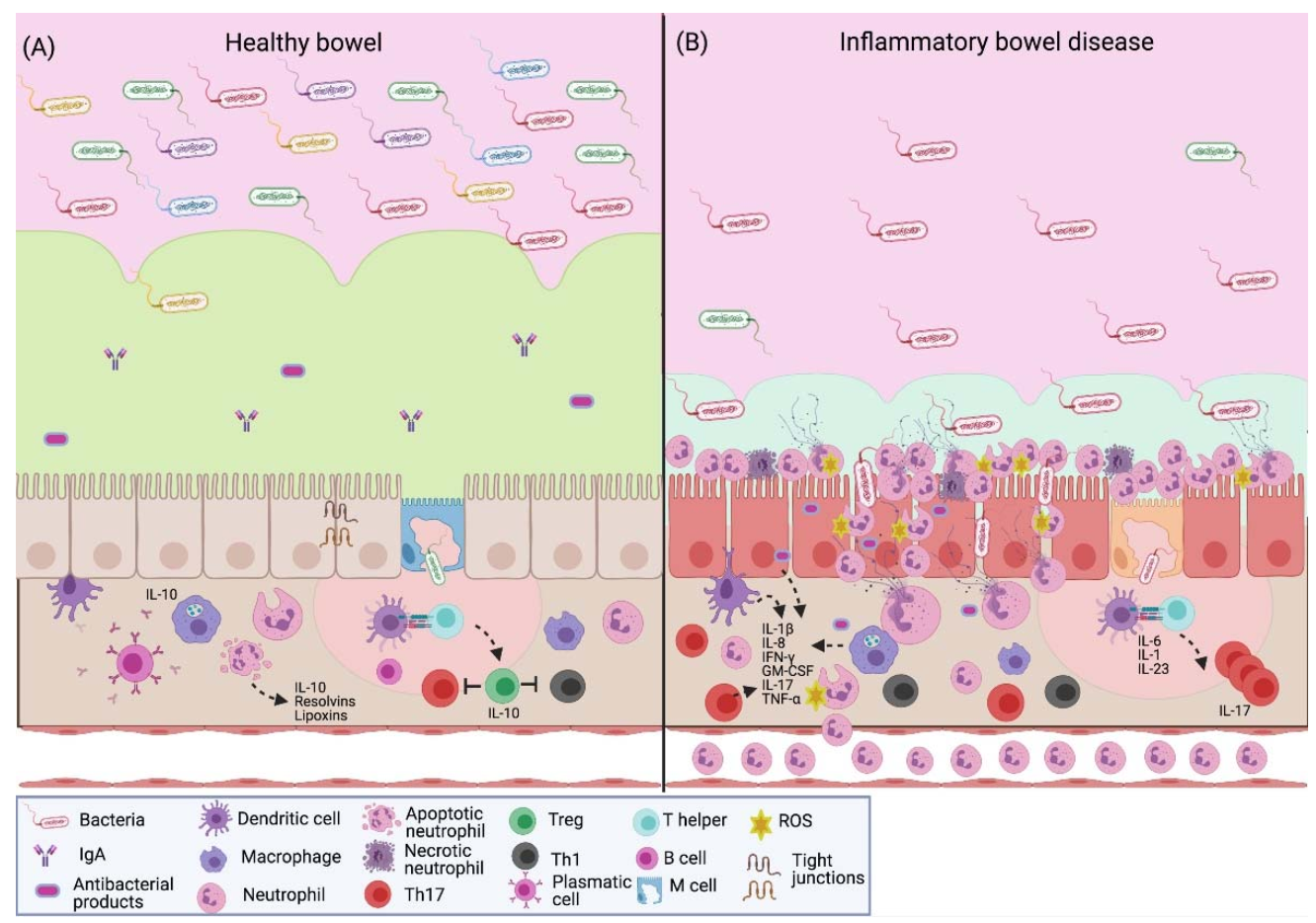
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. NETs in Inflammatory Bowel Disease
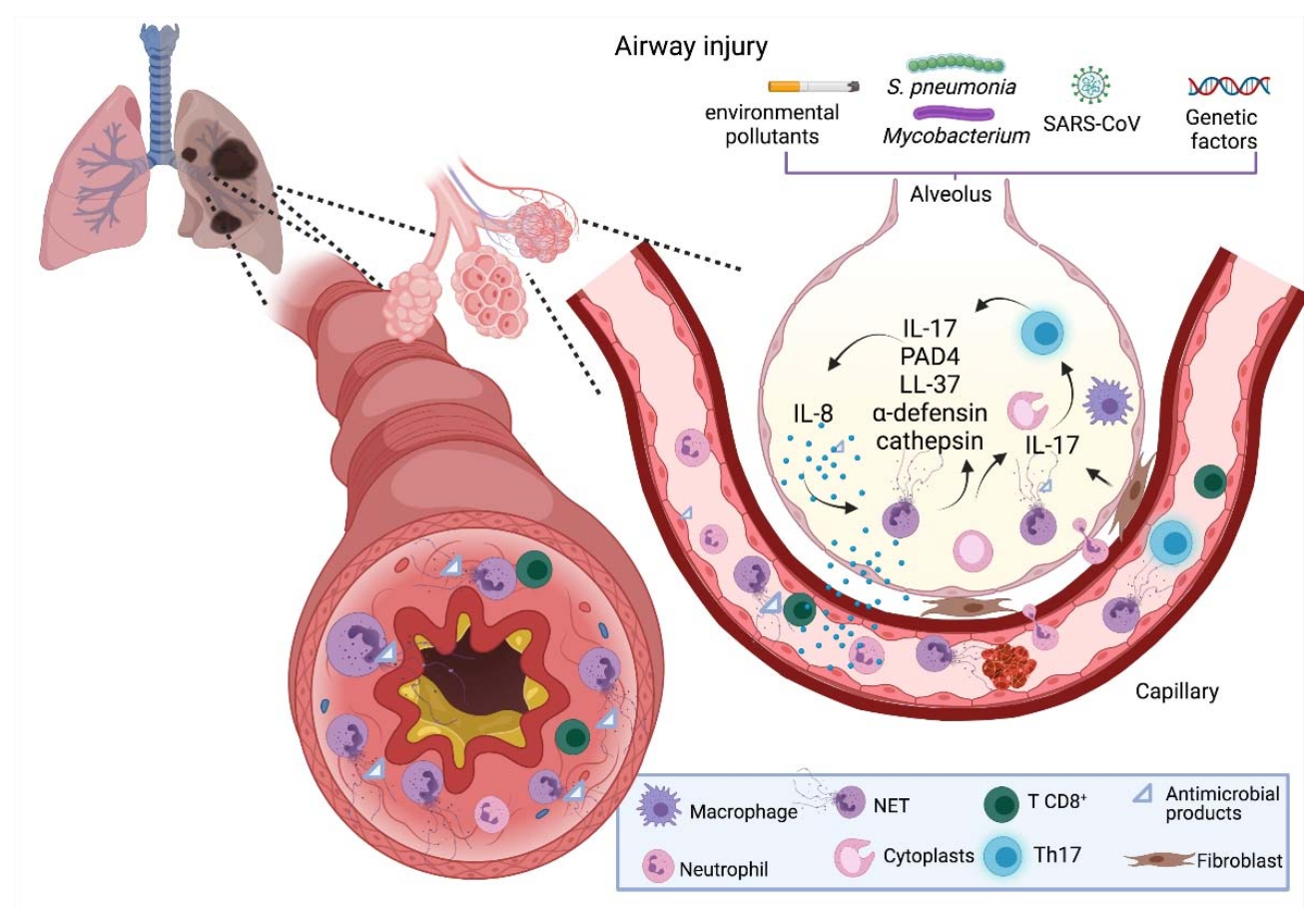
3. NETs and Respiratory Diseases
3.1. Chronic Obstructive Pulmonary Disease
3.2. Asthma
3.3. Pneumonia
3.4. Tuberculosis
3.5. COVID-19
4. NETs in the Genitourinary Tract
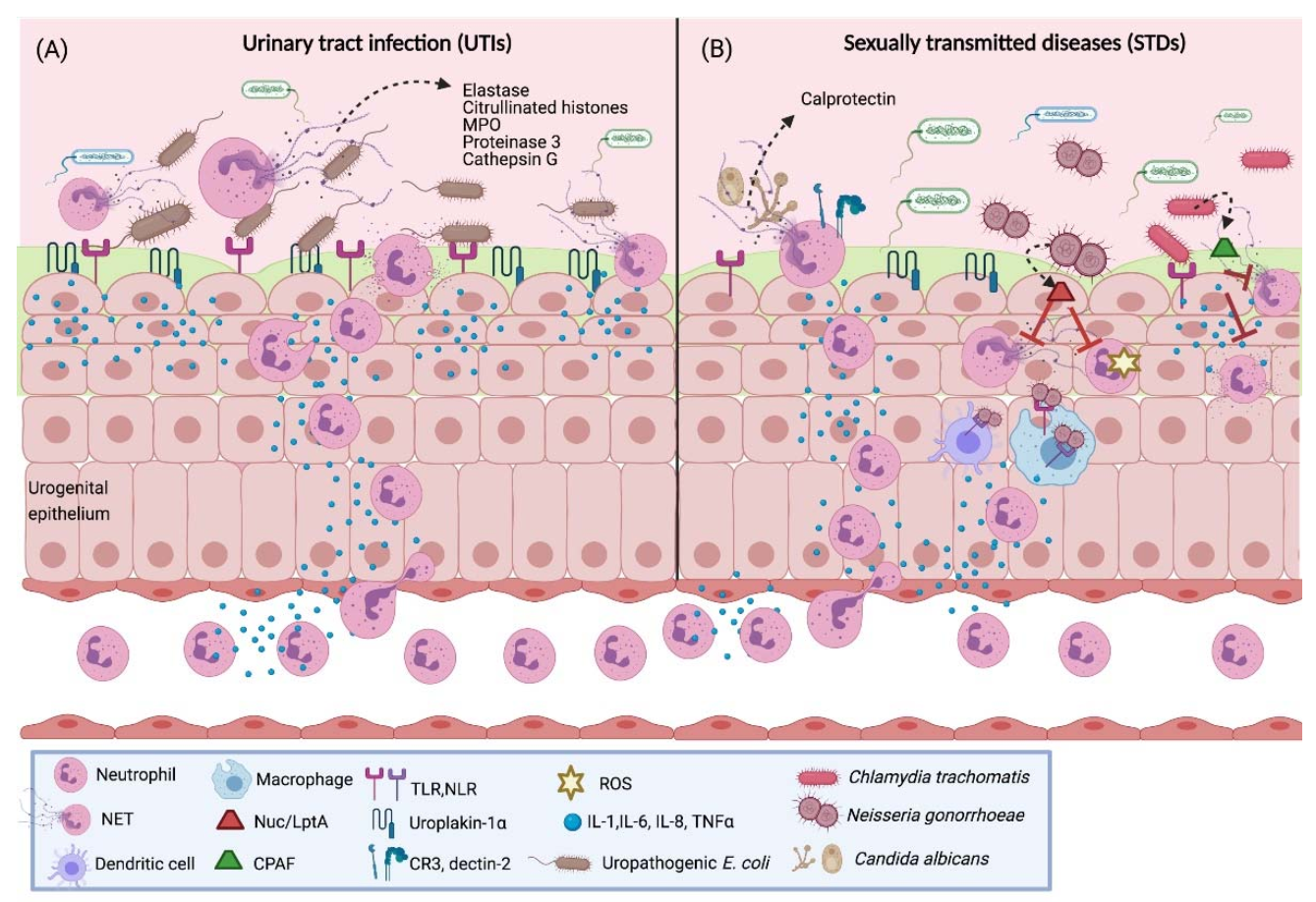
4.1. Urinary Tract Infections (UTIs)
4.2. Candidiasis
4.3. Gonorrhea
4.4. Chlamydia Trachomatis
4.5. Trichomonas Vaginalis
5. NETs and Skin Diseases
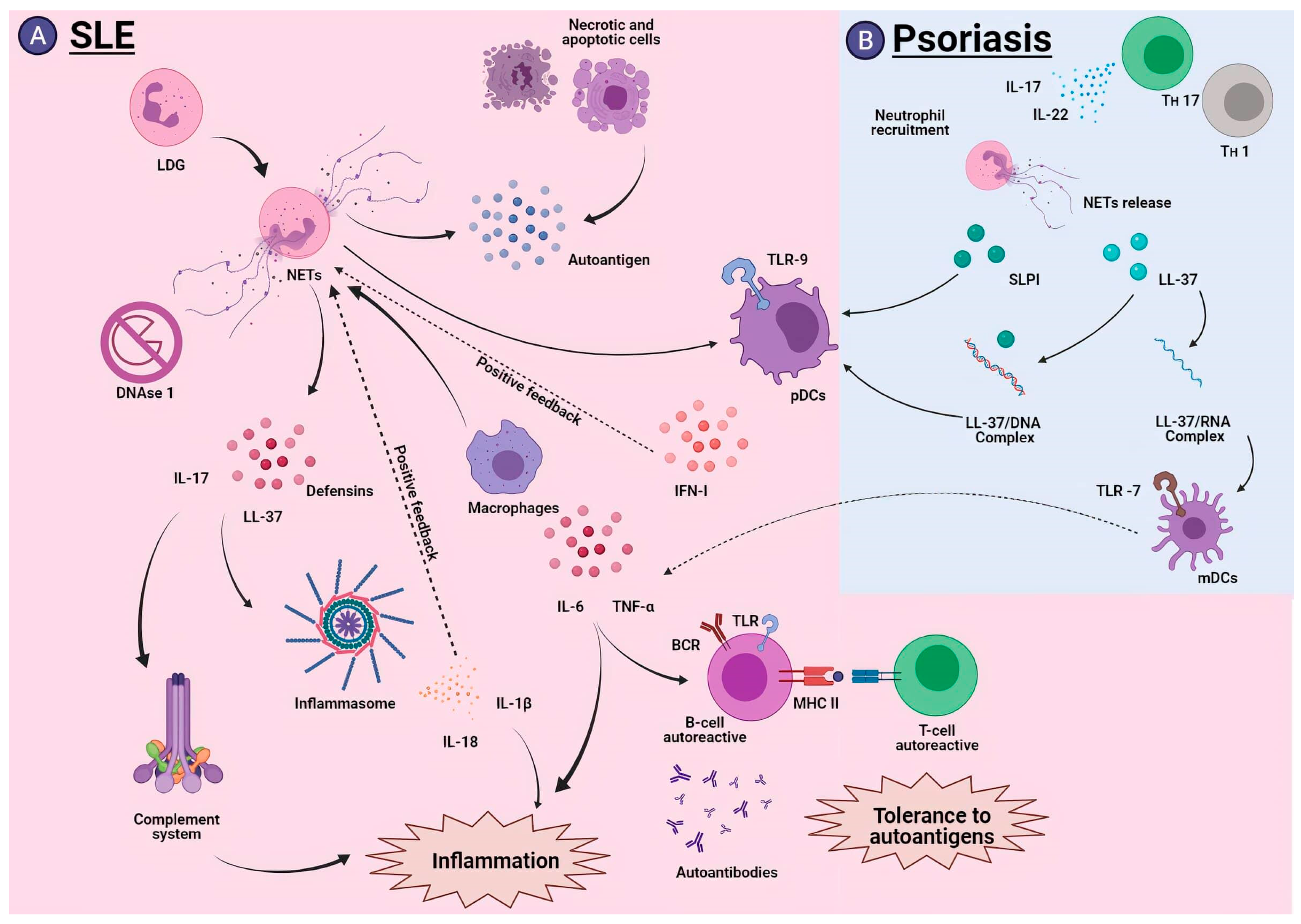
5.1. Systemic Lupus Erythematosus
5.2. Psoriasis
5.3. ANCA-Associated Small Vessel Vasculitis (AAV)
6. NETs in Other Mucosal Surfaces
6.1. Keratitis
6.2. Behçet’s Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef]
- Rojas-Espinosa, O.; Arce-Paredes, P. Fagocitosis: Mecanismos y consecuencias Primera parte. Bioquimia 2003, 28, 19–30. [Google Scholar]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 2014, 5, 448. [Google Scholar] [CrossRef] [Green Version]
- Rørvig, S.; Østergaard, O.; Heegaard, N.H.; Borregaard, N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: Correlation with transcriptome profiling of neutrophil precursors. J. Leukoc. Biol. 2013, 94, 711–721. [Google Scholar] [CrossRef]
- Rørvig, S.; Honore, C.; Larsson, L.I.; Ohlsson, S.; Pedersen, C.C.; Jacobsen, L.C.; Cowland, J.B.; Garred, P.; Borregaard, N. Ficolin-1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fMLP. J. Leukoc. Biol. 2009, 86, 1439–1449. [Google Scholar] [CrossRef] [Green Version]
- Angosto, M.C. Estallido Respiratorio De Los Fagocitos. Anal. Real Acad. Nac. Farm. 2005, 71, 365–386. [Google Scholar]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microbes Infect. 2003, 5, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alasmari, S.Z. In Vivo Imaging of Neutrophil Extracellular Traps (NETs): Visualization Methods and Outcomes. BioMed Res. Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeltz, S.; Amini, P.; Anders, H.-J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T. To NET or not to NET: Current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Biol. Chem. 2010, 191, 677–691. [Google Scholar]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. Int. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Gupta, A.K.; Giaglis, S.; Hasler, P.; Hahn, S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS ONE 2014, 9, e97088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Dicker, A.J.; Crichton, M.L.; Pumphrey, E.G.; Cassidy, A.J.; Suarez-Cuartin, G.; Sibila, O.; Furrie, E.; Fong, C.J.; Ibrahim, W.; Brady, G. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2018, 141, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, P.R.; Palmer, L.J.; Chapple, I.L. Neutrophil extracellular traps as a new paradigm in innate immunity: Friend or foe? Periodontology 2013, 63, 165–197. [Google Scholar] [CrossRef]
- Pullan, J.; Greenwood, H.; Walton, G.M.; Stockley, R.A.; Sapey, E. Neutrophil extracellular traps (NETs) in COPD: A potential novel mechanism for host damage in acute exacerbations. Eur. Respir. Soc. 2015, 46, 5055. [Google Scholar]
- Qin, J.; Fu, S.; Speake, C.; Greenbaum, C.; Odegard, J. NETosis-associated serum biomarkers are reduced in type 1 diabetes in association with neutrophil count. Clin. Exp. Immunol. 2016, 184, 318–322. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryk, A.H.; Prior, S.M.; Plens, K.; Konieczynska, M.; Hohendorff, J.; Malecki, M.T.; Butenas, S.; Undas, A. Predictors of neutrophil extracellular traps markers in type 2 diabetes mellitus: Associations with a prothrombotic state and hypofibrinolysis. Cardiovasc. Diabetol. 2019, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Chiodoni, C.; Guarnotta, C.; Cappetti, B.; Casalini, P.; Piconese, S.; Parenza, M.; Guiducci, C.; Vitali, C. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012, 120, 3007–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabelloni, M.L.; Sabbione, F.; Iula, L.; Keitelman, I.; Jancic, C.; Giordano, M.; Geffner, J.; Trevani, A. Trampas extracelulares de neutrófilos: Una novedosa estrategia antiinfecciosa empleando moléculas antimicrobianas largamente conocidas. Quim. Viva 2013, 12, 3–13. [Google Scholar]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; Von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 2017, 6, e24437. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Silva, F.; Gatica, T.; Pavez, C. Etiología y fisiopatología de la enfermedad inflamatoria intestinal. Rev. Médica Clínica Las Condes 2019, 30, 262–272. [Google Scholar] [CrossRef]
