
Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
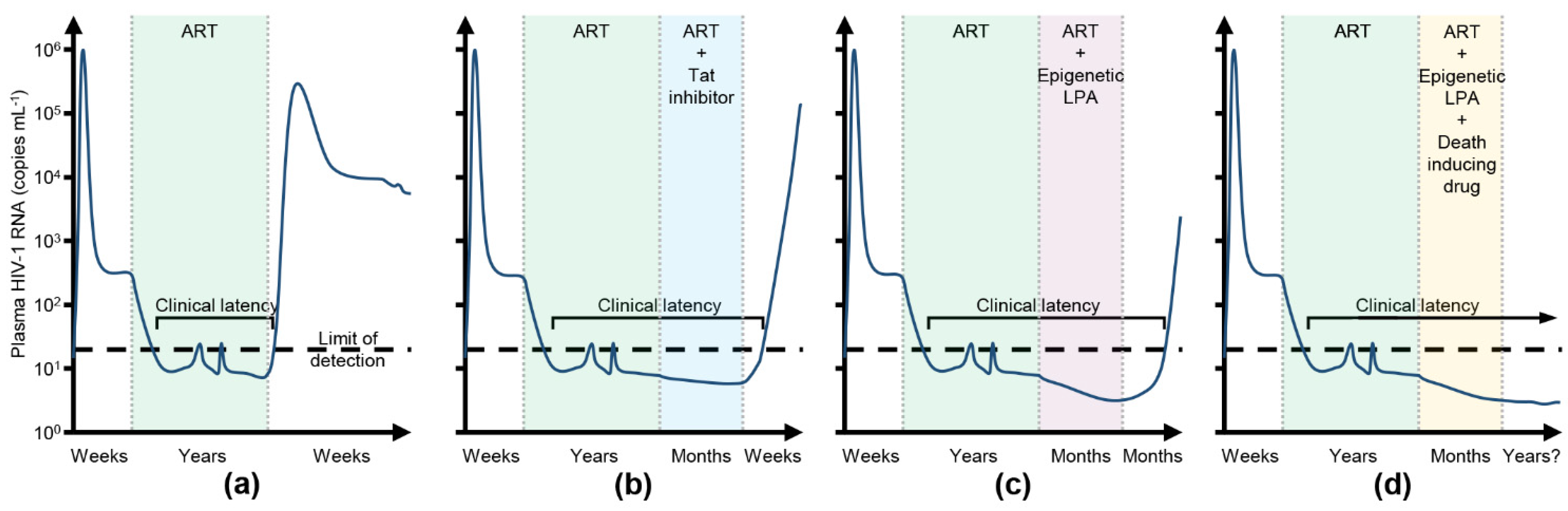
2. The HIV-1 Latent Reservoir
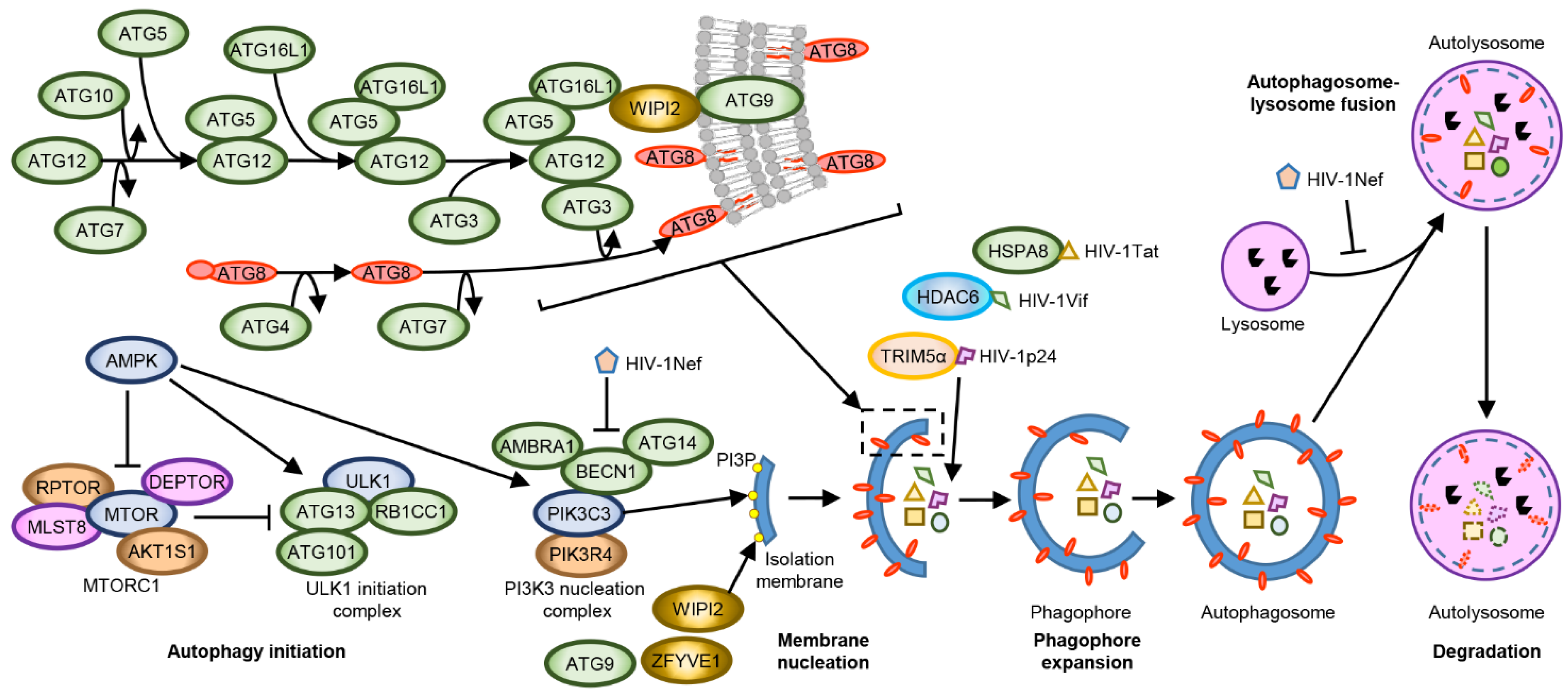
3. Autophagy-Mediated Restriction of HIV-1
4. HIV-1 Cure Strategies
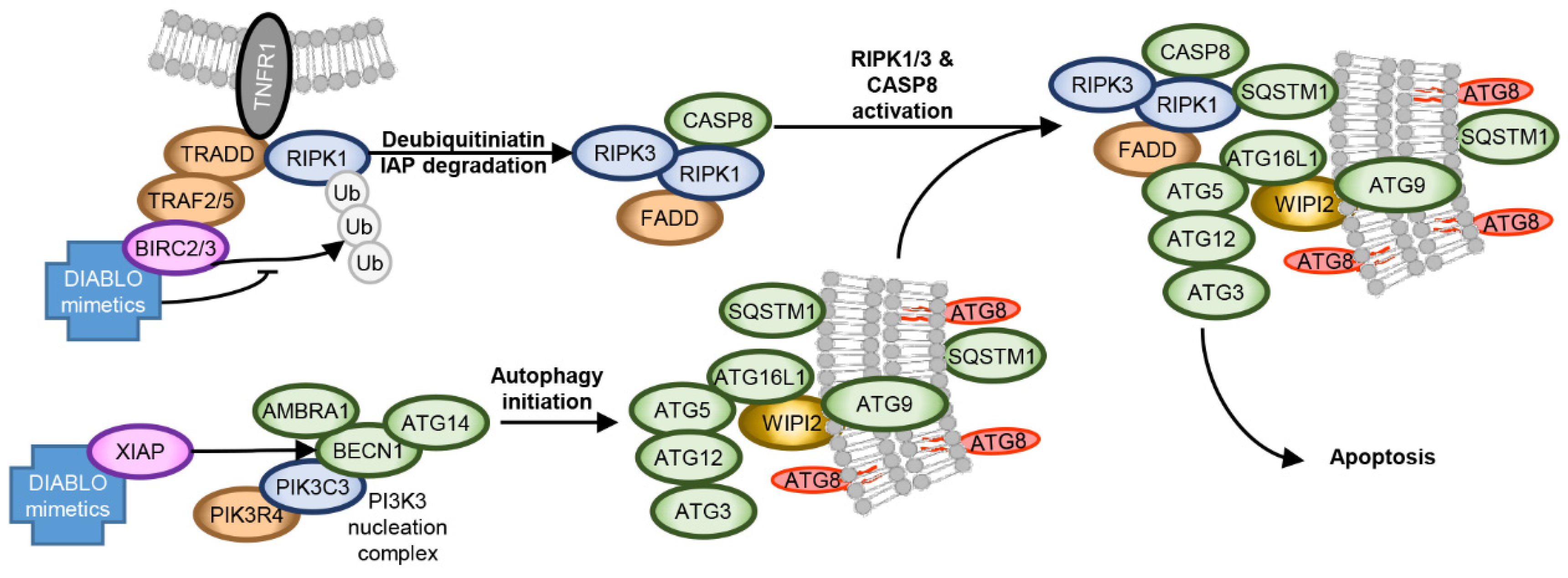
5. Autophagy as Part of a HIV-1 Cure Approach
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 infection. N. Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef] [Green Version]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial cells: The main HIV-1 reservoir in the brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, C.M.; Cordeiro, T.; Gomes, A.P.; Stevenson, M. The interplay of HIV-1 and macrophages in viral persistence. Front. Microbiol. 2021, 12, 646447. [Google Scholar] [CrossRef]
- Valdebenito, S.; Castellano, P.; Ajasin, D.; Eugenin, E.A. Astrocytes are HIV reservoirs in the brain: A cell type with poor HIV infectivity and replication but efficient cell-to-cell viral transfer. J. Neurochem. 2021, 158, 429–443. [Google Scholar] [CrossRef]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Crooks, A.M.; Bateson, R.; Cope, A.B.; Dahl, N.P.; Griggs, M.K.; Kuruc, J.D.; Gay, C.L.; Eron, J.J.; Margolis, D.M.; Bosch, R.J.; et al. Precise quantitation of the latent HIV-1 reservoir: Implications for eradication strategies. J. Infect. Dis. 2015, 212, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, B.; Mittler, J.E.; Zhang, L.; Boden, D.; Hurley, A.; Fang, F.; Macken, C.A.; Perelson, A.S.; Markowitz, M.; Ho, D.D. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 2000, 6, 82–85. [Google Scholar] [CrossRef]
- Hermankova, M.; Ray, S.C.; Ruff, C.; Powell-Davis, M.; Ingersoll, R.; D’Aquila, R.T.; Quinn, T.C.; Siliciano, J.D.; Siliciano, R.F.; Persaud, D. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA 2001, 286, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boritz, E.A.; Darko, S.; Swaszek, L.; Wolf, G.; Wells, D.; Wu, X.; Henry, A.R.; Laboune, F.; Hu, J.; Ambrozak, D.; et al. Multiple origins of virus persistence during natural control of HIV infection. Cell 2016, 166, 1004–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Frange, P.; Faye, A.; Avettand-Fenoel, V.; Bellaton, E.; Descamps, D.; Angin, M.; David, A.; Caillat-Zucman, S.; Peytavin, G.; Dollfus, C.; et al. HIV-1 virological remission lasting more than 12 years after interruption of early antiretroviral therapy in a perinatally infected teenager enrolled in the French ANRS EPF-CO10 paediatric cohort: A case report. Lancet HIV 2016, 3, e49–e54. [Google Scholar] [CrossRef] [Green Version]
- Savini, M.; Zhao, Q.; Wang, M.C. Lysosomes: Signaling hubs for metabolic sensing and longevity. Trends Cell Biol. 2019, 29, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 328–342. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Hall, M.N. Regulation of mTORC2 signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Morozumi, Y.; Shiozaki, K. Conserved and divergent mechanisms that control TORC1 in yeasts and mammals. Genes 2021, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Lu, M.; Wang, J.; Jones, K.T.; Ives, H.E.; Feldman, M.E.; Yao, L.J.; Shokat, K.M.; Ashrafi, K.; Pearce, D. mTOR complex-2 activates ENaC by phosphorylating SGK1. J. Am. Soc. Nephrol. 2010, 21, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Feehan, R.P.; Shantz, L.M. Negative regulation of the FOXO3a transcription factor by mTORC2 induces a pro-survival response following exposure to ultraviolet-B irradiation. Cell. Signal. 2016, 28, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Arora, S.; Ahmed, S.; Banerjea, A.C. Hyperactivation of mammalian target of rapamycin complex 1 by HIV-1 is necessary for virion production and latent viral reactivation. FASEB J. 2017, 31, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR complex controls HIV latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Liao, Q.; Chen, J.; Zhang, L.; He, Q.; Zhu, H.; Zhang, X.; Xu, J. TSC1 and DEPDC5 regulate HIV-1 latency through the mTOR signaling pathway. Emerg. Microbes Infect. 2018, 7, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbay, B.; Shmakova, A.; Vassetzky, Y.; Dokudovskaya, S. Modulation of mTORC1 signaling pathway by HIV-1. Cells 2020, 9, 1090. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Benmoussa, K.; Garaude, J.; Acin-Perez, R. How mitochondrial metabolism contributes to macrophage phenotype and functions. J. Mol. Biol. 2018, 430, 3906–3921. [Google Scholar] [CrossRef]
