
Functional Redundancy of Cyclase-Associated Proteins CAP1 and CAP2 in Differentiating Neurons



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Transgenic Mice
2.2. Cell Culture and Transfection
2.3. Immunocytochemistry
2.4. Growth Cone Morphology
2.5. Live Cell Imaging
2.6. Protein Analysis
2.7. Histology and Immunohistochemistry
2.8. Statistical Analysis
3. Results
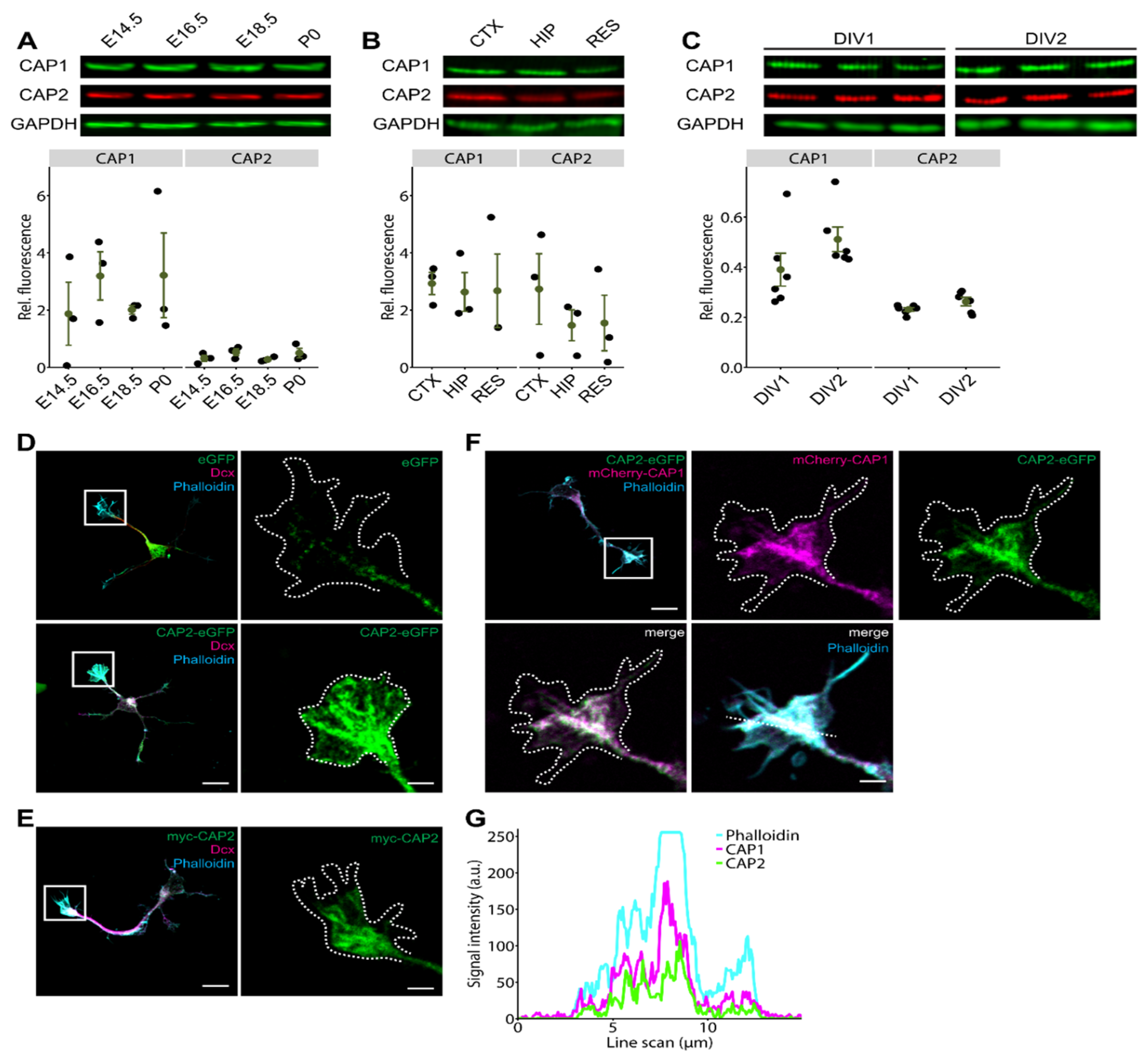
3.1. CAP2 Is Expressed during Neuron Differentiation and Abundant in Growth Cones
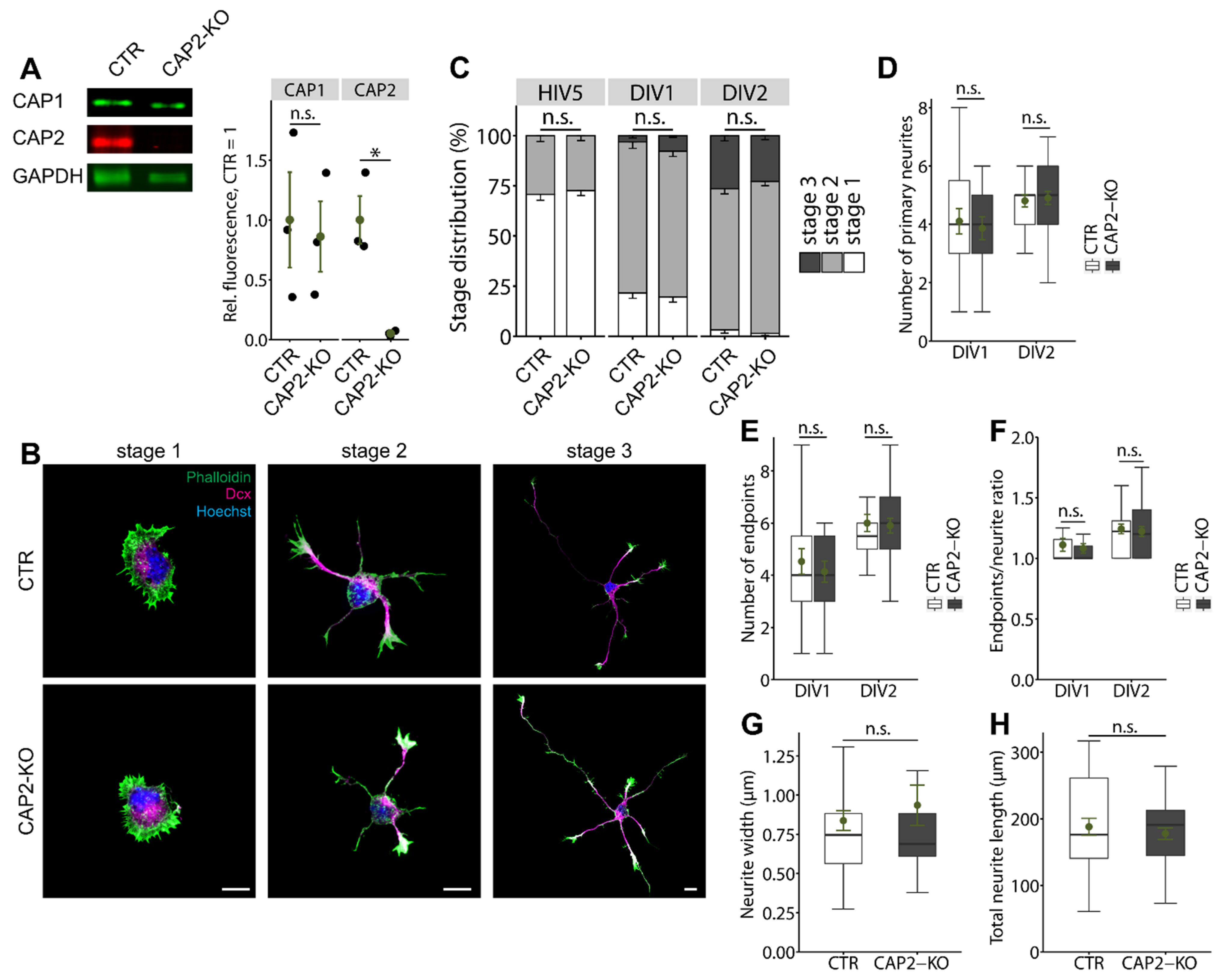
3.1.1. CAP2 Is Not Relevant for Early Neuron Differentiation
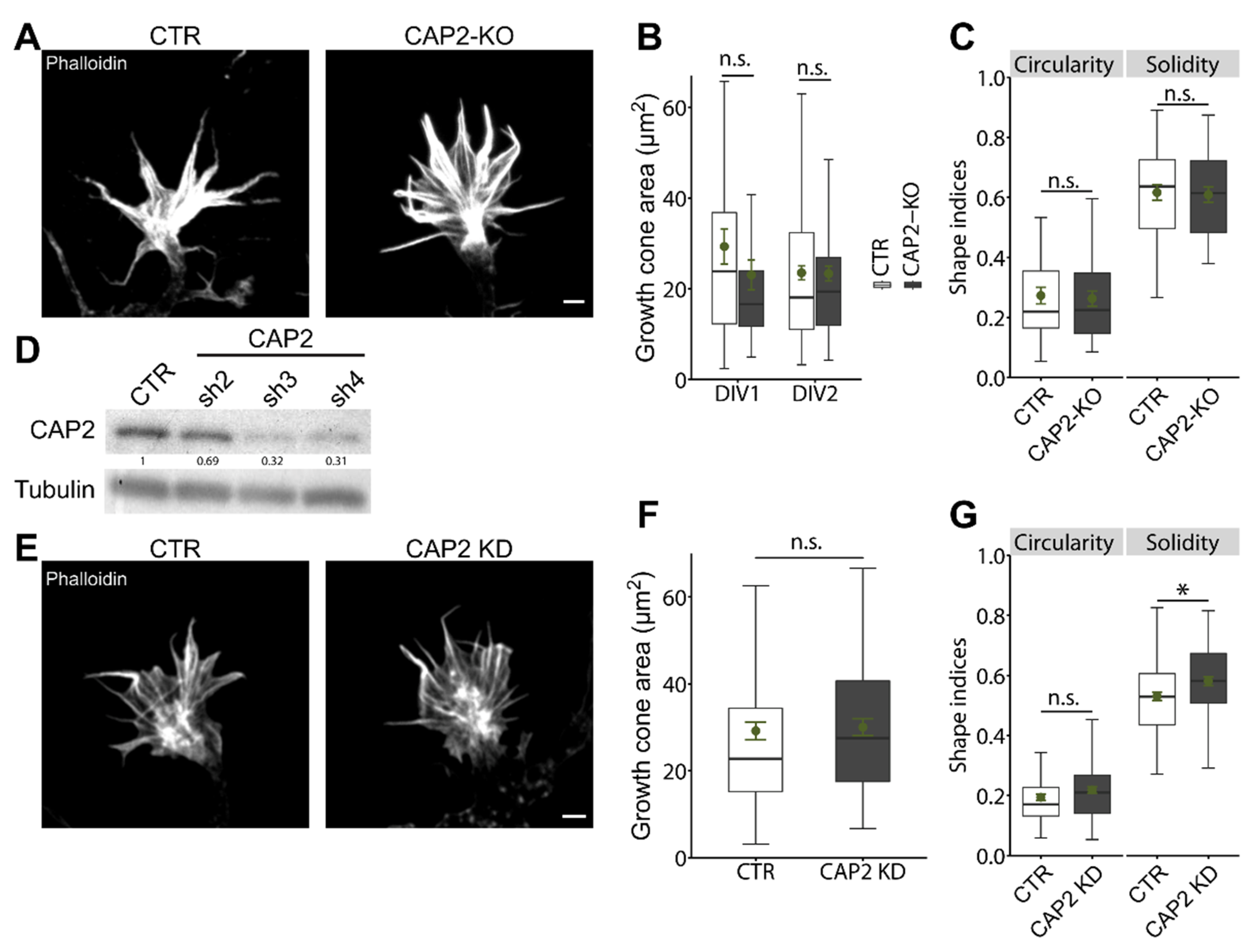
3.1.2. CAP2 Is Dispensable for Growth Cone Size, Morphology, and Motility
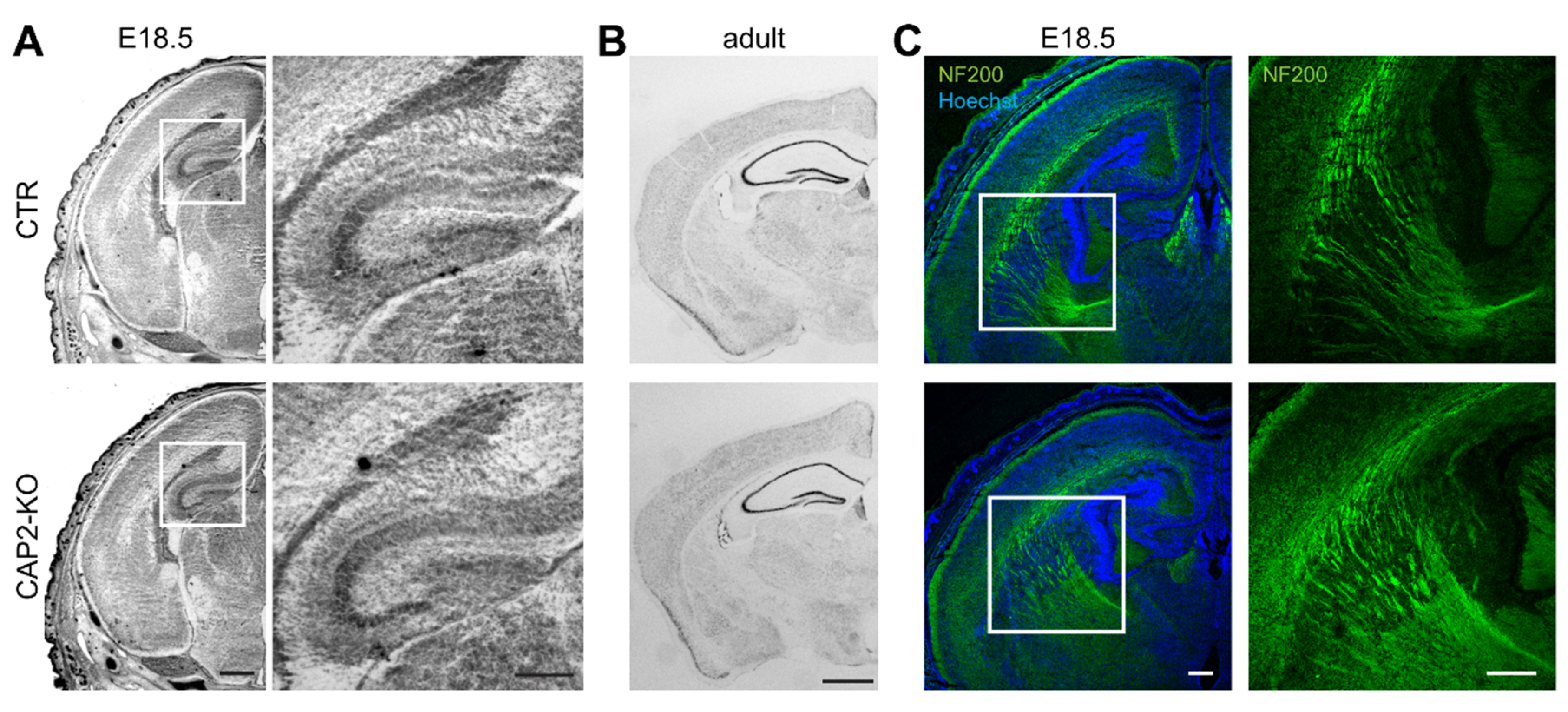
3.1.3. CAP2 Is Dispensable for Brain Development
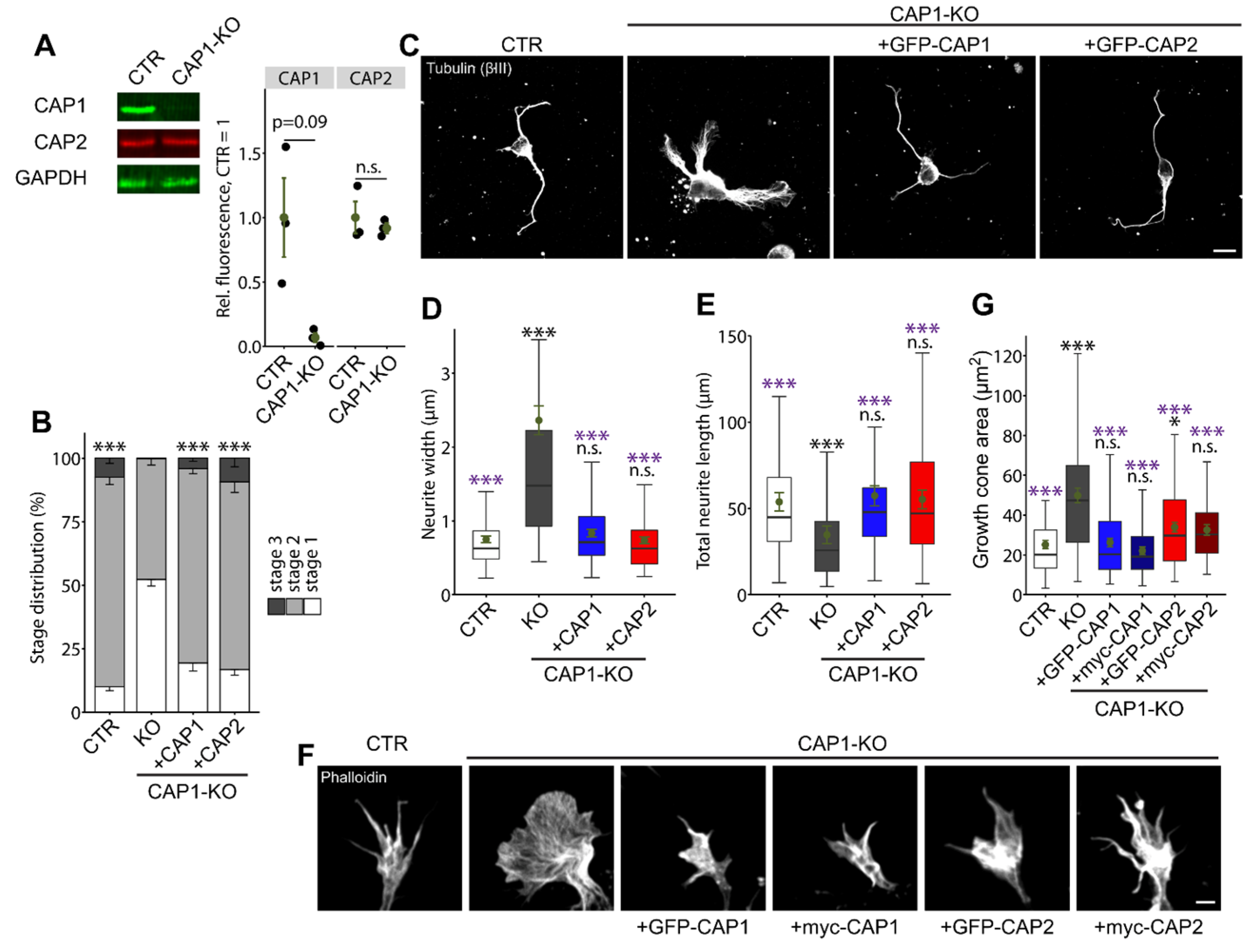
3.1.4. CAP2 Can Rescue Neuron Morphology and Differentiation in CAP1-KO Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ono, S. The role of cyclase-associated protein in regulating actin filament dynamics—More than a monomer-sequestration factor. J. Cell Sci. 2013, 126, 3249–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.B.; Khudayberdiev, S.; Pelucchi, S.; Marcello, E. CAPt’n of Actin Dynamics: Recent Advances in the Molecular, Developmental and Physiological Functions of Cyclase-Associated Protein (CAP). Front. Cell Dev. Biol. 2020, 8, 586631. