
Influence of Subcellular Localization and Functional State on Protein Turnover


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Transfection
2.2. Constructs
2.3. Immunofluorescent Staining
2.4. Imaging and Image Analysis
2.5. Western Blotting and Subcellular Fractionation
2.6. Computation of Biochemical Parameters
2.7. Statistical Analysis
3. Results
3.1. SNAP-Tag Fusion Constructs for Precise Optical Measures of Protein Turnover

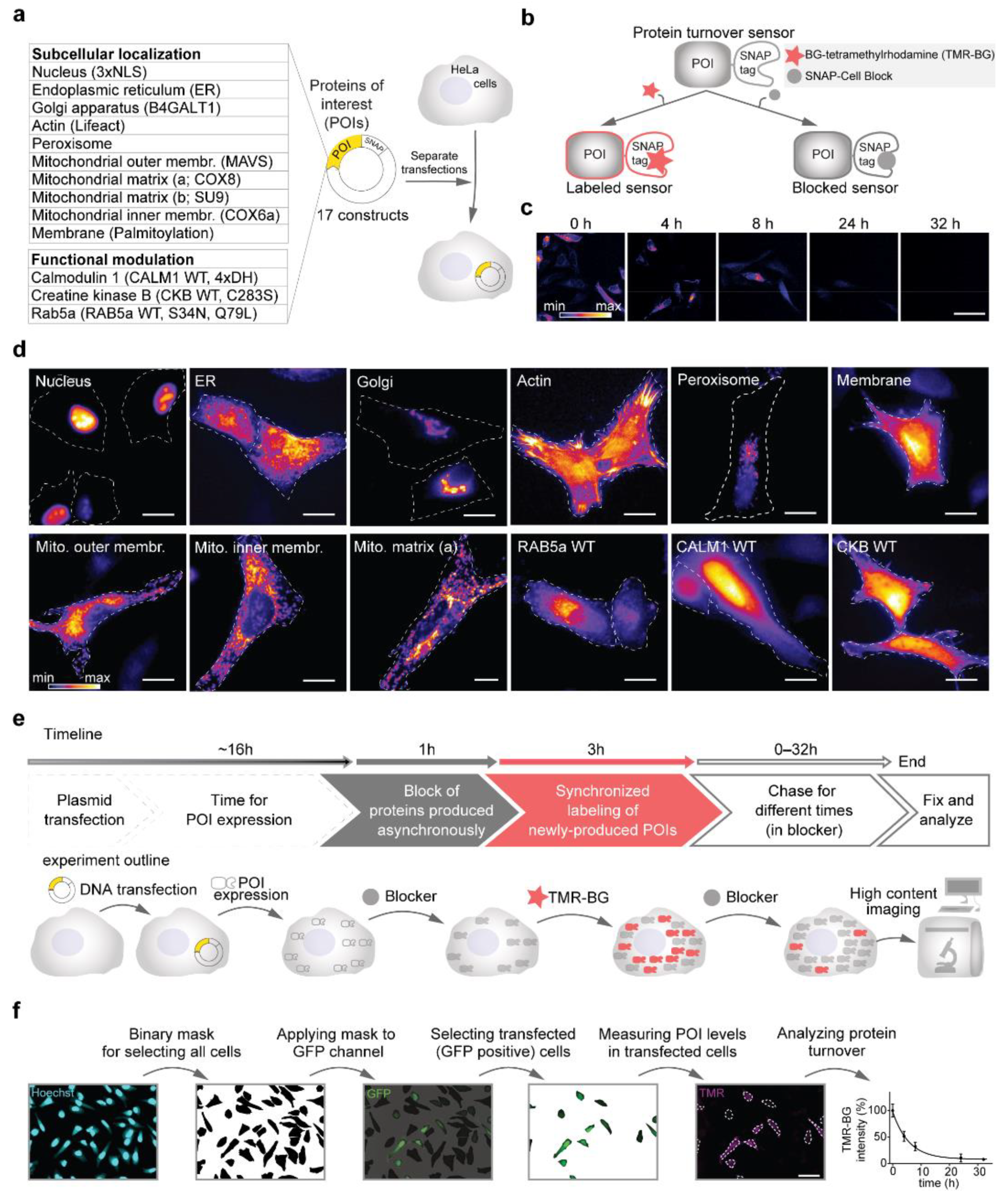{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Description | Construct Name | SNAP Fusion | Protein of Origin | Reference |
|---|---|---|---|---|---|
| 1 | Nucleus (NLS) | SNAP-NLS | N-terminal | SV40 | [38] |
| 2 | Endoplasmic reticulum (ER) | ER-SNAP-KDEL | C-terminal + KDEL | CALR | [39] |
| 3 | Golgi apparatus | Golgi-SNAP | C-terminal | B4GALT1 | [39] |
| 4 | Actin (Lifeact) | Lifeact-SNAP | C-terminal | Abp140 | [40] |
| 5 | Peroxisome | SNAP-PTS | C-terminal | LYKSRL peptide | [39] |
| 6 | Mitochondrial outer membrane | SNAP-MITO-outer | C-terminal | MAVS | [41] |
| 7 | Mitochondrial inner membrane | MITO-inner-SNAP | C-terminal | COX6a | [39] |
| 8 | Mitochondrial matrix (a) | MITO-Matrix-I-SNAP | C-terminal | COX8 | [50] |
| 9 | Mitochondrial matrix (b) | MITO-Matrix-II-SNAP | C-terminal | SU9 | [43] |
| 10 | Membrane | Palmitoyl-SNAP | N-terminal | ARHGEF25 (p63) | [44] |
| 11 | Wild-type calmodulin | SNAP-CaM-WT | N-terminal | CALM1.1 | [45] |
| 12 | Inactive calmodulin | SNAP-CaM-4XDH | N-terminal | CALM1.1 | [45] |
| 13 | Wild-type creatine kinase | CKB-SNAP-WT | C-terminal | CKB | [46] |
| 14 | Kinase-dead creatine kinase | CKB-SNAP-C283S | C-terminal | CKB | [46] |
| 15 | Wild-type Rab5a | SNAP-RAB5A-WT | N-terminal | RAB5a.1 | [47] |
| 16 | Dominant negative Rab5a | SNAP-RAB5A-S34N | N-terminal | RAB5a.1 | [47] |
| 17 | Constitutively active Rab5a | SNAP-RAB5A-Q79L | N-terminal | RAB5a.1 | [47] |
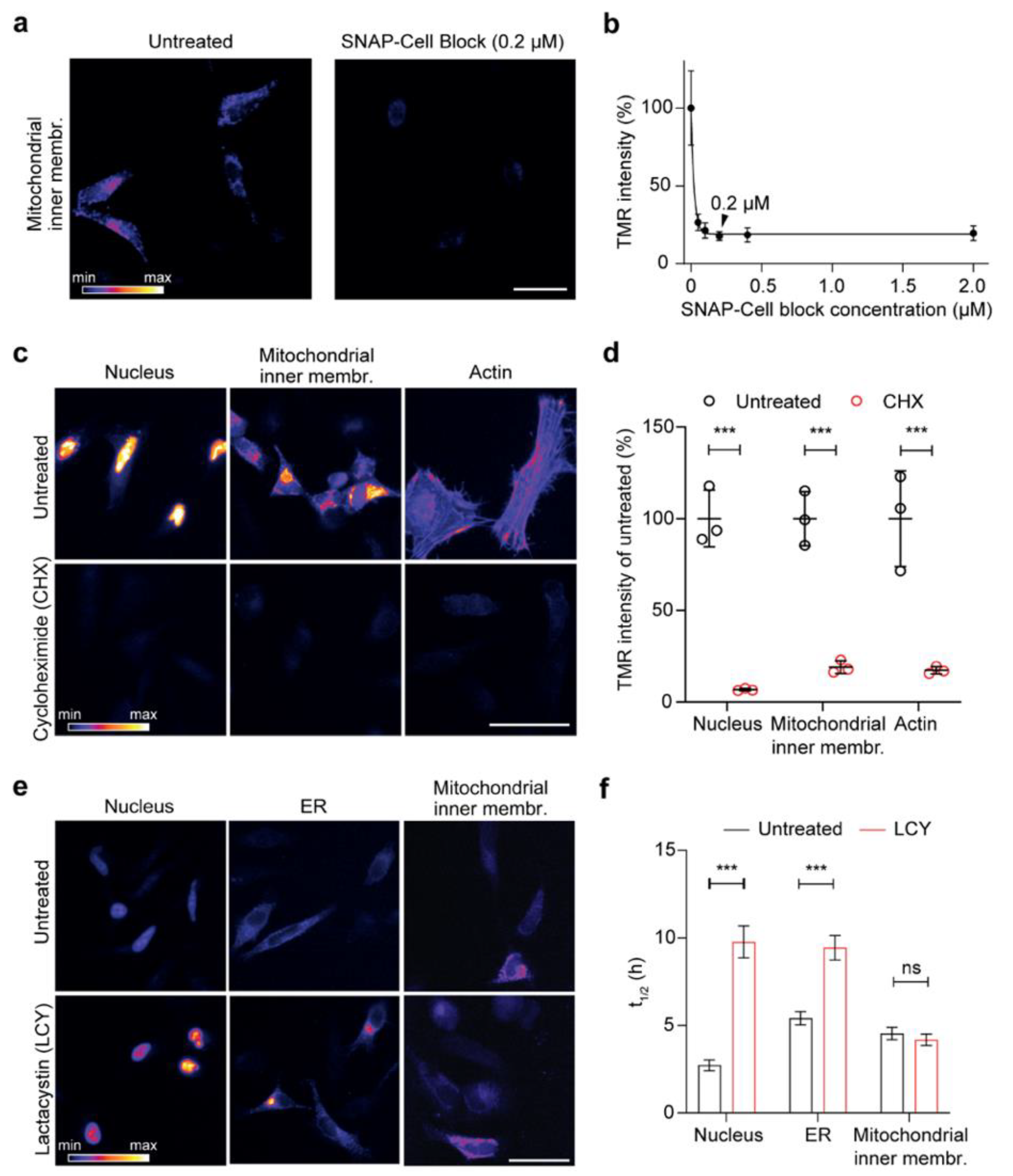
3.2. Workflow Validation
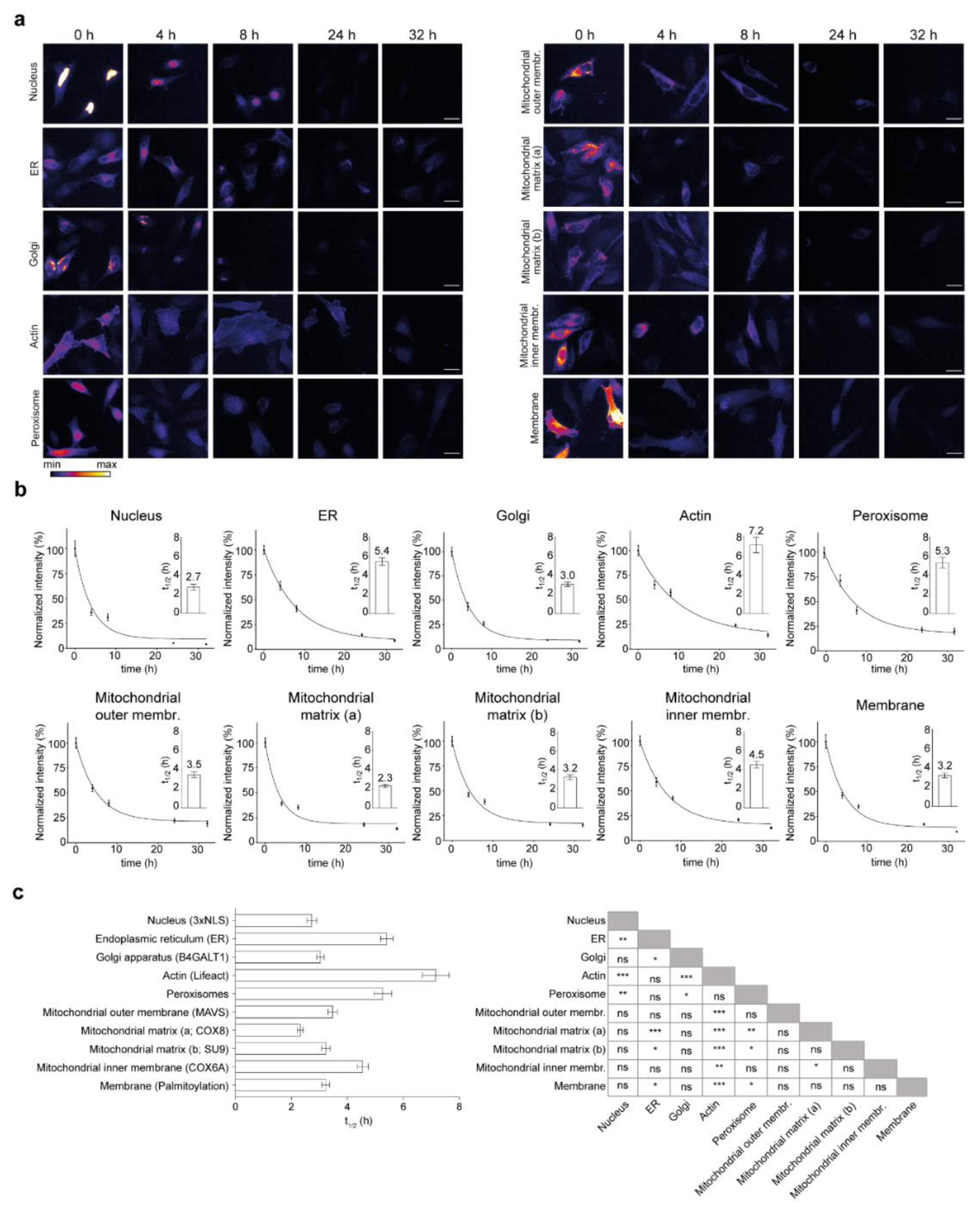
3.3. Influence of Subcellular Localization on Protein Lifetimes
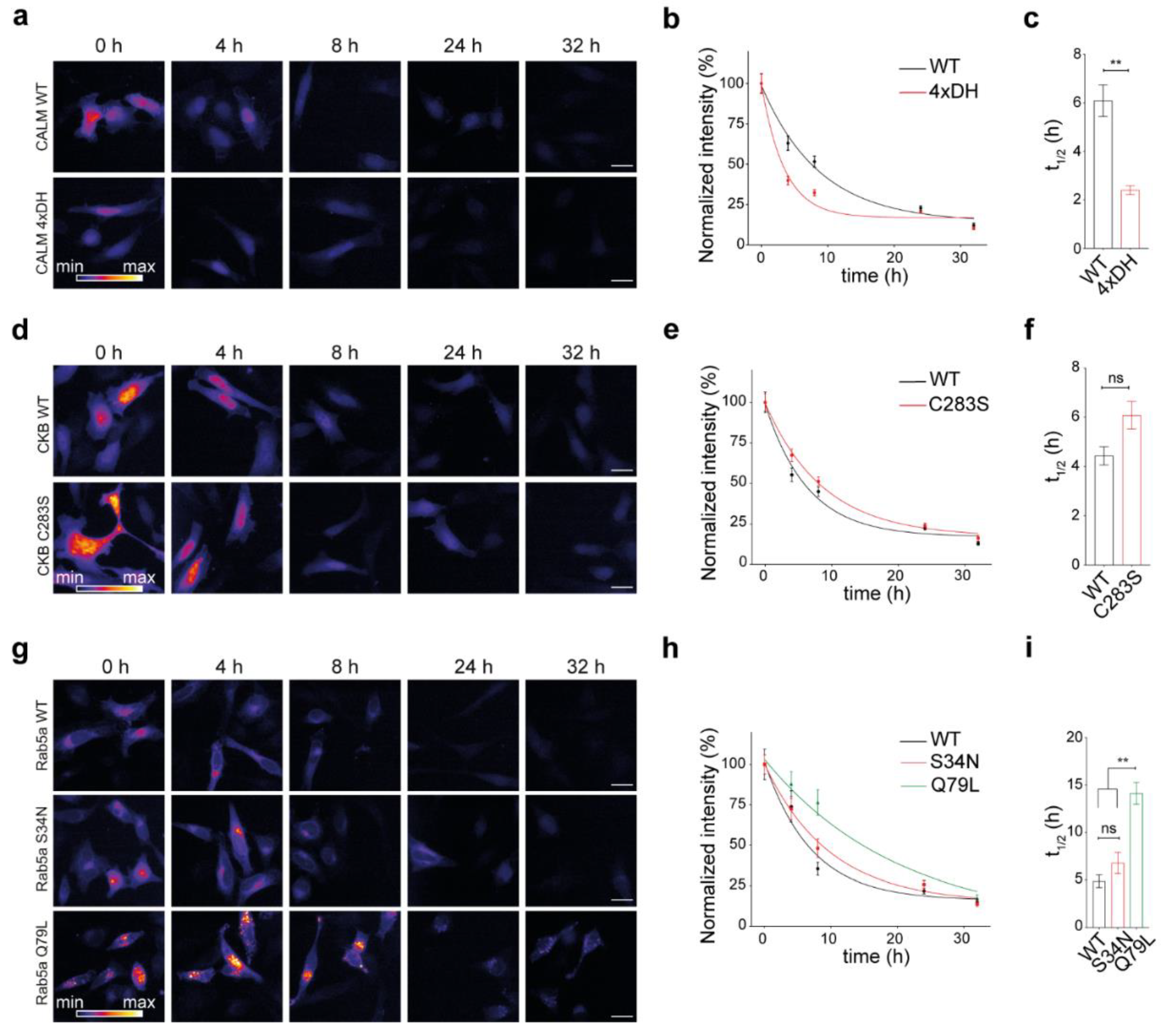
3.4. Influence of Protein Activation State on Protein Lifetimes
