
The Immune System Throws Its Traps: Cells and Their Extracellular Traps in Disease and Protection

 ,
,  ,
, 

Abstract
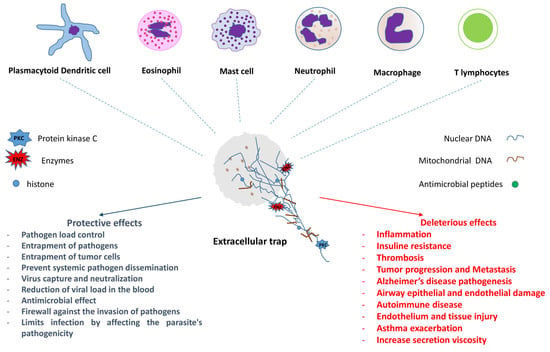
:
1. Preamble
2. General Background
3. Neutrophils
NETs and COVID-19
4. Macrophages
Cooperation between Macrophages and Neutrophils in the Extracellular Traps Context
5. Mast Cells
6. Eosinophils
7. Lymphocytes
8. Other Cells Involved in the Immune Response Whereby the Formation of Extracellular Traps Has Been Identified
8.1. Basophils
8.2. Plamacytoid Dendritic Cells
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hawking, S. The Universe in a Nutshell, 1st ed.; Transworld Publishers Ltd.: London, UK, 2001; ISBN 978-0-593-04815-3. [Google Scholar]
- Tan, C.; Aziz, M.; Wang, P. The Vitals of NETs. J. Leukoc. Biol. 2020, 1–12. [Google Scholar] [CrossRef]
- Brinkmann, V. Neutrophil Extracellular Traps in the Second Decade. J. Innate Immun. 2018, 10, 414–421. [Google Scholar] [CrossRef]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse Stimuli Engage Different Neutrophil Extracellular Trap Pathways. eLife 2017, 6, e24437. [Google Scholar] [CrossRef]
- Burgener, S.S.; Schroder, K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb. Perspect. Biol. 2020, 12, a037028. [Google Scholar] [CrossRef]
- Yousefi, S.; Simon, D.; Stojkov, D.; Karsonova, A.; Karaulov, A.; Simon, H.-U. In Vivo Evidence for Extracellular DNA Trap Formation. Cell Death Dis. 2020, 11, 300. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schuize, I.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophi Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Wartha, F.; Henriques-Normark, B. ETosis: A Novel Cell Death Pathway. Sci. Signal. 2008, 1, pe25. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Rawat, K.; Syeda, S.; Shrivastava, A. Neutrophil-Derived Granule Cargoes: Paving the Way for Tumor Growth and Progression. Cancer Metastasis Rev. 2021, 40, 221–244. [Google Scholar] [CrossRef]
- Wu, M.; Ma, M.; Tan, Z.; Zheng, H.; Liu, X. Neutrophil: A New Player in Metastatic Cancers. Front. Immunol. 2020, 11, 565165. [Google Scholar] [CrossRef]
- Goldmann, O.; Medina, E. The Expanding World of Extracellular Traps: Not Only Neutrophils but Much More. Front. Immunol. 2012, 3, 420. [Google Scholar] [CrossRef] [Green Version]
- Ingelsson, B.; Söderberg, D.; Strid, T.; Söderberg, A.; Bergh, A.-C.; Loitto, V.; Lotfi, K.; Segelmark, M.; Spyrou, G.; Rosén, A. Lymphocytes Eject Interferogenic Mitochondrial DNA Webs in Response to CpG and Non-CpG Oligodeoxynucleotides of Class C. Proc. Natl. Acad. Sci. USA 2018, 115, E478–E487. [Google Scholar] [CrossRef] [Green Version]
- De Bont, C.M.; Stokman, M.E.M.; Faas, P.; Thurlings, R.M.; Boelens, W.C.; Wright, H.L.; Pruijn, G.J.M. Autoantibodies to Neutrophil Extracellular Traps Represent a Potential Serological Biomarker in Rheumatoid Arthritis. J. Autoimmun. 2020, 113, 102484. [Google Scholar] [CrossRef]
- Hofbauer, T.M.; Ondracek, A.S.; Mangold, A.; Scherz, T.; Nechvile, J.; Seidl, V.; Brostjan, C.; Lang, I.M. Neutrophil Extracellular Traps Induce MCP-1 at the Culprit Site in ST-Segment Elevation Myocardial Infarction. Front. Cell Dev. Biol. 2020, 8, 564169. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of Neutrophil Extracellular Traps Promotes Cancer Metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Figueroa, E.; Álvarez-Carrasco, P.; Ortega, E.; Maldonado-Bernal, C. Neutrophils: Many Ways to Die. Front. Immunol. 2021, 12, 631821. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-Induced NETosis Is a Dynamic Process Involving Neutrophil Multitasking in Vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Kubes, P. NETosis: How Vital Is It? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef]
- Ravindran, M.; Khan, M.A.; Palaniyar, N. Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology. Biomolecules 2019, 9, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Daniel, C.; Leppkes, M.; Muñoz, L.E.; Schley, G.; Schett, G.; Herrmann, M. Extracellular DNA Traps in Inflammation, Injury and Healing. Nat. Rev. Nephrol. 2019, 15, 559–575. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA Are Interferogenic and Contribute to Lupus-like Disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in Innate Immune Responses and Inflammatory Pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Baker, V.S.; Imade, G.E.; Molta, N.B.; Tawde, P.; Pam, S.D.; Obadofin, M.O.; Sagay, S.A.; Egah, D.Z.; Iya, D.; Afolabi, B.B.; et al. Cytokine-Associated Neutrophil Extracellular Traps and Antinuclear Antibodies in Plasmodium Falciparum Infected Children under Six Years of Age. Malar. J. 2008, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi Abdallah, D.S.; Lin, C.; Ball, C.J.; King, M.R.; Duhamel, G.E.; Denkers, E.Y. Toxoplasma Gondii Triggers Release of Human and Mouse Neutrophil Extracellular Traps. Infect. Immun. 2012, 80, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Morgado, F.N.; Nascimento, M.T.C.; Saraiva, E.M.; de Oliveira-Ribeiro, C.; de Fátima Madeira, M.; da Costa-Santos, M.; Vasconcellos, E.C.F.; Pimentel, M.I.F.; Rosandiski Lyra, M.; Schubach, A.d.O.; et al. Are Neutrophil Extracellular Traps Playing a Role in the Parasite Control in Active American Tegumentary Leishmaniasis Lesions? PLoS ONE 2015, 10, e0133063. [Google Scholar] [CrossRef] [Green Version]