- Zhang, S.; Carriere, J.; Lin, X.; Xie, N.; Feng, P. Interplay between cellular metabolism and cytokine responses during viral infection. Viruses 2018, 10, 521. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, J.; Saraber, D.L. Metabolic regulation of macrophages in tissues. Cell. Immunol. 2018, 330, 54–59. [Google Scholar] [CrossRef]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Gabandé-Rodríguez, E.; Gómez de Las Heras, M.M.; Mittelbrunn, M. Control of inflammation by calorie restriction mimetics: On the crossroad of autophagy and mitochondria. Cells 2019, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Spector, S.A. Inhibition of human immunodeficiency virus type-1 through autophagy. Curr. Opin. Microbiol. 2013, 16, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Leymarie, O.; Lepont, L.; Berlioz-Torrent, C. Canonical and non-canonical autophagy in HIV-1 replication cycle. Viruses 2017, 9, 270. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, E.; Barbeau, B. Implication of different HIV-1 genes in the modulation of autophagy. Viruses 2017, 9, 389. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Estévez-Herrera, J.; Márquez-Arce, D.; Cabrera, C.; Espert, L.; Blanco, J.; Valenzuela-Fernández, A. The interplay of HIV and autophagy in early infection. Front. Microbiol. 2021, 12, 661446. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Paras, P., Jr.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol. Cell 2017, 67, 1001–1012.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Ping, Y.H.; Rana, T.M. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef] [Green Version]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell. Biol. 2014, 34, 1911–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef] [Green Version]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Mori, L.; Valente, S.T. Key players in HIV-1 transcriptional regulation: Targets for a functional cure. Viruses 2020, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, C.F.; Kim, Y.K.; Churcher, M.J.; West, M.J.; Karn, J. Spt5 cooperates with human immunodeficiency virus type 1 Tat by preventing premature RNA release at terminator sequences. Mol. Cell. Biol. 2002, 22, 1079–1093. [Google Scholar] [CrossRef] [Green Version]
- Parada, C.A.; Roeder, R.G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 1996, 384, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Col, E.; Caron, C.; Seigneurin-Berny, D.; Gracia, J.; Favier, A.; Khochbin, S. The histone acetyltransferase, hGCN5, interacts with and acetylates the HIV transactivator, Tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar] [CrossRef] [Green Version]
- Tréand, C.; du Chéné, I.; Brès, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenet. Chromatin 2019, 12, 23. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Cherrier, T.; Le Douce, V.; Eilebrecht, S.; Riclet, R.; Marban, C.; Dequiedt, F.; Goumon, Y.; Paillart, J.C.; Mericskay, M.; Parlakian, A.; et al. CTIP2 is a negative regulator of P-TEFb. Proc. Natl. Acad. Sci. USA 2013, 110, 12655–12660. [Google Scholar] [CrossRef] [Green Version]
- Eilebrecht, S.; Le Douce, V.; Riclet, R.; Targat, B.; Hallay, H.; Van Driessche, B.; Schwartz, C.; Robette, G.; Van Lint, C.; Rohr, O.; et al. HMGA1 recruits CTIP2-repressed P-TEFb to the HIV-1 and cellular target promoters. Nucleic Acids Res. 2014, 42, 4962–4971. [Google Scholar] [CrossRef]
- Forouzanfar, F.; Ali, S.; Wallet, C.; De Rovere, M.; Ducloy, C.; El Mekdad, H.; El Maassarani, M.; Ait-Ammar, A.; Van Assche, J.; Boutant, E.; et al. HIV-1 Vpr mediates the depletion of the cellular repressor CTIP2 to counteract viral gene silencing. Sci. Rep. 2019, 9, 13154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Moresco, J.J.; Chang, M.; Mukim, A.; Smith, D.; Diedrich, J.K.; Yates, J.R., 3rd; Jones, K.A. SHMT2 and the BRCC36/BRISC deubiquitinase regulate HIV-1 Tat K63-ubiquitylation and destruction by autophagy. PLoS Pathog. 2018, 14, e1007071. [Google Scholar] [CrossRef]
- Brès, V.; Kiernan, R.E.; Linares, L.K.; Chable-Bessia, C.; Plechakova, O.; Tréand, C.; Emiliani, S.; Péloponèse, J.M.; Jeang, K.T.; Coux, O.; et al. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003, 5, 754–761. [Google Scholar] [CrossRef]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making sense of multifunctional proteins: Human immunodeficiency virus type 1 accessory and regulatory proteins and connections to transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Qin, J.; Li, Y.; Wang, J.; He, Q.; Zhou, J.; Liu, M.; Li, D. Modulation of the stability and activities of HIV-1 Tat by its ubiquitination and carboxyl-terminal region. Cell Biosci. 2014, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef]
- Lange, U.C.; Verdikt, R.; Ait-Ammar, A.; Van Lint, C. Epigenetic crosstalk in chronic infection with HIV-1. Semin. Immunopathol. 2020, 42, 187–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamer, E.; Zhong, C.; Hajnik, R.; Soong, L.; Hu, H. Modulation of BRD4 in HIV epigenetic regulation: Implications for finding an HIV cure. Retrovirology 2021, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell. Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [Green Version]
- Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; Van Lint, C.; Schwartz, C. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res. 2012, 40, 1904–1915. [Google Scholar] [CrossRef]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Irwin, D.; Kanazawa, S.; Huang, L.; Romeo, J.; Yen, T.S.; Peterlin, B.M. Transcriptional profiles of latent human immunodeficiency virus in infected individuals: Effects of Tat on the host and reservoir. J. Virol. 2003, 77, 8227–8236. [Google Scholar] [CrossRef] [Green Version]
- Telwatte, S.; Lee, S.; Somsouk, M.; Hatano, H.; Baker, C.; Kaiser, P.; Kim, P.; Chen, T.H.; Milush, J.; Hunt, P.W.; et al. Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS Pathog. 2018, 14, e1007357. [Google Scholar] [CrossRef] [Green Version]
- Sarracino, A.; Gharu, L.; Kula, A.; Pasternak, A.O.; Avettand-Fenoel, V.; Rouzioux, C.; Bardina, M.; De Wit, S.; Benkirane, M.; Berkhout, B.; et al. Posttranscriptional regulation of HIV-1 gene expression during replication and reactivation from latency by nuclear matrix protein MATR3. mBio 2018, 9, e02158-18. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Sung, T.L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chable-Bessia, C.; Meziane, O.; Latreille, D.; Triboulet, R.; Zamborlini, A.; Wagschal, A.; Jacquet, J.M.; Reynes, J.; Levy, Y.; Saib, A.; et al. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 2009, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Knorr, R.L.; Lipowsky, R.; Dimova, R. Autophagosome closure requires membrane scission. Autophagy 2015, 11, 2134–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.J.; Hellman, J.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef] [Green Version]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Coulon, P.G.; Richetta, C.; Rouers, A.; Blanchet, F.P.; Urrutia, A.; Guerbois, M.; Piguet, V.; Theodorou, I.; Bet, A.; Schwartz, O.; et al. HIV-infected dendritic cells present endogenous MHC class II-restricted antigens to HIV-specific CD4+ T cells. J. Immunol. 2016, 197, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, T.K.; Mickelson, D.J.; Fink, J.; Solberg, J.C.; Inglefield, J.R.; Hook, D.; Gupta, S.K.; Gibson, S.; Alkan, S.S. Toll-like receptor (TLR) 2-9 agonists-induced cytokines and chemokines: I. Comparison with T cell receptor-induced responses. Cell. Immunol. 2006, 243, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Bang, C.; Rosigkeit, H.; Schmitz, R.A.; Heine, H. The human-associated archaeon Methanosphaera stadtmanae is recognized through its RNA and induces TLR8-dependent NLRP3 inflammasome activation. Front. Immunol. 2017, 8, 1535. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; To, R.K.; Hanna, J.; Spector, S.A. SARS-CoV-2, SARS-CoV-1, and HIV-1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non-classical pathway. iScience 2021, 24, 102295. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Kang, K.H.; Spector, S.A. Production of interferon α by human immunodeficiency virus type 1 in human plasmacytoid dendritic cells is dependent on induction of autophagy. J. Infect. Dis. 2012, 205, 1258–1267. [Google Scholar] [CrossRef]
- Schlaepfer, E.; Audige, A.; Joller, H.; Speck, R.F. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J. Immunol. 2006, 176, 2888–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, G.R.; Spector, S.A. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog. 2012, 8, e1003017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, S.; De Souza, G.A.; Salte, T.; Wiker, H.G.; Asjo, B. HIV induces both a down-regulation of IRAK-4 that impairs TLR signalling and an up-regulation of the antibiotic peptide dermcidin in monocytic cells. Scand. J. Immunol. 2009, 70, 264–276. [Google Scholar] [CrossRef]
- Haug, C.; Müller, F.; Aukrust, P.; Frøland, S.S. Subnormal serum concentration of 1,25-vitamin D in human immunodeficiency virus infection: Correlation with degree of immune deficiency and survival. J. Infect. Dis. 1994, 169, 889–893. [Google Scholar] [CrossRef]
- Haug, C.J.; Aukrust, P.; Haug, E.; Mørkrid, L.; Müller, F.; Frøland, S.S. Severe deficiency of 1,25-dihydroxyvitamin D3 in human immunodeficiency virus infection: Association with immunological hyperactivity and only minor changes in calcium homeostasis. J. Clin. Endocrinol. Metab. 1998, 83, 3832–3838. [Google Scholar] [CrossRef]
- Teichmann, J.; Stephan, E.; Discher, T.; Lange, U.; Federlin, K.; Stracke, H.; Friese, G.; Lohmeyer, J.; Bretzel, R.G. Changes in calciotropic hormones and biochemical markers of bone metabolism in patients with human immunodeficiency virus infection. Metabolism 2000, 49, 1134–1139. [Google Scholar] [CrossRef]
- Teichmann, J.; Stephan, E.; Lange, U.; Discher, T.; Friese, G.; Lohmeyer, J.; Stracke, H.; Bretzel, R.G. Osteopenia in HIV-infected women prior to highly active antiretroviral therapy. J. Infect. 2003, 46, 221–227. [Google Scholar] [CrossRef]