3.5. SNAP-Tag Rab5a-Q79L Is More Efficiently Associated to the Heavy Membrane Fraction and Reflect Both Changes in Activity and in Subcellular Localization and Membrane Interaction Properties
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. Ser. A Biolmed. Sci. Med. Sci. 2014, 69 (Suppl. S1), S33–S38. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, J.C.; Guan, S.; Burlingame, A.; Prusiner, S.B.; Ghaemmaghami, S. Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14508–14513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, J.N.; Toyama, B.H.; Xu, T.; Yates, J.R.; Hetzer, M.W. Extremely long-lived nuclear pore proteins in the rat brain. Science 2012, 335, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieson, T.; Franken, H.; Kosinski, J.; Kurzawa, N.; Zinn, N.; Sweetman, G.; Poeckel, D.; Ratnu, V.S.; Schramm, M.; Becher, I.; et al. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018, 9, 689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitski, M.M.; Zinn, N.; Faelth-Savitski, M.; Poeckel, D.; Gade, S.; Becher, I.; Muelbaier, M.; Wagner, A.J.; Strohmer, K.; Werner, T.; et al. Multiplexed Proteome Dynamics Profiling Reveals Mechanisms Controlling Protein Homeostasis. Cell 2018, 173, 260–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.; Diering, G.H.; Na, C.H.; Nirujogi, R.S.; Bachman, J.L.; Pandey, A.; Huganir, R.L. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E3827–E3836. [Google Scholar] [CrossRef] [Green Version]
- Fornasiero, E.F.; Mandad, S.; Wildhagen, H.; Alevra, M.; Rammner, B.; Keihani, S.; Opazo, F.; Urban, I.; Ischebeck, T.; Sakib, M.S.; et al. Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun. 2018, 9, 4230. [Google Scholar] [CrossRef] [Green Version]
- Alevra, M.; Mandad, S.; Ischebeck, T.; Urlaub, H.; Rizzoli, S.O.; Fornasiero, E.F. A mass spectrometry workflow for measuring protein turnover rates in vivo. Nat. Protoc. 2019, 14, 3333–3365. [Google Scholar] [CrossRef] [Green Version]
- Borzou, A.; Sadygov, V.R.; Zhang, W.; Sadygov, R.G. Proteome Dynamics from Heavy Water Metabolic Labeling and Peptide Tandem Mass Spectrometry. Int. J. Mass Spectrom. 2019, 445. [Google Scholar] [CrossRef]