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.B.; Collins, A.; Goode, B.L. High-speed depolymerization at actin filament ends jointly catalysed by Twinfilin and Srv2/CAP. Nat. Cell Biol. 2015, 17, 1504–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotila, T.; Kogan, K.; Enkavi, G.; Guo, S.; Vattulainen, I.; Goode, B.L.; Lappalainen, P. Structural basis of actin monomer re-charging by cyclase-associated protein. Nat. Commun. 2018, 9, 1892. [Google Scholar] [CrossRef] [PubMed]
- Kotila, T.; Wioland, H.; Enkavi, G.; Kogan, K.; Vattulainen, I.; Jegou, A.; Romet-Lemonne, G.; Lappalainen, P. Mechanism of synergistic actin filament pointed end depolymerization by cyclase-associated protein and cofilin. Nat. Commun. 2019, 10, 5320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, A.; Fung, T.S.; Kettenbach, A.N.; Chakrabarti, R.; Higgs, H.N. A complex containing lysine-acetylated actin inhibits the formin INF2. Nat. Cell Biol. 2019, 21, 592–602. [Google Scholar]
- Shekhar, S.; Chung, J.; Kondev, J.; Gelles, J.; Goode, B.L. Synergy between Cyclase-associated protein and Cofilin accelerates actin filament depolymerization by two orders of magnitude. Nat. Commun. 2019, 10, 5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, A.; Fung, T.S.; Francomacaro, L.M.; Huynh, T.; Kotila, T.; Svindrych, Z.; Higgs, H.N. Regulation of INF2-mediated actin polymerization through site-specific lysine acetylation of actin itself. Proc. Natl. Acad. Sci. USA 2020, 117, 439–447. [Google Scholar]