- Morgado, F.N.; Schubach, A.O.; Barros, M.B.L.; Conceição-Silva, F. The in Situ Inflammatory Profile of Lymphocutaneous and Fixed Forms of Human Sporotrichosis. Med. Mycol. 2011, 49, 612–620. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil Extracellular Traps Mediate a Host Defense Response to Human Immunodeficiency Virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.C.; Wardini, A.B.; Vieira, M.; Passos, L.S.A.; Martinelli, P.M.; Neves, E.G.A.; do Vale Antonelli, L.R.; Barbosa, D.F.; Velikkakam, T.; Gutseit, E.; et al. Human CD8+ T Cells Release Extracellular Traps Co-Localized with Cytotoxic Vesicles That Are Associated with Lesion Progression and Severity in Human Leishmaniasis. Front. Immunol. 2020, 11, 594581. [Google Scholar] [CrossRef] [PubMed]
- Von Köckritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-Independent Antimicrobial Activity of Mast Cells by Means of Extracellular Trap Formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef]
- Hellebrekers, P.; Vrisekoop, N.; Koenderman, L. Neutrophil Phenotypes in Health and Disease. Eur. J. Clin. Investig. 2018, 48, e12943. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role during Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Pillay, J.; den Braber, I.; Vrisekoop, N.; Kwast, L.M.; de Boer, R.J.; Borghans, J.A.M.; Tesselaar, K.; Koenderman, L. In Vivo Labeling with 2H2O Reveals a Human Neutrophil Lifespan of 5.4 Days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Beneficial Suicide: Why Neutrophils Die to Make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil Function: From Mechanisms to Disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Cruvinel, W.D.M.; Mesquita, D., Jr.; Araújo, J.A.P.; Catelan, T.T.T.; De Souza, A.W.S.; Da Silva, N.P.; Andrade, L.E.C. Immune System-Part I Fundamentals of Innate Immunity with Emphasis on Molecular and Cellular Mechanisms of Inflammatory Response. Bras. J. Rheumatol. 2010, 50, 434–461. [Google Scholar]
- Mócsai, A. Diverse Novel Functions of Neutrophils in Immunity, Inflammation, and Beyond. J. Exp. Med. 2013, 210, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E. Endothelial Cell–Cell Junctions: Happy Together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of Neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [Green Version]
- Palomino, D.C.T.; Marti, L.C. Chemokines and Immunity. Einstein (São Paulo) 2015, 13, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Circulação de Leucócitos e migração para os tecidos. In Imunologia Celular E Molecular; Koogan, G.G., Ed.; Elsevier: Rio de Janeiro, Brazil, 2019; pp. 39–56. ISBN 13: 9788535290745. [Google Scholar]
- Faurschou, M.; Borregaard, N. Neutrophil Granules and Secretory Vesicles in Inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from Marrow to Microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, A.W. How Neutrophils Kill Microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordenfelt, P.; Tapper, H. Phagosome Dynamics during Phagocytosis by Neutrophils. J. Leukoc. Biol. 2011, 90, 271–284. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid Killing of Human Neutrophils by the Potent Activator Phorbol 12-Myristate 13-Acetate (PMA) Accompanied by Changes Different from Typical Apoptosis or Necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Cahilog, Z.; Zhao, H.; Wu, L.; Alam, A.; Eguchi, S.; Weng, H.; Ma, D. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 2020, 43, 2021–2032. [Google Scholar] [CrossRef]
- Kim, S.-W.; Lee, J.-K. Role of HMGB1 in the Interplay between NETosis and Thrombosis in Ischemic Stroke: A Review. Cells 2020, 9, 1794. [Google Scholar] [CrossRef]
- Rada, B. Neutrophil Extracellular Traps and Microcrystals. J. Immunol. Res. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Rosazza, T.; Warner, J.; Sollberger, G. NET Formation-Mechanisms and How They Relate to Other Cell Death Pathways. FEBS J. 2020, 288, 3334–3350. [Google Scholar] [CrossRef] [PubMed]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil Extracellular Traps (NET) Induced by Different Stimuli: A Comparative Proteomic Analysis. PLoS ONE 2019, 14, e0218946. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Naffah de Souza, C.; Breda, L.C.D.; Khan, M.A.; de Almeida, S.R.; Câmara, N.O.S.; Sweezey, N.; Palaniyar, N. Alkaline PH Promotes NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation: A Matter of Mitochondrial Reactive Oxygen Species Generation and Citrullination and Cleavage of Histone. Front. Immunol. 2018, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Maueröder, C.; Hirth, S.; Nowecki, S.; Günther, C.; Billmeier, U.; Paulus, S.; Biermann, M.; Munoz, L.E.; Hoffmann, M.; et al. Externalized Decondensed Neutrophil Chromatin Occludes Pancreatic Ducts and Drives Pancreatitis. Nat. Commun. 2016, 7, 10973. [Google Scholar] [CrossRef]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases Prevent Vascular Occlusion by Neutrophil Extracellular Traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 Channel and Mitochondrial ROS Mediate NADPH Oxidase-Independent NETosis Induced by Calcium Influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [Green Version]
- Cristinziano, L.; Modestino, L.; Loffredo, S.; Varricchi, G.; Braile, M.; Ferrara, A.L.; de Paulis, A.; Antonelli, A.; Marone, G.; Galdiero, M.R. Anaplastic Thyroid Cancer Cells Induce the Release of Mitochondrial Extracellular DNA Traps by Viable Neutrophils. J. Immunol. 2020, 204, 1362–1372. [Google Scholar] [CrossRef]