- Müller, N.J.; Fux, C.A.; Ledergerber, B.; Elzi, L.; Schmid, P.; Dang, T.; Magenta, L.; Calmy, A.; Vergopoulos, A.; Bischoff-Ferrari, H.A. High prevalence of severe vitamin D deficiency in combined antiretroviral therapy-naive and successfully treated Swiss HIV patients. AIDS 2010, 24, 1127–1134. [Google Scholar] [CrossRef]
- Pinzone, M.R.; Di Rosa, M.; Malaguarnera, M.; Madeddu, G.; Foca, E.; Ceccarelli, G.; d’Ettorre, G.; Vullo, V.; Fisichella, R.; Cacopardo, B.; et al. Vitamin D deficiency in HIV infection: An underestimated and undertreated epidemic. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1218–1232. [Google Scholar]
- Viard, J.P.; Souberbielle, J.C.; Kirk, O.; Reekie, J.; Knysz, B.; Losso, M.; Gatell, J.; Pedersen, C.; Bogner, J.R.; Lundgren, J.D.; et al. Vitamin D and clinical disease progression in HIV infection: Results from the EuroSIDA study. AIDS 2011, 25, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, J.E.; Adams, J.S. Vitamin D in HIV-infected patients. Curr. HIV/AIDS Rep. 2011, 8, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, N.; Aguilar-Jiménez, W.; Rugeles, M.T. The potential protective role of vitamin D supplementation on HIV-1 infection. Front. Immunol. 2019, 10, 2291. [Google Scholar] [CrossRef] [PubMed]
- Currò, M.; Visalli, G.; Pellicanò, G.F.; Ferlazzo, N.; Costanzo, M.G.; D’Andrea, F.; Caccamo, D.; Nunnari, G.; Ientile, R. Vitamin D status modulates inflammatory response in HIV+ subjects: Evidence for involvement of autophagy and TG2 expression in PBMC. Int. J. Mol. Sci. 2020, 21, 7558. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Hormonally active vitamin D3 (1α,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J. Biol. Chem. 2011, 286, 18890–18902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [Green Version]
- Black, L.R.; Aiken, C. TRIM5α disrupts the structure of assembled HIV-1 capsid complexes in vitro. J. Virol. 2010, 84, 6564–6569. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Pertel, T.; Gray, S.; Robia, S.L.; Bakowska, J.C.; Luban, J.; Campbell, E.M. p62/sequestosome-1 associates with and sustains the expression of retroviral restriction factor TRIM5α. J. Virol. 2010, 84, 5997–6006. [Google Scholar] [CrossRef] [Green Version]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Munch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Keown, J.R.; Black, M.M.; Ferron, A.; Yap, M.; Barnett, M.J.; Pearce, F.G.; Stoye, J.P.; Goldstone, D.C. A helical LC3-interacting region mediates the interaction between the retroviral restriction factor Trim5α and mammalian autophagy-related ATG8 proteins. J. Biol. Chem. 2018, 293, 18378–18386. [Google Scholar] [CrossRef] [Green Version]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, B.; Chisholm, D.; Kell, A.M.; Mandell, M.A. A non-canonical role for the autophagy machinery in anti-retroviral signaling mediated by TRIM5α. PLoS Pathog. 2020, 16, e1009017. [Google Scholar] [CrossRef] [PubMed]
- Ciccosanti, F.; Corazzari, M.; Casetti, R.; Amendola, A.; Collalto, D.; Refolo, G.; Vergori, A.; Taibi, C.; D’Offizi, G.; Antinori, A.; et al. High levels of TRIM5α are associated with xenophagy in HIV-1-infected long-term nonprogressors. Cells 2021, 10, 1207. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Erpapazoglou, Z.; Walker, O.; Haguenauer-Tsapis, R. Versatile roles of K63-linked ubiquitin chains in trafficking. Cells 2014, 3, 1027–1088. [Google Scholar] [CrossRef] [Green Version]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-González, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernández, S.; Borel, S.; Garcia-Expósito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef counteracts autophagy restriction by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent manner. Autophagy 2021, 17, 553–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antivir. Res. 2019, 166, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [Green Version]
- Pace, M.; Williams, J.; Kurioka, A.; Gerry, A.B.; Jakobsen, B.; Klenerman, P.; Nwokolo, N.; Fox, J.; Fidler, S.; Frater, J.; et al. Histone deacetylase inhibitors enhance CD4 T cell susceptibility to NK cell killing but reduce NK cell function. PLoS Pathog. 2016, 12, e1005782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucera, M.; Tilton, C.A.; Mao, H.; Dobrowolski, C.; Tabler, C.; Haqqani, A.A.; Karn, J.; Tilton, J.C. The histone deacetylase inhibitor vorinostat (SAHA) increases the susceptibility of uninfected CD4+ T cells to HIV by increasing the kinetics and efficiency of post-entry viral events. J. Virol. 2014, 88, 10803–10812. [Google Scholar] [CrossRef] [Green Version]
- Berro, R.; de la Fuente, C.; Klase, Z.; Kehn, K.; Parvin, L.; Pumfery, A.; Agbottah, E.; Vertes, A.; Nekhai, S.; Kashanchi, F. Identifying the membrane proteome of HIV-1 latently infected cells. J. Biol. Chem. 2007, 282, 8207–8218. [Google Scholar] [CrossRef] [Green Version]