- Wu, C.; Ba, Q.; Lu, D.; Li, W.; Salovska, B.; Hou, P.; Mueller, T.; Rosenberger, G.; Gao, E.; Di, Y.; et al. Global and Site-Specific Effect of Phosphorylation on Protein Turnover. Dev. Cell 2021, 56, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Holtz, A.; Schilling, B. Accumulation of “Old Proteins” and the Critical Need for MS-based Protein Turnover Measurements in Aging and Longevity. Proteomics 2020, 20, e1800403. [Google Scholar] [CrossRef]
- Ross, A.B.; Langer, J.D.; Jovanovic, M. Proteome Turnover in the Spotlight: Approaches, Applications, and Perspectives. Mol. Cell. Proteom. 2020, 20, 100016. [Google Scholar] [CrossRef] [PubMed]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Schoenheimer, R. The dynamic state of body constituents. Dyn. State Body Const. 1946, 14, 667. [Google Scholar]
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R.; Hetzer, M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Mandad, S.; Rahman, R.-U.; Centeno, T.P.; Vidal, R.O.; Wildhagen, H.; Rammner, B.; Keihani, S.; Opazo, F.; Urban, I.; Ischebeck, T.; et al. The codon sequences predict protein lifetimes and other parameters of the protein life cycle in the mouse brain. Sci. Rep. 2018, 8, 16913. [Google Scholar] [CrossRef]
- Guharoy, M.; Bhowmick, P.; Sallam, M.; Tompa, P. Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system. Nat. Commun. 2016, 7, 10239. [Google Scholar] [CrossRef] [Green Version]
- Despa, F.; Orgill, D.P.; Lee, R.C. Molecular crowding effects on protein stability. Ann. N. Y. Acad. Sci. 2005, 1066, 54–66. [Google Scholar] [CrossRef]
- Dao, T.P.; Castañeda, C.A. Ubiquitin-Modulated Phase Separation of Shuttle Proteins: Does Condensate Formation Promote Protein Degradation? Bioessays 2020, 42, e2000036. [Google Scholar] [CrossRef]
- Reichmann, D.; Voth, W.; Jakob, U. Maintaining a Healthy Proteome during Oxidative Stress. Mol. Cell 2018, 69, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins de Oliveira, V.; de Godoi Contessoto, V.; Bruno da Silva, F.; Zago Caetano, D.L.; Jurado de Carvalho, S.; Pereira Leite, V.B. Effects of pH and Salt Concentration on Stability of a Protein G Variant Using Coarse-Grained Models. Biophys. J. 2018, 114, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Ota, N.; Kurahashi, R.; Sano, S.; Takano, K. The direction of protein