- Schneider, F.; Duong, T.A.; Metz, I.; Winkelmeier, J.; Hübner, C.A.; Endesfelder, U.; Rust, M.B. Mutual functional dependence of cyclase-associated protein 1 (CAP1) and cofilin1 in neuronal actin dynamics and growth cone function. Prog. Neurobiol. 2021, 202, 102050. [Google Scholar] [CrossRef] [PubMed]
- Bertling, E.; Hotulainen, P.; Mattila, P.K.; Matilainen, T.; Salminen, M.; Lappalainen, P. Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol. Biol. Cell 2004, 15, 2324–2334. [Google Scholar] [CrossRef] [Green Version]
- Peche, V.; Shekar, S.; Leichter, M.; Korte, H.; Schroder, R.; Schleicher, M.; Holak, T.A.; Clemen, C.S.; Ramanath, Y.B.; Pfitzer, G.; et al. CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell Mol. Life Sci. 2007, 64, 2702–2715. [Google Scholar] [CrossRef]
- Kepser, L.J.; Damar, F.; De Cicco, T.; Chaponnier, C.; Proszynski, T.J.; Pagenstecher, A.; Rust, M.B. CAP2 deficiency delays myofibril actin cytoskeleton differentiation and disturbs skeletal muscle architecture and function. Proc. Natl. Acad. Sci. USA 2019, 116, 8397–8402. [Google Scholar] [CrossRef] [Green Version]
- Colpan, M.; Iwanski, J.; Gregorio, C.C. CAP2 is a regulator of actin pointed end dynamics and myofibrillogenesis in cardiac muscle. Commun. Biol. 2021, 4, 365. [Google Scholar] [CrossRef]