- McIlroy, D.J.; Jarnicki, A.G.; Au, G.G.; Lott, N.; Smith, D.W.; Hansbro, P.M.; Balogh, Z.J. Mitochondrial DNA Neutrophil Extracellular Traps Are Formed after Trauma and Subsequent Surgery. J. Crit. Care 2014, 29, 1133.e1–1133.e5. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Rochael, N.C.; Guimarães-Costa, A.B.; Nascimento, M.T.C.; DeSouza-Vieira, T.S.; Oliveira, M.P.; Garcia e Souza, L.F.; Oliveira, M.F.; Saraiva, E.M. Classical ROS-Dependent and Early/Rapid ROS-Independent Release of Neutrophil Extracellular Traps Triggered by Leishmania Parasites. Sci. Rep. 2016, 5, 18302. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An Extracellular Matrix–Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida Albicans. J. Immunol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, S.; Kniemeyer, O.; Hasenberg, M.; Aimanianda, V.; Nietzsche, S.; Thywißen, A.; Jeron, A.; Latgé, J.-P.; Brakhage, A.A.; Gunzer, M. Production of Extracellular Traps against Aspergillus Fumigatus In Vitro and in Infected Lung Tissue Is Dependent on Invading Neutrophils and Influenced by Hydrophobin RodA. PLoS Pathog. 2010, 6, e1000873. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes-Costa, A.B.; Nascimento, M.T.C.; Froment, G.S.; Soares, R.P.P.; Morgado, F.N.; Conceicao-Silva, F.; Saraiva, E.M. Leishmania Amazonensis Promastigotes Induce and Are Killed by Neutrophil Extracellular Traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, C.; McMaster, W.R.; Girard, D.; Descoteaux, A. Leishmania Donovani Promastigotes Evade the Antimicrobial Activity of Neutrophil Extracellular Traps. J. Immunol. 2010, 185, 4319–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regli, I.B.; Passelli, K.; Hurrell, B.P.; Tacchini-Cottier, F. Survival Mechanisms Used by Some Leishmania Species to Escape Neutrophil Killing. Front. Immunol. 2017, 8, 1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passelli, K.; Billion, O.; Tacchini-Cottier, F. The Impact of Neutrophil Recruitment to the Skin on the Pathology Induced by Leishmania Infection. Front. Immunol. 2021, 12, 649348. [Google Scholar] [CrossRef]
- Da Fonseca-Martins, A.M.; de Souza Lima-Gomes, P.; Antunes, M.M.; de Moura, R.G.; Covre, L.P.; Calôba, C.; Rocha, V.G.; Pereira, R.M.; Menezes, G.B.; Gomes, D.C.O.; et al. Leishmania Parasites Drive PD-L1 Expression in Mice and Human Neutrophils with Suppressor Capacity. Front. Immunol. 2021, 12, 598943. [Google Scholar] [CrossRef]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated Neutrophil Extracellular Traps Resolve Inflammation by Proteolysis of Cytokines and Chemokines and Protection from Antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef]
- Knackstedt, S.L.; Georgiadou, A.; Apel, F.; Abu-Abed, U.; Moxon, C.A.; Cunnington, A.J.; Raupach, B.; Cunningham, D.; Langhorne, J.; Krüger, R.; et al. Neutrophil Extracellular Traps Drive Inflammatory Pathogenesis in Malaria. Sci. Immunol. 2019, 4, eaaw0336. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.C.; Kubes, P. Platelets, Neutrophils, and Neutrophil Extracellular Traps (NETs) in Sepsis. J. Thromb. Haemost. 2008, 6, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets Induce Neutrophil Extracellular Traps in Transfusion-Related Acute Lung Injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.K.; Kaplan, M.J. The Role of Neutrophils in the Pathogenesis of Systemic Lupus Erythematosus. Curr. Opin. Rheumatol. 2015, 27, 448–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tydén, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil Extracellular Traps That Are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hally, K.E.; Parker, O.M.; Brunton-O’Sullivan, M.M.; Harding, S.A.; Larsen, P.D. Linking Neutrophil Extracellular Traps and Platelet Activation: A Composite Biomarker Score for Predicting Outcomes after Acute Myocardial Infarction. Thromb. Haemost. 2021. [Google Scholar] [CrossRef]
- Stakos, D.; Skendros, P.; Konstantinides, S.; Ritis, K. Traps N’ Clots: NET-Mediated Thrombosis and Related Diseases. Thromb. Haemost. 2020, 120, 373–383. [Google Scholar] [CrossRef]
- Skendros, P.; Mitroulis, I. Host Cell Autophagy in Immune Response to Zoonotic Infections. Clin. Dev. Immunol. 2012, 2012, 910525. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Moschonas, I.C.; Tselepis, A.D. The Pathway of Neutrophil Extracellular Traps towards Atherosclerosis and Thrombosis. Atherosclerosis 2019, 288, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhang, H.; Qu, M.; Nan, K.; Cao, H.; Cata, J.P.; Chen, W.; Miao, C. Review: The Emerging Role of Neutrophil Extracellular Traps in Sepsis and Sepsis-Associated Thrombosis. Front. Cell. Infect. Microbiol. 2021, 11, 653228. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Holy, E.W.; Akhmedov, A.; Bonetti, N.R.; Nietlispach, F.; Matter, C.M.; Mach, F.; Montecucco, F.; Beer, J.H.; Paneni, F.; et al. Interleukin-1β Mediates Arterial Thrombus Formation via NET-Associated Tissue Factor. J. Clin. Med. 2019, 8, 2072. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Murakami, T.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Tabe, Y.; Nagaoka, I. Neutrophil Extracellular Traps Induce IL-1β Production by Macrophages in Combination with Lipopolysaccharide. Int. J. Mol. Med. 2017, 39, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arter. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Campos, J.; Ponomaryov, T.; De Prendergast, A.; Whitworth, K.; Smith, C.W.; Khan, A.O.; Kavanagh, D.; Brill, A. Neutrophil Extracellular Traps and Inflammasomes Cooperatively Promote Venous Thrombosis in Mice. Blood Adv. 2021, 5, 2319–2324. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil Extracellular Trap Formation Is Associated with IL-1β and Autophagy-Related Signaling in Gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil Extracellular Traps Promote Differentiation and Function of Fibroblasts: NETs Induce Fibrosis via Differentiation of Fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer Cells Induce Metastasis-Supporting Neutrophil Extracellular DNA Traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [Green Version]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; dos Reis, M.C.; de Castro, G.M.M.; da Silva Fontes, Y.; Todeschini, A.R.; Freire-de-Lima, L.; Decoté-Ricardo, D.; et al. The Emerging Role of Neutrophil Extracellular Traps in Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2–Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Godement, M.; Zhu, J.; Cerf, C.; Vieillard-Baron, A.; Maillon, A.; Zuber, B.; Bardet, V.; Geri, G. Neutrophil Extracellular Traps in SARS-CoV2 Related Pneumonia in ICU Patients: The NETCOV2 Study. Front. Med. 2021, 8, 615984. [Google Scholar] [CrossRef] [PubMed]
- Guéant, J.; Guéant-Rodriguez, R.; Fromonot, J.; Oussalah, A.; Louis, H.; Chery, C.; Gette, M.; Gleye, S.; Callet, J.; Raso, J.; et al. Elastase and Exacerbation of Neutrophil Innate Immunity Are Involved in Multi-visceral Manifestations of COVID-19. Allergy 2021, 76, 1846–1858. [Google Scholar] [CrossRef]
- Petito, E.; Falcinelli, E.; Paliani, U.; Cesari, E.; Vaudo, G.; Sebastiano, M.; Cerotto, V.; Guglielmini, G.; Gori, F.; Malvestiti, M.; et al. Association of Neutrophil Activation, More than Platelet Activation, With Thrombotic Complications in Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 933–944. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and Its Implications for Thrombosis and Anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.X.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-Lymphocyte Ratio as an Independent Risk Factor for Mortality in Hospitalized Patients with COVID-19. J. Infect. 2020, 81, e6–e12. [Google Scholar] [CrossRef]
- Ackermann, M.; Anders, H.-J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the Dark-NETs of Neutrophils. Cell Death Differ. 2021. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and Tissue Factor–Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID-19 Immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Morrissey, S.M.; Geller, A.E.; Hu, X.; Tieri, D.; Ding, C.; Klaes, C.K.; Cooke, E.A.; Woeste, M.R.; Martin, Z.C.; Chen, O.; et al. A Specific Low-Density Neutrophil Population Correlates with Hypercoagulation and Disease Severity in Hospitalized COVID-19 Patients. JCI Insight 2021, 6, e148435. [Google Scholar] [CrossRef]
- Yang, C.; Montgomery, M. Dornase Alfa for Cystic Fibrosis. Cochrane Database Syst. Rev. 2018, 6, CD001127. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 25 June 2021).
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil Extracellular Traps Sequester Circulating Tumor Cells and Promote Metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef]
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil Extracellular Traps Sequester Circulating Tumor Cells via B1-Integrin Mediated Interactions. Int. J. Cancer 2017, 140, 2321–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; van der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019, 79, 5626–5639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic Cancer-Induced Neutrophil Extracellular Traps: A Potential Contributor to Cancer-Associated Thrombosis. Int. J. Mol. Sci. 2017, 18, 487. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, T.; Fisher, J.; Bakochi, A.; Neumann, A.; Cardoso, J.F.P.; Karlsson, C.A.Q.; Pavan, C.; Lundgaard, I.; Nilson, B.; Reinstrup, P.; et al. Neutrophil Extracellular Traps in the Central Nervous System Hinder Bacterial Clearance during Pneumococcal Meningitis. Nat. Commun. 2019, 10, 1667. [Google Scholar] [CrossRef]
- De Buhr, N.; Reuner, F.; Neumann, A.; Stump-Guthier, C.; Tenenbaum, T.; Schroten, H.; Ishikawa, H.; Müller, K.; Beineke, A.; Hennig-Pauka, I.; et al. Neutrophil Extracellular Trap Formation in the Streptococcus Suis-Infected Cerebrospinal Fluid Compartment: NETs in Strep. Suis-Infected Cerebrospinal Fluid. Cell. Microbiol. 2017, 19, e12649. [Google Scholar] [CrossRef] [Green Version]
- Laridan, E.; Denorme, F.; Desender, L.; François, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Ischemic Stroke Thrombi: NETs in Stroke. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef]
- Novotny, J.; Oberdieck, P.; Titova, A.; Pelisek, J.; Chandraratne, S.; Nicol, P.; Hapfelmeier, A.; Joner, M.; Maegdefessel, L.; Poppert, H.; et al. Thrombus NET Content Is Associated with Clinical Outcome in Stroke and Myocardial Infarction. Neurology 2020, 94, e2346–e2360. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Bianca, V.D.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils Promote Alzheimer’s Disease–like Pathology and Cognitive Decline via LFA-1 Integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Lachowicz-Scroggins, M.E.; Dunican, E.M.; Charbit, A.R.; Raymond, W.; Looney, M.R.; Peters, M.C.; Gordon, E.D.; Woodruff, P.G.; Lefrançais, E.; Phillips, B.R.; et al. Extracellular DNA, Neutrophil Extracellular Traps, and Inflammasome Activation in Severe Asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Obermayer, A.; Stoiber, W.; Krautgartner, W.-D.; Klappacher, M.; Kofler, B.; Steinbacher, P.; Vitkov, L.; Grabcanovic-Musija, F.; Studnicka, M. New Aspects on the Structure of Neutrophil Extracellular Traps from Chronic Obstructive Pulmonary Disease and In Vitro Generation. PLoS ONE 2014, 9, e97784. [Google Scholar] [CrossRef]