evolution is destined by the stability. Biochimie 2018, 150, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Rutter, J. Ubiquitin-dependent mitochondrial protein degradation. Int. J. Biochem. Cell Biol. 2011, 43, 1422–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juszkiewicz, S.; Hegde, R.S. Quality Control of Orphaned Proteins. Mol. Cell 2018, 71, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capecchi, M.R.; Capecchi, N.E.; Hughes, S.H.; Wahl, G.M. Selective degradation of abnormal proteins in mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA 1974, 71, 4732–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef]
- Bodor, D.L.; Rodríguez, M.G.; Moreno, N.; Jansen, L.E.T. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr. Protoc. Cell Biol. 2012, 55, 8. [Google Scholar] [CrossRef] [Green Version]
- Pacheu-Grau, D.; Callegari, S.; Emperador, S.; Thompson, K.; Aich, A.; Topol, S.E.; Spencer, E.G.; McFarland, R.; Ruiz-Pesini, E.; Torkamani, A.; et al. Mutations of the mitochondrial carrier translocase channel subunit TIM22 cause early-onset mitochondrial myopathy. Hum. Mol. Genet. 2018, 27, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Zizioli, D.; Lausmann, S.; Eskelinen, E.L.; Hamann, J.; Saftig, P.; von Figura, K.; Schu, P. mu1A-adaptin-deficient mice: Lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 2000, 19, 2193–2203. [Google Scholar] [CrossRef] [Green Version]
- Gomkale, R.; Cruz-Zaragoza, L.D.; Suppanz, I.; Guiard, B.; Montoya, J.; Callegari, S.; Pacheu-Grau, D.; Warscheid, B.; Rehling, P. Defining the Substrate Spectrum of the TIM22 Complex Identifies Pyruvate Carrier Subunits as Unconventional Cargos. Curr. Biol. 2020, 30, 1119–1127. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R.; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W.J.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.; Crevenna, A.H.; Kessenbrock, K.; Yu, J.H.; Neukirchen, D.; Bista, M.; Bradke, F.; Jenne, D.; Holak, T.A.; Werb, Z.; et al. Lifeact: A versatile marker to visualize F-actin. Nat. Methods 2008, 5, 605–607. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-$κ$B and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Bastianutto, C.; Brini, M.; Murgia, M.; Pozzan, T. Mitochondrial Ca2+ homeostasis in intact cells. J. Cell Biol. 1994, 126, 1183–1194. [Google Scholar] [CrossRef]