- Peche, V.S.; Holak, T.A.; Burgute, B.D.; Kosmas, K.; Kale, S.P.; Wunderlich, F.T.; Elhamine, F.; Stehle, R.; Pfitzer, G.; Nohroudi, K.; et al. Ablation of cyclase-associated protein 2 (CAP2) leads to cardiomyopathy. Cell Mol. Life Sci. 2013, 70, 527–543. [Google Scholar] [CrossRef] [Green Version]
- Field, J.; Ye, D.Z.; Shinde, M.; Liu, F.; Schillinger, K.J.; Lu, M.; Wang, T.; Skettini, M.; Xiong, Y.; Brice, A.K.; et al. CAP2 in cardiac conduction, sudden cardiac death and eye development. Sci. Rep. 2015, 5, 17256. [Google Scholar] [CrossRef] [Green Version]
- Stockigt, F.; Peche, V.S.; Linhart, M.; Nickenig, G.; Noegel, A.A.; Schrickel, J.W. Deficiency of cyclase-associated protein 2 promotes arrhythmias associated with connexin43 maldistribution and fibrosis. Arch. Med. Sci. 2016, 12, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Bedi, K.; Berritt, S.; Attipoe, B.K.; Brooks, T.G.; Wang, K.; Margulies, K.B.; Field, J. Targeting MRTF/SRF in CAP2-dependent dilated cardiomyopathy delays disease onset. JCI Insight 2019, 4, e124629. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.D.; Lee, S.E.; Yang, J.; Lee, H.C.; Shin, D.; Lee, H.; Lee, J.; Jin, S.; Kim, S.; Lee, S.J.; et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur. Heart J. 2019, 41, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Pelucchi, S.; Vandermeulen, L.; Pizzamiglio, L.; Aksan, B.; Yan, J.; Konietzny, A.; Bonomi, E.; Borroni, B.; Padovani, A.; Rust, M.B.; et al. Cyclase-associated protein 2 dimerization regulates cofilin in synaptic plasticity and Alzheimer’s disease. Brain Commun. 2020, 2, fcaa086. [Google Scholar] [CrossRef]
- Bosch, M.; Hayashi, Y. Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 2012, 22, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.B. ADF/cofilin: A crucial regulator of synapse physiology and behavior. Cell Mol. Life Sci. 2015, 72, 3521–3529. [Google Scholar] [CrossRef] [PubMed]
- Dent, E.W.; Gupton, S.L.; Gertler, F.B. The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb. Perspect. Biol. 2011, 3, a001800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schutz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Khudayberdiev, S.; Idziak, A.; Bicker, S.; Jacob, R.; Schratt, G. The dynamic recruitment of TRBP to neuronal membranes mediates dendritogenesis during development. EMBO Rep. 2018, 19, e44853. [Google Scholar] [CrossRef]
- Schneider, F.; Duong, T.A.; Rust, M.B. Neuron replating—A powerful and versatile approach to study early aspects of neuron differentiation. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Bicker, S.; Khudayberdiev, S.; Weiß, K.; Zocher, K.; Baumeister, S.; Schratt, G. The DEAH-box helicase DHX36 mediates dendritic localization of the neuronal precursor-microRNA-134. Genes Dev. 2013, 27, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, J.A.; Neumeyer, A.; Gurniak, C.B.; Friauf, E.; Witke, W.; Rust, M.B. Profilin1 is required for glial cell adhesion and radial migration of cerebellar granule neurons. EMBO Rep. 2012, 13, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, J.A.; Meyer, S.; Pipicelli, F.; Kyrousi, C.; Schneider, F.; Bartels, N.; Cappello, S.; Rust, M.B. Profilin1-Dependent F-Actin Assembly Controls Division of Apical Radial Glia and Neocortex Development. Cereb. Cortex 2019, 30, 3467–3482. [Google Scholar] [CrossRef]
- Kepser, L.J.; Khudayberdiev, S.; Hinojosa, L.S.; Macchi, C.; Ruscica, M.; Marcello, E.; Culmsee, C.; Grosse, R.; Rust, M.B. Cyclase-associated protein 2 (CAP2) controls MRTF-A localization and SRF activity in mouse embryonic fibroblasts. Sci. Rep. 2021, 11, 4789. [Google Scholar] [CrossRef]
- Dotti, C.G.; Sullivan, C.A.; Banker, G.A. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 1988, 8, 1454–1468. [Google Scholar] [CrossRef] [Green Version]
- Nozumi, M.; Togano, T.; Takahashi-Niki, K.; Lu, J.; Honda, A.; Taoka, M.; Shinkawa, T.; Koga, H.; Takeuchi, K.; Isobe, T.; et al. Identification of functional marker proteins in the mammalian growth cone. Proc. Natl. Acad. Sci. USA 2009, 106, 17211–17216. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Nozumi, M.; Takeuchi, K.; Abe, H.; Igarashi, M. Expression and function of neuronal growth-associated proteins (nGAPs) in PC12 cells. Neurosci. Res. 2011, 70, 85–90. [Google Scholar] [CrossRef]
- Hotulainen, P.; Llano, O.; Smirnov, S.; Tanhuanpaa, K.; Faix, J.; Rivera, C.; Lappalainen, P. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J. Cell Biol. 2009, 185, 323–339. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.B.; Gurniak, C.B.; Renner, M.; Vara, H.; Morando, L.; Gorlich, A.; Sassoe-Pognetto, M.; Banchaabouchi, M.A.; Giustetto, M.; Triller, A.; et al. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010, 29, 1889–1902. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Lee, C.W.; Fan, Y.; Komlos, D.; Tang, X.; Sun, C.; Yu, K.; Hartzell, H.C.; Chen, G.; Bamburg, J.R.; et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci. 2010, 13, 1208–1215. [Google Scholar] [CrossRef] [Green Version]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Zimmermann, A.M.; Gorlich, A.; Gurniak, C.B.; Sassoe-Pognetto, M.; Friauf, E.; Witke, W.; Rust, M.B. ADF/Cofilin Controls Synaptic Actin Dynamics and Regulates Synaptic Vesicle Mobilization and Exocytosis. Cereb. Cortex 2015, 25, 2863–2875. [Google Scholar] [CrossRef]