- Ginsburg, I.; Korem, M.; Koren, E.; Varani, J. Pro-inflammatory agents released by pathogens, dying host cells, and neutrophils act synergistically to destroy host tissues: A working hypothesis. J. Inflamm. Res. 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Ma, X. Role of Nod2 in the development of Crohn’s disease. Microbes Infect. 2009, 11, 912–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, D.L.R.; Castañeda, N.M.S.; Sánchez, H.V. Una mirada actualizada a la patogenia de la enfermedad inflamatoria intestinal. Arch. Cuba. Gastroenterol. 2021, 1, 3. [Google Scholar]
- Rubin, D.T.; Ananthakrishnan, A.N.; Siegel, C.A.; Sauer, B.G.; Long, M.D. ACG clinical guideline: Ulcerative colitis in adults. Am. J. Gastroenterol. 2019, 114, 384–413. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.L. Harrison: Principios de Medicina Interna (18a); McGraw Hill: Ciudad de México, México, 2012. [Google Scholar]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Wéra, O.; Lancellotti, P.; Oury, C. The dual role of neutrophils in inflammatory bowel diseases. J. Clin. Med. 2016, 5, 118. [Google Scholar] [CrossRef]
- Dinallo, V.; Marafini, I.; Di Fusco, D.; Laudisi, F.; Franzè, E.; Di Grazia, A.; Figliuzzi, M.M.; Caprioli, F.; Stolfi, C.; Monteleone, I. Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis. J. Crohn’s Colitis 2019, 13, 772–784. [Google Scholar] [CrossRef]
- Mohamed, A.S.; Shehta, A.; Sheta, H.A.E.A.S. Neutrophil extracellular traps-associated protein peptidyl arginine deaminase 4 immunohistochemical expression in ulcerative colitis and its association with the prognostic predictors. Pathol. Res. Pract. 2020, 216, 153102. [Google Scholar]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J. Neutrophil extracellular traps induce intestinal damage and thrombotic tendency in inflammatory bowel disease. J. Crohn’s Colitis 2020, 14, 240–253. [Google Scholar] [CrossRef]
- Angelidou, I.; Chrysanthopoulou, A.; Mitsios, A.; Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Ritis, D.; Tsironidou, V.; Moschos, I.; Dalla, V. REDD1/autophagy pathway is associated with neutrophil-driven IL-1β inflammatory response in active ulcerative colitis. J. Immnol. 2018, 200, 3950–3961. [Google Scholar] [CrossRef] [PubMed]
- Drury, B.; Hardisty, G.; Gray, R.D.; Ho, G.-T. Neutrophil Extracellular Traps in Inflammatory Bowel Disease: Pathogenic Mechanisms and Clinical Translation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Forum of International Respiratory Societies. The Global Impact of Respiratory Disease, 2nd ed.; European Respiratory Society: Sheffield, UK, 2017. [Google Scholar]
- C.S.G. of the International. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. 2020. Available online: https://covid19.who.int/ (accessed on 22 April 2021).
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.-D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rabe, K.F.; Watz, H. Chronic Obstructive Pulmonary Disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Singh, D.; Edwards, L.; Tal-Singer, R.; Rennard, S. Sputum neutrophils as a biomarker in COPD: Findings from the ECLIPSE study. Respir. Res. 2010, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Baraldo, S.; Turato, G.; Badin, C.; Bazzan, E.; Beghé, B.; Zuin, R.; Calabrese, F.; Casoni, G.; Maestrelli, P.; Papi, A. Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax 2004, 59, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Caramori, G.; Gnemmi, I.; Contoli, M.; Bristot, L.; Capelli, A.; Ricciardolo, F.L.M.; Magno, F.; D’Anna, S.E.; Zanini, A. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax 2009, 64, 968–975. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, F.; Marwitz, S.; Holz, O.; Kirsten, A.; Bahmer, T.; Waschki, B.; Magnussen, H.; Rabe, K.F.; Goldmann, T.; Uddin, M. Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir. Med. 2015, 109, 1360–1362. [Google Scholar] [CrossRef] [Green Version]