- John, G.B.; Anjum, R.; Khar, A.; Nagaraj, R. Subcellular localization and physiological consequences of introducing a mitochondrial matrix targeting signal sequence in bax and its mutants. Exp. Cell Res. 2002, 278, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Aittaleb, M.; Nishimura, A.; Linder, M.E.; Tesmer, J.J.G. Plasma Membrane Association of p63 Rho Guanine Nucleotide Exchange Factor (p63RhoGEF) Is Mediated by Palmitoylation and Is Required for Basal Activity in Cells. J. Biol. Chem. 2011, 286, 34448–34456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.J.; Heim, R.; Lu, P.; Pu, Y.; Tsien, R.Y.; Chang, D.C. Dynamic redistribution of calmodulin in HeLa cells during cell division as revealed by a GFP-calmodulin fusion protein technique. J. Cell Sci. 1999, 112, 1567–1577. [Google Scholar] [CrossRef]
- Lin, L.; Perryman, M.B.; Friedman, D.; Roberts, R.; Ma, T.S. Determination of the catalytic site of creatine kinase by site-directed mutagenesis. Biochim. Biophys. Acta 1994, 1206, 97–104. [Google Scholar] [CrossRef]
- Stenmark, H.; Parton, R.G.; Steele-Mortimer, O.; Lütcke, A.; Gruenberg, J.; Zerial, M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994, 13, 1287–1296. [Google Scholar] [CrossRef]
- Lee, A.; Wong, S.T.; Gallagher, D.; Li, B.; Storm, D.R.; Scheuer, T.; Catterall, W.A. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 1999, 399, 155–159. [Google Scholar] [CrossRef]
- Mori, Y.; Yoshida, Y.; Satoh, A.; Moriya, H. Development of an experimental method of systematically estimating protein expression limits in HEK293 cells. Sci. Rep. 2020, 10, 4798. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Pozzan, T. Targeting recombinant aequorin to specific intracellular organelles. Methods Cell Biol. 1994, 40, 339–358. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Leonhard, K.; Guiard, B.; Pellecchia, G.; Tzagoloff, A.; Neupert, W.; Langer, T. Membrane protein degradation by AAA proteases in mitochondria: Extraction of substrates from either membrane surface. Mol. Cell 2000, 5, 629–638. [Google Scholar] [CrossRef]