- Swingler, S.; Mann, A.M.; Zhou, J.; Swingler, C.; Stevenson, M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007, 3, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.L.; Chunduri, H.; Wise, J.; Mindley, R.; Rimland, D. Replication-independent expression of anti-apoptosis marker genes in human peripheral blood mononuclear cells infected with the wild-type HIV-1 and reverse transcriptase variants. Viral Immunol. 2012, 25, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Trout, R.N.; Spector, S.A. SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected resting memory CD4+ T cells. Cell Host Microbe 2018, 24, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 is a negative regulator of HIV-1 transcription and can be targeted by Smac mimetics to promote reversal of viral latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.H.; Du, C. Smac/DIABLO selectively reduces the levels of c-IAP1 and c-IAP2 but not that of XIAP and livin in HeLa cells. J. Biol. Chem. 2004, 279, 16963–16970. [Google Scholar] [CrossRef] [Green Version]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; To, R.K.; Zhang, G.; Spector, S.A. SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected macrophages. Cell Death Dis. 2020, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Dashti, A.; Waller, C.; Mavigner, M.; Schoof, N.; Bar, K.J.; Shaw, G.M.; Vanderford, T.H.; Liang, S.; Lifson, J.D.; Dunham, R.M.; et al. SMAC mimetic plus triple-combination bispecific HIVxCD3 retargeting molecules in SHIV.C.CH505-infected, antiretroviral therapy-suppressed rhesus macaques. J. Virol. 2020, 94, e00793-20. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.A.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological activation of non-canonical NF-κB signaling activates latent HIV-1 reservoirs in vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef]
- Sarabia, I.; Huang, S.H.; Ward, A.R.; Jones, R.B.; Bosque, A. The intact non-inducible latent HIV-1 reservoir is established in an in vitro primary TCM cell model of latency. J. Virol. 2021, 95, e01297-20. [Google Scholar] [CrossRef]
- Mavigner, M.; Liao, L.E.; Brooks, A.D.; Ke, R.; Mattingly, C.; Schoof, N.; McBrien, J.; Carnathan, D.; Liang, S.; Vanderford, T.H.; et al. CD8 lymphocyte depletion enhances the latency reversal activity of the SMAC mimetic AZD5582 in ART-suppressed SIV-infected rhesus macaques. J. Virol. 2021, 95, e01429-20. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting cellular and tissue HIV reservoirs with Toll-like receptor agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef] [Green Version]
- Lore, K.; Betts, M.R.; Brenchley, J.M.; Kuruppu, J.; Khojasteh, S.; Perfetto, S.; Roederer, M.; Seder, R.A.; Koup, R.A. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J. Immunol. 2003, 171, 4320–4328. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The TLR and IL-1 signalling network at a glance. J. Cell Sci. 2014, 127, 2383–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatica, A.; Schwarzer, R.; Brantley, W.; Varco-Merth, B.; Sperber, H.S.; Hull, P.A.; Montano, M.; Migueles, S.A.; Rosenthal, D.; Hogan, L.E.; et al. Evaluating a new class of AKT/mTOR activators for HIV latency reversing activity ex vivo and in vivo. J. Virol. 2021, 95, e02393-20. [Google Scholar] [CrossRef]
- Feorino, P.M.; Butera, S.T.; Folks, T.M.; Schinazi, R.F. Prevention of activation of HIV-1 by antiviral agents in OM-10.1 cells. Antivir. Chem. Chemother. 1993, 4, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Patzold, S.; Schneider, J.; Rudolph, C.; Marme, D.; Schachtele, C. Novel indolocarbazole protein kinase C inhibitors prevent reactivation of HIV-1 in latently infected cells. Antivir. Res. 1993, 22, 273–283. [Google Scholar] [CrossRef]
- Namazi, G.; Fajnzylber, J.M.; Aga, E.; Bosch, R.J.; Acosta, E.P.; Sharaf, R.; Hartogensis, W.; Jacobson, J.M.; Connick, E.; Volberding, P.; et al. The control of HIV after antiretroviral medication pause (CHAMP) study: Posttreatment controllers identified from 14 clinical studies. J. Infect. Dis. 2018, 218, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Violari, A.; Cotton, M.F.; Kuhn, L.; Schramm, D.B.; Paximadis, M.; Loubser, S.; Shalekoff, S.; Da Costa Dias, B.; Otwombe, K.; Liberty, A.; et al. A child with perinatal HIV infection and long-term sustained virological control following antiretroviral treatment cessation. Nat. Commun. 2019, 10, 412. [Google Scholar] [CrossRef]
- Kamori, D.; Ueno, T. HIV-1 Tat and viral latency: What we can learn from naturally occurring sequence variations. Front. Microbiol. 2017, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Esquieu, D.; Péloponèse, J.M.; Opi, S.; Grégoire, C.; de Mareuil, J.; Watkins, J.; Campbell, G.; Dunot, J.P.; Sturgis, J.; Witvrouw, M.; et al. Discovery of a Tat HIV-1 inhibitor through computer-aided drug design. Spectroscopy 2003, 17, 639–645. [Google Scholar] [CrossRef]