- Stockley, J.A.; Walton, G.M.; Lord, J.M.; Sapey, E. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: The neutrophil as an immunotherapeutic target. Int. Immunopharmacol. 2013, 17, 1211–1217. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef]
- Lugli, E.B.; Correia, R.E.; Fischer, R.; Lundberg, K.; Bracke, K.R.; Montgomery, A.B.; Kessler, B.M.; Brusselle, G.G.; Venables, P.J. Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: Implications for autoimmunity in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, T.K.; Gibson, P.G.; Simpson, J.L.; McDonald, V.M.; Wood, L.G.; Baines, K.J. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology 2016, 21, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locksley, R.M. Asthma and allergic inflammation. Cell 2010, 140, 777–783. [Google Scholar] [CrossRef] [Green Version]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; DuPont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immnol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, B.N.; Stein, R.T. Neutrophil extracellular traps in pulmonary diseases: Too much of a good thing? Front. Immunol. 2016, 7, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, K.J.; Simpson, J.L.; Wood, L.G.; Scott, R.J.; Gibson, P.G. Systemic upregulation of neutrophil α-defensins and serine proteases in neutrophilic asthma. Thorax 2011, 66, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldar, P.; Pavord, I.D. Noneosinophilic asthma: A distinct clinical and pathologic phenotype. J. Allergy Clin. Immunol. 2007, 119, 1043–1052. [Google Scholar] [CrossRef]
- Pham, D.; Ban, G.Y.; Kim, S.H.; Shin, Y.; Ye, Y.M.; Chwae, Y.J.; Park, H.S. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin. Exp. Allergy 2017, 47, 57–70. [Google Scholar] [CrossRef]
- Uddin, M.; Watz, H.; Malmgren, A.; Pedersen, F. NETopathic inflammation in chronic obstructive pulmonary disease and severe asthma. Front. Immunol. 2019, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Douda, D.N.; Brüggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.-E.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3, 4747. [Google Scholar] [CrossRef] [Green Version]
- Welte, T.; Torres, A.; Nathwani, D. Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax 2012, 67, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, F.; Giaglis, S.; Hahn, S.; Blum, C.A.; Baumgartner, C.; Kutz, A.; van Breda, S.V.; Mueller, B.; Schuetz, P.; Christ-Crain, M. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: Secondary analysis of a randomised controlled trial. Eur. Respir. J. 2018, 51, 1701389. [Google Scholar] [CrossRef]
- Narayana Moorthy, A.; Narasaraju, T.; Rai, P.; Perumalsamy, R.; Tan, K.; Wang, S.; Engelward, B.; Chow, V.T. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front. Immunol. 2013, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef] [Green Version]
- Gray, R.D. NETs in pneumonia: Is just enough the right amount? Eur. Respir. Soc. 2018, 51, 1800619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, J.T.; Simpson, A.J.; Aziz, R.K.; Liu, G.Y.; Kristian, S.A.; Kotb, M.; Feramisco, J.; Nizet, V. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 2006, 16, 396–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Köckritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storisteanu, D.M.; Pocock, J.M.; Cowburn, A.S.; Juss, J.K.; Nadesalingam, A.; Nizet, V.; Chilvers, E.R. Evasion of neutrophil extracellular traps by respiratory pathogens. Am. J. Respir. Cell Mol. Biol. 2017, 56, 423–431. [Google Scholar] [CrossRef]
- Kiran, D.; Podell, B.K.; Chambers, M.; Basaraba, R.J. Host-directed therapy targeting the Mycobacterium tuberculosis granuloma: A review. Proc. Semin. Immunopathol. 2016, 38, 167–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, P.; Aichele, P.; Bandermann, S.; Hauser, A.E.; Lu, B.; Gerard, N.P.; Gerard, C.; Ehlers, S.; Mollenkopf, H.J.; Kaufmann, S.H. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur. J. Immunol. 2003, 33, 2676–2686. [Google Scholar] [CrossRef]
- Ramos-Kichik, V.; Mondragón-Flores, R.; Mondragón-Castelán, M.; Gonzalez-Pozos, S.; Muñiz-Hernandez, S.; Rojas-Espinosa, O.; Chacón-Salinas, R.; Estrada-Parra, S.; Estrada-García, I. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis 2009, 89, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Filio-Rodríguez, G.; Estrada-García, I.; Arce-Paredes, P.; Moreno-Altamirano, M.M.; Islas-Trujillo, S.; Ponce-Regalado, M.D.; Rojas-Espinosa, O. In vivo induction of neutrophil extracellular traps by Mycobacterium tuberculosis in a guinea pig model. Innate Immun. 2017, 23, 625–637. [Google Scholar] [CrossRef] [PubMed]