- Zimmermann, A.M.; Jene, T.; Wolf, M.; Gorlich, A.; Gurniak, C.B.; Sassoe-Pognetto, M.; Witke, W.; Friauf, E.; Rust, M.B. Attention-Deficit/Hyperactivity Disorder-like Phenotype in a Mouse Model with Impaired Actin Dynamics. Biol. Psychiatry 2015, 78, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Paeger, L.; Kosmas, K.; Kloppenburg, P.; Noegel, A.A.; Peche, V.S. Neuronal Actin Dynamics, Spine Density and Neuronal Dendritic Complexity Are Regulated by CAP2. Front. Cell Neurosci. 2016, 10, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, F.; Metz, I.; Khudayberdiev, S.; Rust, M.B. Functional Redundancy of Cyclase-Associated Proteins CAP1 and CAP2 in Differentiating Neurons. Cells 2021, 10, 1525. https://doi.org/10.3390/cells10061525
Schneider F, Metz I, Khudayberdiev S, Rust MB. Functional Redundancy of Cyclase-Associated Proteins CAP1 and CAP2 in Differentiating Neurons. Cells. 2021; 10(6):1525. https://doi.org/10.3390/cells10061525
Chicago/Turabian StyleSchneider, Felix, Isabell Metz, Sharof Khudayberdiev, and Marco B. Rust. 2021. "Functional Redundancy of Cyclase-Associated Proteins CAP1 and CAP2 in Differentiating Neurons" Cells 10, no. 6: 1525. https://doi.org/10.3390/cells10061525
APA StyleSchneider, F., Metz, I., Khudayberdiev, S., & Rust, M. B. (2021). Functional Redundancy of Cyclase-Associated Proteins CAP1 and CAP2 in Differentiating Neurons. Cells, 10(6), 1525. https://doi.org/10.3390/cells10061525