- Moorthy, A.N.; Rai, P.; Jiao, H.; Wang, S.; Tan, K.B.; Qin, L.; Watanabe, H.; Zhang, Y.; Teluguakula, N.; Chow, V.T.K. Capsules of Virulent Pneumococcal Serotypes Enhance Formation of Neutrophil Extracellular Traps during in vivo Pathogenesis of Pneumonia. Oncotarget 2016, 7, 19327–19340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.-D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil Extracellular Trap (NET) Formation Characterises Stable and Exacerbated COPD and Correlates with Airflow Limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN Exacerbates Disease in Tuberculosis-Susceptible Mice by Inducing Neutrophil-Mediated Lung Inflammation and NETosis. Nat. Commun. 2020, 11, 5566. [Google Scholar] [CrossRef]
- Toussaint, M.; Jackson, D.J.; Swieboda, D.; Guedán, A.; Tsourouktsoglou, T.-D.; Ching, Y.M.; Radermecker, C.; Makrinioti, H.; Aniscenko, J.; Bartlett, N.W.; et al. Host DNA Released by NETosis Promotes Rhinovirus-Induced Type-2 Allergic Asthma Exacerbation. Nat. Med. 2017, 23, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnen, M.; Leschczyk, C.; Möller, S.; Batel, T.; Klinger, M.; Solbach, W.; Laskay, T. Immobilized Immune Complexes Induce Neutrophil Extracellular Trap Release by Human Neutrophil Granulocytes via FcγRIIIB and Mac-1. J. Immunol. 2014, 193, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.J.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA Traps Promote Thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil Extracellular Traps License Macrophages for Cytokine Production in Atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.-A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, C.H.; Toller-Kawahisa, J.E.; Fumagalli, M.J.; Colon, D.F.; Figueiredo, L.T.M.; Fonseca, B.A.L.D.; Franca, R.F.O.; Cunha, F.Q. Neutrophil Extracellular Traps Effectively Control Acute Chikungunya Virus Infection. Front. Immunol. 2020, 10, 3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiducci, E.; Lemberg, C.; Küng, N.; Schraner, E.; Theocharides, A.P.A.; LeibundGut-Landmann, S. Candida Albicans-Induced NETosis Is Independent of Peptidylarginine Deiminase 4. Front. Immunol. 2018, 9, 1573. [Google Scholar] [CrossRef]
- Silva, J.C.; Rodrigues, N.C.; Thompson-Souza, G.A.; Muniz, V.D.S.; Neves, J.S.; Figueiredo, R.T. Mac-1 Triggers Neutrophil DNA Extracellular Trap Formation to Aspergillus fumigatus Independently of PAD4 Histone Citrullination. J. Leukoc. Biol. 2020, 107, 69–83. [Google Scholar] [CrossRef]
- Sousa-Rocha, D.; Thomaz-Tobias, M.; Diniz, L.F.A.; Souza, P.S.S.; Pinge-Filho, P.; Toledo, K.A. Trypanosoma Cruzi and Its Soluble Antigens Induce NET Release by Stimulating Toll-Like Receptors. PLoS ONE 2015, 10, e0139569. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; de Souza Silva, L.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; De Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF Are Required for Neutrophil Extracellular Trap Release Triggered by Plasmodium-Infected Erythrocytes. PLoS Pathog. 2020, 16, e1008230. [Google Scholar] [CrossRef]
- Han, X.; Ding, S.; Jiang, H.; Liu, G. Roles of Macrophages in the Development and Treatment of Gut Inflammation. Front. Cell Dev. Biol. 2021, 9, 625423. [Google Scholar] [CrossRef]
- Haider, P.; Kral-Pointner, J.B.; Mayer, J.; Richter, M.; Kaun, C.; Brostjan, C.; Eilenberg, W.; Fischer, M.B.; Speidl, W.S.; Hengstenberg, C.; et al. Neutrophil Extracellular Trap Degradation by Differently Polarized Macrophage Subsets. Arter. Thromb. Vasc. Biol. 2020, 40, 2265–2278. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, L.; Zhang, G.; Zhang, B. Umbilical Cord-Matrix Stem Cells Induce the Functional Restoration of Vascular Endothelial Cells and Enhance Skin Wound Healing in Diabetic Mice via the Polarized Macrophages. Stem Cell Res. 2020, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wu, X.; Liao, C.; Liu, X.; Du, J.; Shi, H.; Wang, X.; Bai, X.; Peng, P.; Yu, L.; et al. Escherichia Coli and Candida Albicans Induced Macrophage Extracellular Trap-Like Structures with Limited Microbicidal Activity. PLoS ONE 2014, 9, e90042. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, A.; Pais, C.; Sampaio, P. Relevance of Macrophage Extracellular Traps in C. Albicans Killing. Front. Immunol. 2019, 10, 2767. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, S.; Horibata, S.; McElwee, J.L.; Dannenberg, A.J.; Coonrod, S.A. Identification of Macrophage Extracellular Trap-Like Structures in Mammary Gland Adipose Tissue: A Preliminary Study. Front. Immunol. 2013, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Caro, T.; Silva, L.M.R.; Ritter, C.; Taubert, A.; Hermosilla, C. Besnoitia Besnoiti Tachyzoites Induce Monocyte Extracellular Trap Formation. Parasitol. Res. 2014, 113, 4189–4197. [Google Scholar] [CrossRef]
- Pérez, D.; Muñoz, M.C.; Molina, J.M.; Muñoz-Caro, T.; Silva, L.M.R.; Taubert, A.; Hermosilla, C.; Ruiz, A. Eimeria Ninakohlyakimovae Induces NADPH Oxidase-Dependent Monocyte Extracellular Trap Formation and Upregulates IL-12 and TNF-α, IL-6 and CCL2 Gene Transcription. Vet. Parasitol. 2016, 227, 143–150. [Google Scholar] [CrossRef]
- Pertiwi, K.R.; de Boer, O.J.; Mackaaij, C.; Pabittei, D.R.; de Winter, R.J.; Li, X.; van der Wal, A.C. Extracellular Traps Derived from Macrophages, Mast Cells, Eosinophils and Neutrophils Are Generated in a Time-Dependent Manner during Atherothrombosis. J. Pathol. 2019, 247, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambrano, F.; Schulz, M.; Pilatz, A.; Wagenlehner, F.; Schuppe, H.-C.; Conejeros, I.; Uribe, P.; Taubert, A.; Sánchez, R.; Hermosilla, C. Increase of Leucocyte-Derived Extracellular Traps (ETs) in Semen Samples from Human Acute Epididymitis Patients—a Pilot Study. J. Assist. Reprod. Genet. 2020, 37, 2223–2231. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Tan, Y.; Liu, Y.; Li, J.; Deng, Q.; Yan, S.; Zhang, W.; Han, L.; Zhong, M. Hepcidin Gene Silencing Ameliorated Inflammation and Insulin Resistance in Adipose Tissue of Db/Db Mice via Inhibiting METs Formation. Mol. Immunol. 2021, 133, 110–121. [Google Scholar] [CrossRef]