- de Melo, M.G.M.; Mesquita, E.D.D.; Oliveira, M.M.; Silva-Monteiro, C.D.; Silveira, A.K.; Malaquias, T.S.; Dutra, T.C.; Galliez, R.M.; Kritski, A.L.; Silva, E.C. Imbalance of NET and alpha-1-antitrypsin in tuberculosis patients is related with hyper inflammation and severe lung tissue damage. Front. Immunol. 2019, 9, 3147. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Peng, Y.-P.; Deng, Z.; Deng, Y.-T.; Ye, J.-Q.; Guo, Y.; Huang, Z.-K.; Luo, Q.; Jiang, H.; Li, J.-M. Mycobacterium tuberculosis infection induces low-density granulocyte generation by promoting neutrophil extracellular trap formation via ROS pathway. Front. Microbiol. 2019, 10, 1468. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Stratton, C.W.; Tang, Y.W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Gao, D.; Li, Y.; Zhuang, Z.; Cao, P.; Lou, Y.; Yang, L. An Updated Comparison of COVID-19 and Influenza. 2020. Available online: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3573503 (accessed on 21 April 2021).
- Chu, H.; Chan, J.F.-W.; Wang, Y.; Yuen, T.T.-T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; He, J.; Wu, S. The definition and risks of cytokine release syndrome-like in 11 COVID-19-infected pneumonia critically ill patients: Disease characteristics and retrospective analysis. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Kaveri, S.V.; Sakuntabhai, A.; Gilardin, L.; Bayry, J. Adjunct immunotherapies for the management of severely Ill COVID-19 patients. Cell Rep. Med. 2020, 1, 100016. [Google Scholar] [CrossRef]
- Rodriguez, L.; Pekkarinen, P.T.; Lakshmikanth, T.; Tan, Z.; Consiglio, C.R.; Pou, C.; Chen, Y.; Mugabo, C.H.; Nguyen, N.A.; Nowlan, K. Systems-level immunomonitoring from acute to recovery phase of severe COVID-19. Cell Rep. Med. 2020, 1, 100078. [Google Scholar] [CrossRef]
- Schönrich, G.; Raftery, M.J.; Samstag, Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar] [CrossRef]
- Guan, J.; Wei, X.; Qin, S.; Liu, X.; Jiang, Y.; Chen, Y.; Chen, Y.; Lu, H.; Qian, J.; Wang, Z. Continuous tracking of COVID-19 patients’ immune status. Int. Immunopharmacol. 2020, 89, 107034. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Xiang, P.; Pu, L.; Xiong, H.; Li, C.; Zhang, M.; Tan, J.; Xu, Y.; Song, R. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M.; Wolf, D.A.; Zhao, B.; Akkanti, B.; McDonald, M.; Lelenwa, L.; Reilly, N.; Ottaviani, G.; Elghetany, M.T.; Trujillo, D.O. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): Report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc. Pathol. 2020, 48, 107233. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Golonka, R.M.; Saha, P.; Yeoh, B.S.; Chattopadhyay, S.; Gewirtz, A.T.; Joe, B.; Vijay-Kumar, M. Harnessing Innate Immunity to Eliminate SARS-CoV-2 and Ameliorate COVID-19 Disease. Physiol. Genom. 2020, 52, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Iijima, N.; Thompson, J.; Iwasaki, A. Dendritic cells and macrophages in the genitourinary tract. Mucosal Immunol. 2008, 1, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laupland, K.; Ross, T.; Pitout, J.; Church, D.; Gregson, D. Community-onset urinary tract infections: A population-based assessment. Infection 2007, 35, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Nielubowicz, G.R.; Mobley, H.L. Host–pathogen interactions in urinary tract infection. Nat. Rev. Urol. 2010, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kwon, K.; Tsitrin, T.; Bekele, S.; Sikorski, P.; Nelson, K.E.; Pieper, R. Characterization of early phase neutrophil extracellular traps in urinary tract infections. PLoS Pathog. 2017, 13, e1006151. [Google Scholar] [CrossRef]
- Bauters, T.G.; Dhont, M.A.; Temmerman, M.I.; Nelis, H.J. Prevalence of vulvovaginal candidiasis and susceptibility to fluconazole in women. Am. J. Obstet. Gynecol. 2002, 187, 569–574. [Google Scholar] [CrossRef]
- Beigi, R.H.; Meyn, L.A.; Moore, D.M.; Krohn, M.A.; Hillier, S.L. Vaginal yeast colonization in nonpregnant women: A longitudinal study. Obstet. Gynecol. 2004, 104, 926–930. [Google Scholar] [CrossRef]