- Montembault, M.; Vo-Thanh, G.; Deyine, A.; Fargeas, V.; Villiéras, M.; Adjou, A.; Dubreuil, D.; Esquieu, D.; Grégoire, C.; Opi, S.; et al. A possible improvement for structure-based drug design illustrated by the discovery of a Tat HIV-1 inhibitor. Bioorg. Med. Chem. Lett. 2004, 14, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.N.; Palu, G. Inhibitors of HIV-1 Tat-mediated transactivation. Curr. Med. Chem. 2006, 13, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.D.; Campbell, G.R.; Halimi, H.; Loret, E.P. Homonuclear 1H NMR and circular dichroism study of the HIV-1 Tat Eli variant. Retrovirology 2008, 5, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Li, D.; Lin, M.H.; Li, L.; Harrich, D. Tat-based therapies as an adjuvant for an HIV-1 functional cure. Viruses 2020, 12, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moranguinho, I.; Valente, S.T. Block-And-Lock: New horizons for a cure for HIV-1. Viruses 2020, 12, 1443. [Google Scholar] [CrossRef]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-cortistatin A inhibits HIV-1 by specifically binding to the unstructured basic region of Tat. mBio 2019, 10, e02662-18. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, L.U.; Rodríguez-González, J. Modeling the effect of tat inhibitors on HIV latency. J. Theor. Biol. 2019, 473, 20–27. [Google Scholar] [CrossRef]
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.P.; Planelles, V.; D’Orso, I. Transcriptional circuit fragility influences HIV proviral fate. Cell Rep. 2019, 27, 154–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacombe, B.; Morel, M.; Margottin-Goguet, F.; Ramirez, B.C. Specific inhibition of HIV infection by the action of spironolactone in T cells. J. Virol. 2016, 90, 10972–10980. [Google Scholar] [CrossRef] [Green Version]
- Leoz, M.; Kukanja, P.; Luo, Z.; Huang, F.; Cary, D.C.; Peterlin, B.M.; Fujinaga, K. HEXIM1-Tat chimera inhibits HIV-1 replication. PLoS Pathog. 2018, 14, e1007402. [Google Scholar] [CrossRef]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373. [Google Scholar] [CrossRef]
- Alamer, E.; Zhong, C.; Liu, Z.; Niu, Q.; Long, F.; Guo, L.; Gelman, B.B.; Soong, L.; Zhou, J.; Hu, H. Epigenetic suppression of HIV in myeloid cells by the BRD4-selective small molecule modulator ZL0580. J. Virol. 2020, 94, e01880-19. [Google Scholar] [CrossRef]
- Meredith, L.W.; Sivakumaran, H.; Major, L.; Suhrbier, A.; Harrich, D. Potent inhibition of HIV-1 replication by a Tat mutant. PLoS ONE 2009, 4, e7769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.H.; Sivakumaran, H.; Apolloni, A.; Wei, T.; Jans, D.A.; Harrich, D. Nullbasic, a potent anti-HIV tat mutant, induces CRM1-dependent disruption of HIV rev trafficking. PLoS ONE 2012, 7, e51466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.H.; Sivakumaran, H.; Jones, A.; Li, D.; Harper, C.; Wei, T.; Jin, H.; Rustanti, L.; Meunier, F.A.; Spann, K.; et al. A HIV-1 Tat mutant protein disrupts HIV-1 Rev function by targeting the DEAD-box RNA helicase DDX1. Retrovirology 2014, 11, 121. [Google Scholar] [CrossRef]
- Lin, M.H.; Apolloni, A.; Cutillas, V.; Sivakumaran, H.; Martin, S.; Li, D.; Wei, T.; Wang, R.; Jin, H.; Spann, K.; et al. A mutant tat protein inhibits HIV-1 reverse transcription by targeting the reverse transcription complex. J. Virol. 2015, 89, 4827–4836. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Z.; Etemad, B.; Ahmed, H.; Aga, E.; Bosch, R.J.; Mellors, J.W.; Kuritzkes, D.R.; Lederman, M.M.; Para, M.; Gandhi, R.T. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 2016, 30, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Kaiser, P.; Lampiris, H.W.; Kim, P.; Yukl, S.A.; Havlir, D.V.; Greene, W.C.; Wong, J.K. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 2016, 22, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vidal, E.; Castellvi, M.; Pujantell, M.; Badia, R.; Jou, A.; Gomez, L.; Puig, T.; Clotet, B.; Ballana, E.; Riveira-Munoz, E.; et al. Evaluation of the innate immune modulator acitretin as a strategy to clear the HIV reservoir. Antimicrob. Agents Chemother. 2017, 61, e01368-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Hayashi, T.; Jean, M.; Kong, W.; Fiches, G.; Biswas, A.; Liu, S.; Yosief, H.O.; Zhang, X.; Bradner, J.; et al. Inhibition of polo-like kinase 1 (PLK1) facilitates the elimination of HIV-1 viral reservoirs in CD4+ T cells ex vivo. Sci. Adv. 2020, 6, eaba1941. [Google Scholar] [CrossRef]