- Mann, D.F.; Shah, K.; Stein, D.; Snead, G.A. Protein hydrophobicity and stability support the thermodynamic theory of protein degradation. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. 1984, 788, 17–22. [Google Scholar] [CrossRef]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef]
- Wegner, C.S.; Wegener, C.S.; Malerød, L.; Pedersen, N.M.; Progida, C.; Prodiga, C.; Bakke, O.; Stenmark, H.; Brech, A. Ultrastructural characterization of giant endosomes induced by GTPase-deficient Rab5. Histochem. Cell Biol. 2010, 133, 41–55. [Google Scholar] [CrossRef]
- Quirós, P.M.; Langer, T.; López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, F.; Tatsuta, T.; Langer, T. Mitochondrial AAA proteases—towards a molecular understanding of membrane-bound proteolytic machines. Biochim. Biophys. Acta 2012, 1823, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halling, D.B.; Liebeskind, B.J.; Hall, A.W.; Aldrich, R.W. Conserved properties of individual Ca2+-binding sites in calmodulin. Proc. Natl. Acad. Sci. USA 2016, 113, E1216–E1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalobo, A.; González-Muñoz, M.; Berchtold, M.W. Proteins with calmodulin-like domains: Structures and functional roles. Cell. Mol. Life Sci. 2019, 76, 2299–2328. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, L.; Petrillo, M.; Sepe, L.; Boccia, A.; D’Agostino, N.; Passamano, M.; Di Nardo, S.; Tasco, G.; Casadio, R.; Paolella, G. Systematic analysis of human kinase genes: A large number of genes and alternative splicing events result in functional and structural diversity. BMC Bioinform. 2005, 6 (Suppl. S4), S20. [Google Scholar] [CrossRef] [Green Version]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-S.; Cheng, T.-H.; Chang, C.-P.; Chen, H.-M.; Chern, Y. Enhancement of brain-type creatine kinase activity ameliorates neuronal deficits in Huntington’s disease. Biochim. Biophys. Acta 2013, 1832, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Dörrbaum, A.R.; Kochen, L.; Langer, J.D.; Schuman, E.M. Local and global influences on protein turnover in neurons and glia. Elife 2018, 7. [Google Scholar] [CrossRef]