- Martins, N.; Ferreira, I.C.; Barros, L.; Silva, S.; Henriques, M. Candidiasis: Predisposing factors, prevention, diagnosis and alternative treatment. Mycopathologia 2014, 177, 223–240. [Google Scholar] [CrossRef]
- Yano, J.; Lilly, E.; Barousse, M.; Fidel, P.L. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect. Immun. 2010, 78, 5126–5137. [Google Scholar] [CrossRef] [Green Version]
- Jabra-Rizk, M.A.; Kong, E.F.; Tsui, C.; Nguyen, M.H.; Clancy, C.J.; Fidel, P.L.; Noverr, M. Candida albicans pathogenesis: Fitting within the host-microbe damage response framework. Infect. Immun. 2016, 84, 2724–2739. [Google Scholar] [CrossRef] [Green Version]
- Fidel, P.L.; Barousse, M.; Espinosa, T.; Ficarra, M.; Sturtevant, J.; Martin, D.H.; Quayle, A.J.; Dunlap, K. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect. Immun. 2004, 72, 2939–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An extracellular matrix–based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immnol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.-Y.; Weng, C.-L.; Jheng, M.-J.; Kan, H.-W.; Hsieh, S.-T.; Liu, F.-T.; Wu-Hsieh, B.A. Candida albicans triggers NADPH oxidase-independent neutrophil extracellular traps through dectin-2. PLoS Pathog. 2019, 15, e1008096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- WHO. Report on Global Sexually Transmitted Infection Surveillance 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Dela, H.; Attram, N.; Behene, E.; Kumordjie, S.; Addo, K.K.; Nyarko, E.O.; Kyei, N.N.; Carroll, J.N.A.; Kwakye, C.; Duplessis, C.A. Risk factors associated with gonorrhea and chlamydia transmission in selected health facilities in Ghana. BMC Infect. Dis. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Wi, T.; Lahra, M.M.; Ndowa, F.; Bala, M.; Dillon, J.-A.R.; Ramon-Pardo, P.; Eremin, S.R.; Bolan, G.; Unemo, M. Antimicrobial resistance in Neisseria gonorrhoeae: Global surveillance and a call for international collaborative action. PLoS Med. 2017, 14, e1002344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criss, A.K.; Seifert, H.S. A bacterial siren song: Intimate interactions between Neisseria and neutrophils. Nat. Rev. Microbiol. 2012, 10, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Handing, J.W.; Criss, A.K. The lipooligosaccharide-modifying enzyme LptA enhances gonococcal defence against human neutrophils. Cell. Microbiol. 2015, 17, 910–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, C.W.; Seifert, H.S. Neisseria gonorrhoeae elicits extracellular traps in primary neutrophil culture while suppressing the oxidative burst. MBio 2015, 6, 245214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juneau, R.A.; Stevens, J.S.; Apicella, M.A.; Criss, A.K. A thermonuclease of Neisseria gonorrhoeae enhances bacterial escape from killing by neutrophil extracellular traps. J. Infect. Dis. 2015, 212, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handing, J.W.; Ragland, S.A.; Bharathan, U.V.; Criss, A.K. The MtrCDE efflux pump contributes to survival of Neisseria gonorrhoeae from human neutrophils and their antimicrobial components. Front. Microbiol. 2018, 9, 2688. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Juneau, R.A.; Criss, A.K.; Cornelissen, C.N. Neisseria gonorrhoeae evades calprotectin-mediated nutritional immunity and survives neutrophil extracellular traps by production of TdfH. Infect. Immun. 2016, 84, 2982–2994. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, M.; Sood, S.; Mukherjee, A.; Muralidhar, S.; Bala, M. Genital Chlamydia trachomatis: An update. Indian J. Med. Res. 2013, 138, 303. [Google Scholar]
- Rajeeve, K.; Das, S.; Prusty, B.K.; Rudel, T. Chlamydia trachomatis paralyses neutrophils to evade the host innate immune response. Nat. Microbiol. 2018, 3, 824–835. [Google Scholar] [CrossRef]
- Kissinger, P. Trichomonas vaginalis: A review of epidemiologic, clinical and treatment issues. BMC Infect. Dis. 2015, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Leitsch, D. Recent advances in the molecular biology of the protist parasite Trichomonas vaginalis. Fac. Rev. 2021, 10, 26. [Google Scholar] [CrossRef]
- Mercer, F.; Ng, S.H.; Brown, T.M.; Boatman, G.; Johnson, P.J. Neutrophils kill the parasite Trichomonas vaginalis using trogocytosis. PLoS Biol. 2018, 16, e2003885. [Google Scholar] [CrossRef]