- Wong, K.-W.; Jacobs, W.R. Mycobacterium Tuberculosis Exploits Human Interferon γ to Stimulate Macrophage Extracellular Trap Formation and Necrosis. J. Infect. Dis. 2013, 208, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haritha, V.H.; Seena, P.; Shaji, B.V.; Nithin, T.U.; Hazeena, V.N.; Anie, Y. Monocyte Clearance of Apoptotic Neutrophils Is Unhindered in the Presence of NETosis, but Proteins of NET Trigger ETosis in Monocytes. Immunol. Lett. 2019, 207, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rayner, B.S.; Jensen, M.; Hawkins, C.L. In Vitro Stimulation and Visualization of Extracellular Trap Release in Differentiated Human Monocyte-Derived Macrophages. J. Vis. Exp. 2019, 1, 60541. [Google Scholar] [CrossRef]
- Pertiwi, K.R.; de Boer, O.J.; Gabriels, P.A.M.; van der Wal, A.C. Etosis, Rather than Apoptosis or Cell Proliferation, Typifies Thrombus Progression—An Immunohistochemical Study of Coronary Aspirates. Int. J. Cardiol. Heart Vasc. 2020, 26, 100439. [Google Scholar] [CrossRef]
- Antunes, S.A.; Canziani, M.E.F. Hepcidin: An Important Iron Metabolism Regulator in Chronic Kidney Disease. J. Bras. Nefrol. 2016, 38, 351–355. [Google Scholar] [CrossRef]
- Josefs, T.; Barrett, T.J.; Brown, E.J.; Quezada, A.; Wu, X.; Voisin, M.; Amengual, J.; Fisher, E.A. Neutrophil Extracellular Traps Promote Macrophage Inflammation and Impair Atherosclerosis Resolution in Diabetic Mice. JCI Insight 2020, 5, e134796. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yu, X.; Liu, J.; Wang, Z.; Li, C.; Shi, J.; Sun, L.; Liu, Y.; Zhang, F.; Chen, H.; et al. Neutrophil Extracellular Traps Promote Aberrant Macrophages Activation in Behçet’s Disease. Front. Immunol. 2021, 11, 590622. [Google Scholar] [CrossRef]
- Eghbalzadeh, K.; Georgi, L.; Louis, T.; Zhao, H.; Keser, U.; Weber, C.; Mollenhauer, M.; Conforti, A.; Wahlers, T.; Paunel-Görgülü, A. Compromised Anti-Inflammatory Action of Neutrophil Extracellular Traps in PAD4-Deficient Mice Contributes to Aggravated Acute Inflammation After Myocardial Infarction. Front. Immunol. 2019, 10, 2313. [Google Scholar] [CrossRef]
- Oishi, S.; Tsukiji, N.; Otake, S.; Oishi, N.; Sasaki, T.; Shirai, T.; Yoshikawa, Y.; Takano, K.; Shinmori, H.; Inukai, T.; et al. Heme Activates Platelets and Exacerbates Rhabdomyolysis-Induced Acute Kidney Injury via CLEC-2 and GPVI/FcRγ. Blood Adv. 2021, 5, 2017–2026. [Google Scholar] [CrossRef]
- Tian, Y.; Russo, R.M.; Li, Y.; Karmakar, M.; Liu, B.; Puskarich, M.A.; Jones, A.E.; Stringer, K.A.; Standiford, T.J.; Alam, H.B. Serum Citrullinated Histone H3 Concentrations Differentiate Patients with Septic Verses Non-Septic Shock and Correlate with Disease Severity. Infection 2021, 49, 83–93. [Google Scholar] [CrossRef]
- Li, L.; Krilis, S. Mast-cell Growth and Differentiation. Allergy 1999, 54, 306–313. [Google Scholar] [CrossRef]
- Féger, F.; Varadaradjalou, S.; Gao, Z.; Abraham, S.N.; Arock, M. The Role of Mast Cells in Host Defense and Their Subversion by Bacterial Pathogens. Trends Immunol. 2002, 23, 151–158. [Google Scholar] [CrossRef]
- Kunder, C.A.; St John, A.L.; Abraham, S.N. Mast Cell Modulation of the Vascular and Lymphatic Endothelium. Blood 2011, 118, 5383–5393. [Google Scholar] [CrossRef]
- Dudeck, A.; Köberle, M.; Goldmann, O.; Meyer, N.; Dudeck, J.; Lemmens, S.; Rohde, M.; Roldán, N.G.; Dietze-Schwonberg, K.; Orinska, Z.; et al. Mast Cells as Protectors of Health. J. Allergy Clin. Immunol. 2019, 144, S4–S18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varricchi, G.; Rossi, F.W.; Galdiero, M.R.; Granata, F.; Criscuolo, G.; Spadaro, G.; de Paulis, A.; Marone, G. Physiological Roles of Mast Cells: Collegium Internationale Allergologicum Update 2019. Int. Arch. Allergy Immunol. 2019, 179, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Tsilioni, I.; Ren, H. Recent Advances in Our Understanding of Mast Cell Activation—or Should It Be Mast Cell Mediator Disorders? Expert Rev. Clin. Immunol. 2019, 15, 639–656. [Google Scholar] [CrossRef] [PubMed]
- John, A.L.S.; Abraham, S.N. Innate Immunity and Its Regulation by Mast Cells. J. Immunol. 2013, 190, 4458–4463. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast Cells as Sources of Cytokines, Chemokines, and Growth Factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef]
- Dawicki, W.; Marshall, J.S. New and Emerging Roles for Mast Cells in Host Defence. Curr. Opin. Immunol. 2007, 19, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Nija, R.J.; Sanju, S.; Sidharthan, N.; Mony, U. Extracellular Trap by Blood Cells: Clinical Implications. Tissue Eng. Regen. Med. 2020, 17, 141–153. [Google Scholar] [CrossRef]