- Dörrbaum, A.R.; Alvarez-Castelao, B.; Nassim-Assir, B.; Langer, J.D.; Schuman, E.M. Proteome dynamics during homeostatic scaling in cultured neurons. Elife 2020, 9. [Google Scholar] [CrossRef]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: An updated view on their regulation and functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef]
- Stevens, F.C. Calmodulin: An introduction. Can. J. Biochem. Cell Biol. 1983, 61, 906–910. [Google Scholar] [CrossRef]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin structure refined at 1.7 A resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Putkey, J.A.; Sweeney, H.L.; Campbell, S.T. Site-directed mutation of the trigger calcium-binding sites in cardiac troponin C. J. Biol. Chem. 1989, 264, 12370–12378. [Google Scholar] [CrossRef]
- Xiong, L.-W.; Kleerekoper, Q.K.; Wang, X.; Putkey, J.A. Intra- and Interdomain Effects Due to Mutation of Calcium-binding Sites in Calmodulin. J. Biol. Chem. 2010, 285, 8094–8103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, P.J. Degradation of mutant proteins, underlying “loss of function” phenotypes, plays a major role in genetic disease. Curr. Issues Mol. Biol. 2001, 3, 57–65. [Google Scholar] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Tamarappoo, B.K.; Verkman, A.S. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J. Clin. Investig. 1998, 101, 2257–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 25710–25718. [Google Scholar] [CrossRef]
- Bass, J.; Turck, C.; Rouard, M.; Steiner, D.F. Furin-mediated processing in the early secretory pathway: Sequential cleavage and degradation of misfolded insulin receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 11905–11909. [Google Scholar] [CrossRef] [Green Version]
- Lukong, K.E.; Landry, K.; Elsliger, M.A.; Chang, Y.; Lefrancois, S.; Morales, C.R.; Pshezhetsky, A.V. Mutations in sialidosis impair sialidase binding to the lysosomal multienzyme complex. J. Biol. Chem. 2001, 276, 17286–17290. [Google Scholar] [CrossRef] [Green Version]