- Midgley, A.; Beresford, M. Cellular localization of nuclear antigen during neutrophil apoptosis: Mechanism for autoantigen exposure? Lupus 2011, 20, 641–646. [Google Scholar] [CrossRef]
- Ren, Y.; Tang, J.; Mok, M.; Chan, A.W.; Wu, A.; Lau, C. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Linden, M.; van den Hoogen, L.L.; Westerlaken, G.H.; Fritsch-Stork, R.D.; van Roon, J.A.; Radstake, T.R.; Meyaard, L. Neutrophil extracellular trap release is associated with antinuclear antibodies in systemic lupus erythematosus and anti-phospholipid syndrome. Rheumatology 2018, 57, 1228–1234. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Pascual, V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Båve, U.; Magnusson, M.; Eloranta, M.-L.; Perers, A.; Alm, G.V.; Rönnblom, L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. J. Immnol. 2003, 171, 3296–3302. [Google Scholar] [CrossRef]
- Means, T.K.; Latz, E.; Hayashi, F.; Murali, M.R.; Golenbock, D.T.; Luster, A.D. Human lupus autoantibody–DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Investig. 2005, 115, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [Green Version]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [Green Version]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immnol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.K.; Kaplan, M.J. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr. Opin. Rheumatol. 2015, 27, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap–associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immnol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera-Vargas, A.; Gómez-Martín, D.; Carmona-Rivera, C.; Merayo-Chalico, J.; Torres-Ruiz, J.; Manna, Z.; Hasni, S.; Alcocer-Varela, J.; Kaplan, M.J. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 944–950. [Google Scholar] [CrossRef]
- Dubois, B.; Massacrier, C.; Vanbervliet, B.; Fayette, J.; Brière, F.; Banchereau, J.; Caux, C. Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J. Immnol. 1998, 161, 2223–2231. [Google Scholar]
- Tillack, K.; Breiden, P.; Martin, R.; Sospedra, M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immnol. 2012, 188, 3150–3159. [Google Scholar] [CrossRef]
- Bendorius, M.; Neeli, I.; Wang, F.; Bonam, S.R.; Dombi, E.; Buron, N.; Borgne-Sanchez, A.; Poulton, J.; Radic, M.; Muller, S. The Mitochondrion-lysosome axis in adaptive and innate immunity: Effect of lupus regulator peptide P140 on mitochondria autophagy and NETosis. Front. Immunol. 2018, 9, 2158. [Google Scholar] [CrossRef]
- Muller, S.; Monneaux, F.; Schall, N.; Rashkov, R.K.; Oparanov, B.A.; Wiesel, P.; Geiger, J.M.; Zimmer, R. Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: Results of an early phase II clinical trial. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2008, 58, 3873–3883. [Google Scholar] [CrossRef]
- Jeremic, I.; Djuric, O.; Nikolic, M.; Vlajnic, M.; Nikolic, A.; Radojkovic, D.; Bonaci-Nikolic, B. Neutrophil extracellular traps-associated markers are elevated in patients with systemic lupus erythematosus. Rheumatol. Int. 2019, 39, 1849–1857. [Google Scholar] [CrossRef]
- Hu, S.C.-S.; Yu, H.-S.; Yen, F.-L.; Lin, C.-L.; Chen, G.-S.; Lan, C.-C.E. Neutrophil extracellular trap formation is increased in psoriasis and induces human β-defensin-2 production in epidermal keratinocytes. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Christophers, E.; Metzler, G.; Röcken, M. Bimodal immune activation in psoriasis. Br. J. Dermatol. 2014, 170, 59–65. [Google Scholar] [CrossRef]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immnol. 2011, 187, 490–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA–antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.-H.; Homey, B.; Cao, W.; Wang, Y.-H.; Su, B.; Nestle, F.O. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzeczynska-Moncznik, J.; Wlodarczyk, A.; Zabieglo, K.; Kapinska-Mrowiecka, M.; Marewicz, E.; Dubin, A.; Potempa, J.; Cichy, J. Secretory leukocyte proteinase inhibitor-competent DNA deposits are potent stimulators of plasmacytoid dendritic cells: Implication for psoriasis. J. Immnol. 