- Alto, A.; Natesampillai, S.; Chandrasekar, A.P.; Krogman, A.; Misra, A.; Shweta, F.; VanLith, C.; Yao, J.D.; Cummins, N.W.; Badley, A.D. The combination of venetoclax and ixazomib selectively and efficiently kills HIV-infected cell lines but has unacceptable toxicity in primary cell models. J. Virol. 2021, 95, e00138-21. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Lungu, C.; Crespo, R.; Steijaert, T.H.; Gorska, A.; Palstra, R.J.; Prins, H.A.B.; van Ijcken, W.; Mueller, Y.M.; van Kampen, J.J.A.; et al. Selective cell death in HIV-1-infected cells by DDX3 inhibitors leads to depletion of the inducible reservoir. Nat. Commun. 2021, 12, 2475. [Google Scholar] [CrossRef]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, J.; Paquette, J.S.; Fortin, J.F.; Tremblay, M.J. The immunosuppressant rapamycin represses human immunodeficiency virus type 1 replication. Antimicrob. Agents Chemother. 2002, 46, 3447–3455. [Google Scholar] [CrossRef] [Green Version]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Cloherty, A.P.M.; van Teijlingen, N.H.; Eisden, T.; van Hamme, J.L.; Rader, A.G.; Geijtenbeek, T.B.H.; Schreurs, R.; Ribeiro, C.M.S. Autophagy-enhancing drugs limit mucosal HIV-1 acquisition and suppress viral replication ex vivo. Sci. Rep. 2021, 11, 4767. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, F.; Di Sandro, S.; De Ruvo, N.; Montalti, R.; Ballarin, R.; Guerrini, G.P.; Spaggiari, M.; Guaraldi, G.; Gerunda, G. First report on a series of HIV patients undergoing rapamycin monotherapy after liver transplantation. Transplantation 2010, 89, 733–738. [Google Scholar] [CrossRef]
- Henrich, T.J.; Schreiner, C.; Cameron, C.; Hogan, L.E.; Richardson, B.; Rutishauser, R.L.; Deitchman, A.N.; Chu, S.; Rogers, R.; Thanh, C.; et al. Everolimus, an mTORC1/2 inhibitor, in ART-suppressed individuals who received solid organ transplantation: A prospective study. Am. J. Transplant. 2020, 21, 1765–1779. [Google Scholar] [CrossRef]
- Nicoletti, F.; Lapenta, C.; Donati, S.; Spada, M.; Ranazzi, A.; Cacopardo, B.; Mangano, K.; Belardelli, F.; Perno, C.; Aquaro, S. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clin. Exp. Immunol. 2009, 155, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Amoroso, A.; Davis, C.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV β-chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef] [Green Version]
- Goodall, M.L.; Cramer, S.D.; Thorburn, A. Autophagy complexes cell death by necroptosis. Oncotarget 2016, 7, 50818–50819. [Google Scholar] [CrossRef]
- Kriel, J.; Loos, B. The good, the bad and the autophagosome: Exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 2019, 26, 640–652. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.O.; DeMarino, C.; Vo, T.T.; Cowen, M.; Kim, Y.; Pleet, M.L.; Barclay, R.A.; Noren Hooten, N.; Evans, M.K.; Heredia, A.; et al. Low-level ionizing radiation induces selective killing of HIV-1-infected cells with reversal of cytokine induction using mTOR inhibitors. Viruses 2020, 12, 885. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Spector, S.A. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Herns, S.D.; Joshi, S.; Durden, D.L.; Spector, S.A. Induction of autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 inhibitors suppresses HIV-1 replication. J. Biol. Chem. 2018, 293, 5808–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, P.; Hon, S.; Teodorof-Diedrich, C.; Spector, S.A. Trehalose inhibits human immunodeficiency virus type 1 infection in primary human macrophages and CD4+ T lymphocytes through two distinct mechanisms. J. Virol. 2020, 94, e00237-20. [Google Scholar] [CrossRef]
- Sharma, V.; Makhdoomi, M.; Singh, L.; Kumar, P.; Khan, N.; Singh, S.; Verma, H.N.; Luthra, K.; Sarkar, S.; Kumar, D. Trehalose limits opportunistic mycobacterial survival during HIV co-infection by reversing HIV-mediated autophagy block. Autophagy 2021, 17, 476–495. [Google Scholar] [CrossRef] [Green Version]
- Brüning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, T.; Goswami, S.; Hu, X.; Laverdure, S.; Yang, J.; Qiu, J.; Chen, Q.; Sherman, B.T.; Chang, W. MicroRNA profiles in monocyte-derived macrophages generated by interleukin-27 and human serum: Identification of a novel HIV-inhibiting and autophagy-inducing microRNA. Int. J. Mol. Sci. 2021, 22, 1290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Luk, B.T.; Wei, X.; Campbell, G.R.; Fang, R.H.; Zhang, L.; Spector, S.A. Selective cell death of latently HIV-infected CD4+ T cells mediated by autosis inducing nanopeptides. Cell Death Dis. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Luk, B.T.; Hamidy, M.; Zhang, L.; Spector, S.A. Induction of a Na+/K+-ATPase-dependent form of autophagy triggers preferential cell death of human immunodeficiency virus type-1-infected macrophages. Autophagy 2018, 14, 1359–1375. [Google Scholar] [CrossRef]
- Adesina, S.K.; Akala, E.O. Nanotechnology approaches for the delivery of exogenous siRNA for HIV therapy. Mol. Pharm. 2015, 12, 4175–4187. [Google Scholar] [CrossRef]
- Bowman, M.C.; Ballard, T.E.; Ackerson, C.J.; Feldheim, D.L.; Margolis, D.M.; Melander, C. Inhibition of HIV fusion with multivalent gold nanoparticles. J. Am. Chem. Soc. 2008, 130, 6896–6897. [Google Scholar] [CrossRef] [Green Version]