- Corydon, T.J.; Bross, P.; Jensen, T.G.; Corydon, M.J.; Lund, T.B.; Jensen, U.B.; Kim, J.J.; Gregersen, N.; Bolund, L. Rapid degradation of short-chain acyl-CoA dehydrogenase variants with temperature-sensitive folding defects occurs after import into mitochondria. J. Biol. Chem. 1998, 273, 13065–13071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, J.; Vashisht, A.A.; Wan, J.; Jen, J.C.; Claypool, S.M.; Wohlschlegel, J.A.; Koehler, C.M. Rapid degradation of mutant SLC25A46 by the ubiquitin-proteasome system results in MFN1/2-mediated hyperfusion of mitochondria. Mol. Biol. Cell 2017, 28, 600–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, B.; Ozhathil, L.C.; Medeiros-Domingo, A.; Gollob, M.H.; Abriel, H. Four TRPM4 Cation Channel Mutations Found in Cardiac Conduction Diseases Lead to Altered Protein Stability. Front. Physiol. 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, F.; Soranzo, M.R.; Borelli, V.; Bertoncin, P.; Zabucchi, G. Subcellular localization of the small GTPase Rab5a in resting and stimulated human neutrophils. Exp. Cell Res. 1996, 227, 367–373. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Musinova, Y.R.; Lisitsyna, O.M.; Golyshev, S.A.; Tuzhikov, A.I.; Polyakov, V.Y.; Sheval, E.V. Nucleolar localization/retention signal is responsible for transient accumulation of histone H2B in the nucleolus through electrostatic interactions. Biochim. Biophys. Acta 2011, 1813, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenworthy, A.K. Fluorescence-based methods to image palmitoylated proteins. Methods 2006, 40, 198–205. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousefi, R.; Jevdokimenko, K.; Kluever, V.; Pacheu-Grau, D.; Fornasiero, E.F. Influence of Subcellular Localization and Functional State on Protein Turnover. Cells 2021, 10, 1747. https://doi.org/10.3390/cells10071747
Yousefi R, Jevdokimenko K, Kluever V, Pacheu-Grau D, Fornasiero EF. Influence of Subcellular Localization and Functional State on Protein Turnover. Cells. 2021; 10(7):1747. https://doi.org/10.3390/cells10071747
Chicago/Turabian StyleYousefi, Roya, Kristina Jevdokimenko, Verena Kluever, David Pacheu-Grau, and Eugenio F. Fornasiero. 2021. "Influence of Subcellular Localization and Functional State on Protein Turnover" Cells 10, no. 7: 1747. https://doi.org/10.3390/cells10071747
APA StyleYousefi, R., Jevdokimenko, K., Kluever, V., Pacheu-Grau, D., & Fornasiero, E. F. (2021). Influence of Subcellular Localization and Functional State on Protein Turnover. Cells, 10(7), 1747. https://doi.org/10.3390/cells10071747