2012, 189, 1611–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabieglo, K.; Majewski, P.; Majchrzak-Gorecka, M.; Wlodarczyk, A.; Grygier, B.; Zegar, A.; Kapinska-Mrowiecka, M.; Naskalska, A.; Pyrc, K.; Dubin, A. The inhibitory effect of secretory leukocyte protease inhibitor (SLPI) on formation of neutrophil extracellular traps. J. Leukoc. Biol. 2015, 98, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Fang, H.; Dang, E.; Xue, K.; Zhang, J.; Li, B.; Qiao, H.; Cao, T.; Zhuang, Y.; Shen, S. Neutrophil extracellular traps promote inflammatory responses in psoriasis via activating epidermal TLR4/IL-36R crosstalk. Front. Immunol. 2019, 10, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2014, 73, 1854–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennette, J.C.; Falk, R.J. Small-vessel vasculitis. N. Engl. J. Med. 1997, 337, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Hunder, G.G.; Arend, W.P.; Bloch, D.A.; Calabrese, L.H.; Fauci, A.S.; Fries, J.F.; Leavitt, R.Y.; Lie, J.; Lightfoot, R.W., Jr.; Masi, A.T. The American College of Rheumatology 1990 criteria for the classification of vasculitis: Introduction. Arthritis Rheum. 1990, 33, 1065–1067. [Google Scholar] [CrossRef]
- Goeser, M.R.; Laniosz, V.; Wetter, D.A. A practical approach to the diagnosis, evaluation, and management of cutaneous small-vessel vasculitis. Am. J. Clin. Dermatol. 2014, 15, 299–306. [Google Scholar] [CrossRef]
- Marzano, A.V.; Raimondo, M.G.; Berti, E.; Meroni, P.L.; Ingegnoli, F. Cutaneous manifestations of ANCA-associated small vessels vasculitis. Clin. Rev. Allergy Immunol. 2017, 53, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Xiao, H.; Jennette, J.C.; Schneider, W.; Luft, F.C.; Kettritz, R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, D.; Tomaru, U.; Yamamoto, C.; Jodo, S.; Ishizu, A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front. Immunol. 2012, 3, 333. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Zhang, Y.; Yin, S.W.; Gao, X.J.; Shi, W.W.; Wang, Y.; Huang, X.; Wang, L.; Zou, L.Y.; Zhao, J.H. Neutrophil extracellular trap formation is associated with autophagy-related signalling in ANCA-associated vasculitis. Clin. Exp. Immunol. 2015, 180, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Wang, C.-Y.; Chen, J.; Gong, Q.; Zhu, P.; Zheng, F.; Tan, Z.; Gong, F.; Fang, M. High-mobility group box 1 promotes early acute allograft rejection by enhancing IL-6-dependent Th17 alloreactive response. Lab. Investig. 2011, 91, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Brunini, F.; Page, T.H.; Gallieni, M.; Pusey, C.D. The role of monocytes in ANCA-associated vasculitides. Autoimmun. Rev. 2016, 15, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-H.; Ma, T.-T.; Wang, C.; Wang, H.; Chang, D.-Y.; Chen, M.; Zhao, M.-H. High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res. Ther. 2016, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, A.; Grüneboom, A.; Petru, L.; Podolska, M.J.; Kling, L.; Maueröder, C.; Dahms, F.; Christiansen, S.; Günter, L.; Krenn, V. Frontline Science: Aggregated neutrophil extracellular traps prevent inflammation on the neutrophil-rich ocular surface. J. Leukoc. Biol. 2019, 105, 1087–1098. [Google Scholar] [CrossRef]
- Feller, L.; Altini, M.; Khammissa, R.; Chandran, R.; Bouckaert, M.; Lemmer, J. Oral mucosal immunity. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 576–583. [Google Scholar] [CrossRef]
- Leppkes, M.; Schick, M.; Hohberger, B.; Mahajan, A.; Knopf, J.; Schett, G.; Muñoz, L.E.; Herrmann, M. Updates on NET formation in health and disease. Proc. Semin. Arthritis Rheum. 2019, 49, S43–S48. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Scott, B.N.V.; Peiseler, M.; Willson, M.E.; Zeng, Z.; Warrener, P.; Keller, A.E.; Surewaard, B.G.J.; Dozier, E.A.; Korhonen, J.T. Neutrophil extracellular traps confine Pseudomonas aeruginosa ocular biofilms and restrict brain invasion. Cell Host Microbe 2019, 25, 526–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koganti, R.; Yadavalli, T.; Naqvi, R.A.; Shukla, D.; Naqvi, A.R. Pathobiology and treatment of viral keratitis. Exp. Eye Res. 2021, 205, 108483. [Google Scholar] [CrossRef]
- Jin, X.; Zhao, Y.; Zhang, F.; Wan, T.; Fan, F.; Xie, X.; Lin, Z. Neutrophil extracellular traps involvement in corneal fungal infection. Mol. Vis. 2016, 22, 944. [Google Scholar] [PubMed]