- Glass, J.J.; Yuen, D.; Rae, J.; Johnston, A.P.; Parton, R.G.; Kent, S.J.; De Rose, R. Human immune cell targeting of protein nanoparticles--caveospheres. Nanoscale 2016, 8, 8255–8265. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhang, G.; Ran, D.; Krishnan, N.; Fang, R.H.; Gao, W.; Spector, S.A.; Zhang, L. T-cell-mimicking nanoparticles can neutralize HIV infectivity. Adv. Mater. 2018, 30, e1802233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Campbell, G.R.; Zhang, Q.; Maule, E.; Hanna, J.; Gao, W.; Zhang, L.; Spector, S.A. CD4+ T cell-mimicking nanoparticles broadly neutralize HIV-1 and suppress viral replication through autophagy. mBio 2020, 11, e00903-20. [Google Scholar] [CrossRef]
- Fang, R.H.; Kroll, A.V.; Zhang, L. Nanoparticle-based manipulation of antigen-presenting cells for cancer immunotherapy. Small 2015, 11, 5483–5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teleanu, D.M.; Negut, I.; Grumezescu, V.; Grumezescu, A.M.; Teleanu, R.I. Nanomaterials for drug delivery to the central nervous system. Nanomaterials 2019, 9, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Lucas, A.; Kim, Y.; Rivera-Pabon, O.; Chae, S.; Kim, D.H.; Kim, B. Targeting the PI3K/Akt cell survival pathway to induce cell death of HIV-1 infected macrophages with alkylphospholipid compounds. PLoS ONE 2010, 5, e13121. [Google Scholar] [CrossRef]
- Kim, Y.; Hollenbaugh, J.A.; Kim, D.H.; Kim, B. Novel PI3K/Akt inhibitors screened by the cytoprotective function of human immunodeficiency virus type 1 Tat. PLoS ONE 2011, 6, e21781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Guillemard, E.; Jacquemot, C.; Aillet, F.; Schmitt, N.; Barre-Sinoussi, F.; Israel, N. Human immunodeficiency virus 1 favors the persistence of infection by activating macrophages through TNF. Virology 2004, 329, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; To, R.K.; Spector, S.A. TREM-1 protects HIV-1-infected macrophages from apoptosis through maintenance of mitochondrial function. mBio 2019, 10, e02638-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubrez, L.; Berthelet, J.; Glorian, V. IAP proteins as targets for drug development in oncology. OncoTargets Ther. 2013, 9, 1285–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obexer, P.; Ausserlechner, M.J. X-linked inhibitor of apoptosis protein—A critical death resistance regulator and therapeutic target for personalized cancer therapy. Front. Oncol. 2014, 4, 197. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhao, H.; Jin, M.; Zhu, H.; Shan, B.; Geng, J.; Dziedzic, S.A.; Amin, P.; Mifflin, L.; Naito, M.G.; et al. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature 2020, 587, 133–138. [Google Scholar] [CrossRef]
- Dumétier, B.; Zadoroznyj, A.; Dubrez, L. IAP-mediated protein ubiquitination in regulating cell signaling. Cells 2020, 9, 1118. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Najafov, A.; Luu, H.S.; Mookhtiar, A.K.; Mifflin, L.; Xia, H.G.; Amin, P.P.; Ordureau, A.; Wang, H.; Yuan, J. RIPK1 promotes energy sensing by the mTORC1 pathway. Mol. Cell 2021, 81, 370–385.e7. [Google Scholar] [CrossRef]
- Gao, Z.; Tian, Y.; Wang, J.; Yin, Q.; Wu, H.; Li, Y.M.; Jiang, X. A dimeric Smac/diablo peptide directly relieves caspase-3 inhibition by XIAP. Dynamic and cooperative regulation of XIAP by Smac/Diablo. J. Biol. Chem. 2007, 282, 30718–30727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, S.I.; Matsuda, K.; Tsuchiya, K.; Gatanaga, H.; Oka, S.; Yoshimura, K.; Mitsuya, H.; Maeda, K. Combination of a latency-reversing agent with a Smac mimetic minimizes secondary HIV-1 infection in vitro. Front. Microbiol. 2018, 9, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, G.; Preston, S.; Allison, C.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Scott, H.W.; Baschuk, N.; Nachbur, U.; et al. Cellular inhibitor of apoptosis proteins prevent clearance of hepatitis B virus. Proc. Natl. Acad. Sci. USA 2015, 112, 5797–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, G.; Allison, C.; Preston, S.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Baschuk, N.; Nachbur, U.; Torresi, J.; et al. Eliminating hepatitis B by antagonizing cellular inhibitors of apoptosis. Proc. Natl. Acad. Sci. USA 2015, 112, 5803–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, E.; Mackiewicz, L.; Silke, N.; Pellegrini, M.; Silke, J.; Brumatti, G.; Ebert, G. Combinatorial treatment of birinapant and zosuquidar enhances effective control of HBV replication in vivo. Viruses 2020, 12, 901. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, G.R.; Spector, S.A. Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure. Cells 2021, 10, 1798. https://doi.org/10.3390/cells10071798
Campbell GR, Spector SA. Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure. Cells. 2021; 10(7):1798. https://doi.org/10.3390/cells10071798
Chicago/Turabian StyleCampbell, Grant R., and Stephen A. Spector. 2021. "Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure" Cells 10, no. 7: 1798. https://doi.org/10.3390/cells10071798
APA StyleCampbell, G. R., & Spector, S. A. (2021). Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure. Cells, 10(7), 1798. https://doi.org/10.3390/cells10071798