- Shan, Q.; Dwyer, M.; Rahman, S.; Gadjeva, M. Distinct susceptibilities of corneal Pseudomonas aeruginosa clinical isolates to neutrophil extracellular trap-mediated immunity. Infect. Immun. 2014, 82, 4135–4143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Zhang, L.; Yuan, K.; Huang, X.; Hu, R.; Jin, X. Neutrophil extracellular traps may have a dual role in Pseudomonas aeruginosa keratitis. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 169–180. [Google Scholar] [CrossRef]
- Kandhavelu, J.; Demonte, N.L.; Namperumalsamy, V.P.; Prajna, L.; Thangavel, C.; Jayapal, J.M.; Kuppamuthu, D. Aspergillus flavus induced alterations in tear protein profile reveal pathogen-induced host response to fungal infection. J. Proteom. 2017, 152, 13–21. [Google Scholar] [CrossRef]
- Clark, H.L.; Abbondante, S.; Minns, M.S.; Greenberg, E.N.; Sun, Y.; Pearlman, E. Protein deiminase 4 and CR3 regulate Aspergillus fumigatus and β-glucan-induced neutrophil extracellular trap formation, but hyphal killing is dependent only on CR3. Front. Immunol. 2018, 9, 1182. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Huang, X.; Yuan, K.; Zhu, B.; Zhao, Y.; Hu, R.; Wan, T.; Zhu, L.; Jin, X. Glucocorticoids may exacerbate fungal keratitis by increasing fungal aggressivity and inhibiting the formation of neutrophil extracellular traps. Curr. Eye Res. 2020, 45, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, T.; Sjögren, J.; Kahn, F.; Abu-Humaidan, A.H.; Fisker, N.; Assing, K.; Mörgelin, M.; Bengtsson, A.A.; Borregaard, N.; Sørensen, O.E. A novel mechanism for NETosis provides antimicrobial defense at the oral mucosa. Blood 2015, 126, 2128–2137. [Google Scholar] [CrossRef] [Green Version]
- Perazzio, S.F.; Soeiro-Pereira, P.V.; Dos Santos, V.C.; de Brito, M.V.; Salu, B.; Oliva, M.L.V.; Stevens, A.M.; de Souza, A.W.S.; Ochs, H.D.; Torgerson, T.R. Soluble CD40L is associated with increased oxidative burst and neutrophil extracellular trap release in Behçet’s disease. Arthritis Res. Ther. 2017, 19, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Safi, R.; Kallas, R.; Bardawil, T.; Mehanna, C.J.; Abbas, O.; Hamam, R.; Uthman, I.; Kibbi, A.-G.; Nassar, D. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behçet’s disease. J. Dermatol. Sci. 2018, 92, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Joncour, A.; Martos, R.; Loyau, S.; Lelay, N.; Dossier, A.; Cazes, A.; Fouret, P.; Domont, F.; Papo, T.; Jandrot-Perrus, M. Critical role of neutrophil extracellular traps (NETs) in patients with Behcet’s disease. Ann. Rheum. Dis. 2019, 78, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, T.; He, J.; Liu, Y. Correspondence on ‘Critical role of neutrophil extracellular traps (NETs) in patients with Behcet’s disease’. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Li, L.; Yu, X.; Liu, J.; Wang, Z.; Li, C.; Shi, J.; Sun, L.; Liu, Y.; Zhang, F.; Chen, H. Neutrophil Extracellular Traps Promote Aberrant Macrophages Activation in Behçet’s Disease. Front. Immunol. 2020, 11, 590622. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez-Díaz, C.; Varela-Trinidad, G.U.; Muñoz-Sánchez, G.; Solórzano-Castanedo, K.; Avila-Arrezola, K.E.; Iñiguez-Gutiérrez, L.; Delgado-Rizo, V.; Fafutis-Morris, M. To Trap a Pathogen: Neutrophil Extracellular Traps and Their Role in Mucosal Epithelial and Skin Diseases. Cells 2021, 10, 1469. https://doi.org/10.3390/cells10061469
Domínguez-Díaz C, Varela-Trinidad GU, Muñoz-Sánchez G, Solórzano-Castanedo K, Avila-Arrezola KE, Iñiguez-Gutiérrez L, Delgado-Rizo V, Fafutis-Morris M. To Trap a Pathogen: Neutrophil Extracellular Traps and Their Role in Mucosal Epithelial and Skin Diseases. Cells. 2021; 10(6):1469. https://doi.org/10.3390/cells10061469
Chicago/Turabian StyleDomínguez-Díaz, Carolina, Gael Urait Varela-Trinidad, Germán Muñoz-Sánchez, Karla Solórzano-Castanedo, Karina Elizabeth Avila-Arrezola, Liliana Iñiguez-Gutiérrez, Vidal Delgado-Rizo, and Mary Fafutis-Morris. 2021. "To Trap a Pathogen: Neutrophil Extracellular Traps and Their Role in Mucosal Epithelial and Skin Diseases" Cells 10, no. 6: 1469. https://doi.org/10.3390/cells10061469
APA StyleDomínguez-Díaz, C., Varela-Trinidad, G. U., Muñoz-Sánchez, G., Solórzano-Castanedo, K., Avila-Arrezola, K. E., Iñiguez-Gutiérrez, L., Delgado-Rizo, V., & Fafutis-Morris, M. (2021). To Trap a Pathogen: Neutrophil Extracellular Traps and Their Role in Mucosal Epithelial and Skin Diseases. Cells, 10(6), 1469. https://doi.org/10.3390/cells10061469