- Di Nardo, A.; Vitiello, A.; Gallo, R.L. Cutting Edge: Mast Cell Antimicrobial Activity Is Mediated by Expression of Cathelicidin Antimicrobial Peptide. J. Immunol. 2003, 170, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Abel, J.; Goldmann, O.; Ziegler, C.; Höltje, C.; Smeltzer, M.S.; Cheung, A.L.; Bruhn, D.; Rohde, M.; Medina, E. Staphylococcus Aureus Evades the Extracellular Antimicrobial Activity of Mast Cells by Promoting Its Own Uptake. J. Innate Immun. 2011, 3, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.M.; Biggs, T.C.; Goldie, S.P.; Harries, P.G.; Walls, A.F.; Allan, R.N.; Pender, S.L.F.; Salib, R.J. Staphylococcus Aureus Internalization in Mast Cells in Nasal Polyps: Characterization of Interactions and Potential Mechanisms. J. Allergy Clin. Immunol. 2020, 145, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branitzki-Heinemann, K.; Okumura, C.Y.; Völlger, L.; Kawakami, Y.; Kawakami, T.; Naim, H.Y.; Nizet, V.; Von Köckritz-Blickwede, M. A Novel Role for the Transcription Factor HIF-1α in the Formation of Mast Cell Extracellular Traps. Biochem. J. 2012, 446, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rodriguez, K.M.; Bahri, R.; Sattentau, C.; Roberts, I.S.; Goenka, A.; Bulfone-Paus, S. Human Mast Cells Exhibit an Individualized Pattern of Antimicrobial Responses. Immun. Inflamm. Dis. 2020, 8, 198–210. [Google Scholar] [CrossRef]
- Campillo-Navarro, M.; Leyva-Paredes, K.; Donis-Maturano, L.; Rodríguez-López, G.M.; Soria-Castro, R.; García-Pérez, B.E.; Puebla-Osorio, N.; Ullrich, S.E.; Luna-Herrera, J.; Flores-Romo, L.; et al. Mycobacterium Tuberculosis Catalase Inhibits the Formation of Mast Cell Extracellular Traps. Front. Immunol. 2018, 9, 1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauth, X.; von Köckritz-Blickwede, M.; McNamara, C.W.; Myskowski, S.; Zinkernagel, A.S.; Beall, B.; Ghosh, P.; Gallo, R.L.; Nizet, V. M1 Protein Allows Group A Streptococcal Survival in Phagocyte Extracellular Traps through Cathelicidin Inhibition. J. Innate Immun. 2009, 1, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Kim, J.; Etesami, N.; Shimamoto, J.; Whalen, R.V.; Martin, G.; Okumura, C.Y.M. Group A Streptococcus Prevents Mast Cell Degranulation to Promote Extracellular Trap Formation. Front. Immunol. 2018, 9, 327. [Google Scholar] [CrossRef] [Green Version]
- Scheb-Wetzel, M.; Rohde, M.; Bravo, A.; Goldmann, O. New Insights into the Antimicrobial Effect of Mast Cells against Enterococcus Faecalis. Infect. Immun. 2014, 82, 4496–4507. [Google Scholar] [CrossRef] [Green Version]
- Lopes, J.P.; Stylianou, M.; Nilsson, G.; Urban, C.F. Opportunistic Pathogen Candida Albicans Elicits a Temporal Response in Primary Human Mast Cells. Sci. Rep. 2015, 5, 12287. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, N.; Ahuja, K.; Selvapandiyan, A.; Dey, R.; Nakhasi, H.; Puri, N. Role of Mast Cells in Clearance of Leishmania through Extracellular Trap Formation. Sci. Rep. 2017, 7, 13240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piliponsky, A.M.; Romani, L. The Contribution of Mast Cells to Bacterial and Fungal Infection Immunity. Immunol. Rev. 2018, 282, 188–197. [Google Scholar] [CrossRef]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast Cells and Neutrophils Release IL-17 through Extracellular Trap Formation in Psoriasis. J. Immunol. 2011, 187, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Campillo-Navarro, M.; Leyva-Paredes, K.; Donis-Maturano, L.; González-Jiménez, M.; Paredes-Vivas, Y.; Cerbulo-Vázquez, A.; Serafín-López, J.; García-Pérez, B.; Ullrich, S.E.; Flores-Romo, L.; et al. Listeria Monocytogenes Induces Mast Cell Extracellular Traps. Immunobiology 2017, 222, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Weller, P.F.; Spencer, L.A. Functions of Tissue-Resident Eosinophils. Nat. Rev. Immunol. 2017, 17, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Yoshikawa, M.; Asaka, D.; Okushi, T.; Matsuwaki, Y.; Otori, N.; Hama, T.; Moriyama, H. Mucosal Eosinophilia and Recurrence of Nasal Polyps—New Classification of Chronic Rhinosinusitis. Rhinology 2011, 49, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Neves, J.S. Eosinophils in Fungal Diseases: An Overview. J. Leukoc. Biol. 2018, 104, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Strandmark, J.; Rausch, S.; Hartmann, S. Eosinophils in Homeostasis and Their Contrasting Roles during Inflammation and Helminth Infections. Crit. Rev. Immunol. 2016, 36, 193–238. [Google Scholar] [CrossRef] [PubMed]
- Gigon, L.; Yousefi, S.; Karaulov, A.; Simon, H.-U. Mechanisms of Toxicity Mediated by Neutrophil and Eosinophil Granule Proteins. Allergol. Int. 2021, 70, 30–38. [Google Scholar] [CrossRef]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like Release of Mitochondrial DNA by Eosinophils Contributes to Antibacterial Defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Morshed, M.; Yousefi, S.; Stöckle, C.; Simon, H.-U.; Simon, D. Thymic Stromal Lymphopoietin Stimulates the Formation of Eosinophil Extracellular Traps. Allergy 2012, 67, 1127–1137. [Google Scholar] [CrossRef]
- Ueki, S.; Melo, R.C.N.; Ghiran, I.; Spencer, L.A.; Dvorak, A.M.; Weller, P.F. Eosinophil Extracellular DNA Trap Cell Death Mediates Lytic Release of Free Secretion-Competent Eosinophil Granules in Humans. Blood 2013, 121, 2074–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.; Radonjic-Hösli, S.; Straumann, A.; Yousefi, S.; Simon, H.-U. Active Eosinophilic Esophagitis Is Characterized by Epithelial Barrier Defects and Eosinophil Extracellular Trap Formation. Allergy 2015, 70, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Ueki, S.; Konno, Y.; Takeda, M.; Moritoki, Y.; Hirokawa, M.; Matsuwaki, Y.; Honda, K.; Ohta, N.; Yamamoto, S.; Takagi, Y.; et al. Eosinophil Extracellular Trap Cell Death-Derived DNA Traps: Their Presence in Secretions and Functional Attributes. J. Allergy Clin. Immunol. 2016, 137, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Le Pham, D.; Lee, D.-H.; Lee, S.-H.; Kim, S.-H.; Park, H.-S. Biological Function of Eosinophil Extracellular Traps in Patients with Severe Eosinophilic Asthma. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Muniz, V.S.; Silva, J.C.; Braga, Y.A.V.; Melo, R.C.N.; Ueki, S.; Takeda, M.; Hebisawa, A.; Asano, K.; Figueiredo, R.T.; Neves, J.S. Eosinophils Release Extracellular DNA Traps in Response to Aspergillus Fumigatus. J. Allergy Clin. Immunol. 2018, 141, 571–585.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, C.S.; Park, S.C.; Cho, H.-J.; Park, D.-J.; Yoon, J.-H.; Kim, C.-H. Eosinophil Extracellular Trap Formation Is Closely Associated with Disease Severity in Chronic Rhinosinusitis Regardless of Nasal Polyp Status. Sci. Rep. 2019, 9, 8061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrens, A.; Lenz, B.; Neumann, A.-L.; Giarrizzo, S.; Reichwald, J.J.; Frohberger, S.J.; Stamminger, W.; Buerfent, B.C.; Fercoq, F.; Martin, C.; et al. Microfilariae Trigger Eosinophil Extracellular DNA Traps in a Dectin-1-Dependent Manner. Cell Rep. 2021, 34, 108621. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, A.A.; Nuñez, N.K.; de Souza, R.G.; Moraes Vargas, M.H.; Silveira, J.S.; Antunes, G.L.; da Silva Durante, L.; Porto, B.N.; Marczak, E.S.; Jones, M.H.; et al. Recombinant Human Deoxyribonuclease Therapy Improves Airway Resistance and Reduces DNA Extracellular Traps in a Murine Acute Asthma Model. Exp. Lung Res. 2016, 42, 66–74. [Google Scholar] [CrossRef]
- Yousefi, S.; Sharma, S.K.; Stojkov, D.; Germic, N.; Aeschlimann, S.; Ge, M.Q.; Flayer, C.H.; Larson, E.D.; Redai, I.G.; Zhang, S.; et al. Oxidative Damage of SP-D Abolishes Control of Eosinophil Extracellular DNA Trap Formation. J. Leukoc. Biol. 2018, 104, 205–214. [Google Scholar] [CrossRef]
- Ohta, N.; Ueki, S.; Konno, Y.; Hirokawa, M.; Kubota, T.; Tomioka-Matsutani, S.; Suzuki, T.; Ishida, Y.; Kawano, T.; Miyasaka, T.; et al. ETosis-Derived DNA Trap Production in Middle Ear Effusion Is a Common Feature of Eosinophilic Otitis Media. Allergol. Int. 2018, 67, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, E.; Zhang, N.; Krysko, O.; Lan, F.; Holtappels, G.; De Ruyck, N.; Nauwynck, H.; Yousefi, S.; Simon, H.-U.; Bachert, C. Extracellular Eosinophilic Traps in Association with Staphylococcus Aureus at the Site of Epithelial Barrier Defects in Patients with Severe Airway Inflammation. J. Allergy Clin. Immunol. 2017, 139, 1849–1860.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omokawa, A.; Ueki, S.; Kikuchi, Y.; Takeda, M.; Asano, M.; Sato, K.; Sano, M.; Ito, H.; Hirokawa, M. Mucus Plugging in Allergic Bronchopulmonary Aspergillos is: Implication of the Eosinophil DNA Traps. Allergol. Int. 2018, 67, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.V.; Gropillo, I.; Detoni, M.A.A.; Thompson-Souza, G.A.; Muniz, V.S.; Vasconcelos, C.R.I.; Figueiredo, R.T.; Melo, R.C.N.; Neves, J.S. Structural and Signaling Events Driving Aspergillus Fumigatus-Induced Human Eosinophil Extracellular Trap Release. Front. Microbiol. 2021, 12, 633696. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.; Novotny, J.; Salbeck, D.; Zellner, K.R.; Nicolai, L.; Pekayvaz, K.; Kilani, B.; Stockhausen, S.; Bürgener, N.; Kupka, D.; et al. Eosinophil-Platelet Interactions Promote Atherosclerosis and Stabilize Thrombosis with Eosinophil Extracellular Traps. Blood 2019, 134, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.; Hoesli, S.; Roth, N.; Staedler, S.; Yousefi, S.; Simon, H.-U. Eosinophil Extracellular DNA Traps in Skin Diseases. J. Allergy Clin. Immunol. 2011, 127, 194–199. [Google Scholar] [CrossRef]
- Jones, V.A.; Patel, P.M.; Amber, K.T. Eosinophils in Bullous Pemphigoid. Panminerva Med. 2020. [Google Scholar] [CrossRef]
- Kerstan, A.; Simon, H.-U.; Yousefi, S.; Leverkus, M. Extensive Accumulation of Eosinophil Extracellular Traps in Bullous Delayed-Pressure Urticaria: A Pathophysiological Link? Br. J. Derm. 2012, 166, 1151–1152. [Google Scholar] [CrossRef] [PubMed]
- Rocha Arrieta, Y.C.; Rojas, M.; Vasquez, G.; Lopez, J. The Lymphocytes Stimulation Induced DNA Release, a Phenomenon Similar to NETosis. Scand. J. Immunol. 2017, 86, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Costanza, M.; Poliani, P.L.; Portararo, P.; Cappetti, B.; Musio, S.; Pagani, F.; Steinman, L.; Colombo, M.P.; Pedotti, R.; Sangaletti, S. DNA Threads Released by Activated CD4(+) T Lymphocytes Provide Autocrine Costimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 8985–8994. [Google Scholar] [CrossRef] [Green Version]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.-U.; et al. NADPH Oxidase–Independent Formation of Extracellular DNA Traps by Basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorn, C.; Janko, C.; Latzko, M.; Chaurio, R.; Schett, G.; Herrmann, M. Monosodium Urate Crystals Induce Extracellular DNA Traps in Neutrophils, Eosinophils, and Basophils but Not in Mononuclear Cells. Front. Immunol. 2012, 3, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Morshed, M.; Amini, P.; Stojkov, D.; Simon, D.; von Gunten, S.; Kaufmann, T.; Simon, H.-U. Basophils Exhibit Antibacterial Activity through Extracellular Trap Formation. Allergy 2015, 70, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Loures, F.V.; Röhm, M.; Lee, C.K.; Santos, E.; Wang, J.P.; Specht, C.A.; Calich, V.L.G.; Urban, C.F.; Levitz, S.M. Recognition of Aspergillus Fumigatus Hyphae by Human Plasmacytoid Dendritic Cells Is Mediated by Dectin-2 and Results in Formation of Extracellular Traps. PLoS Pathog. 2015, 11, e1004643. [Google Scholar] [CrossRef]
- Karasuyama, H.; Miyake, K.; Yoshikawa, S.; Yamanishi, Y. Multifaceted Roles of Basophils in Health and Disease. J. Allergy Clin. Immunol. 2018, 142, 370–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]



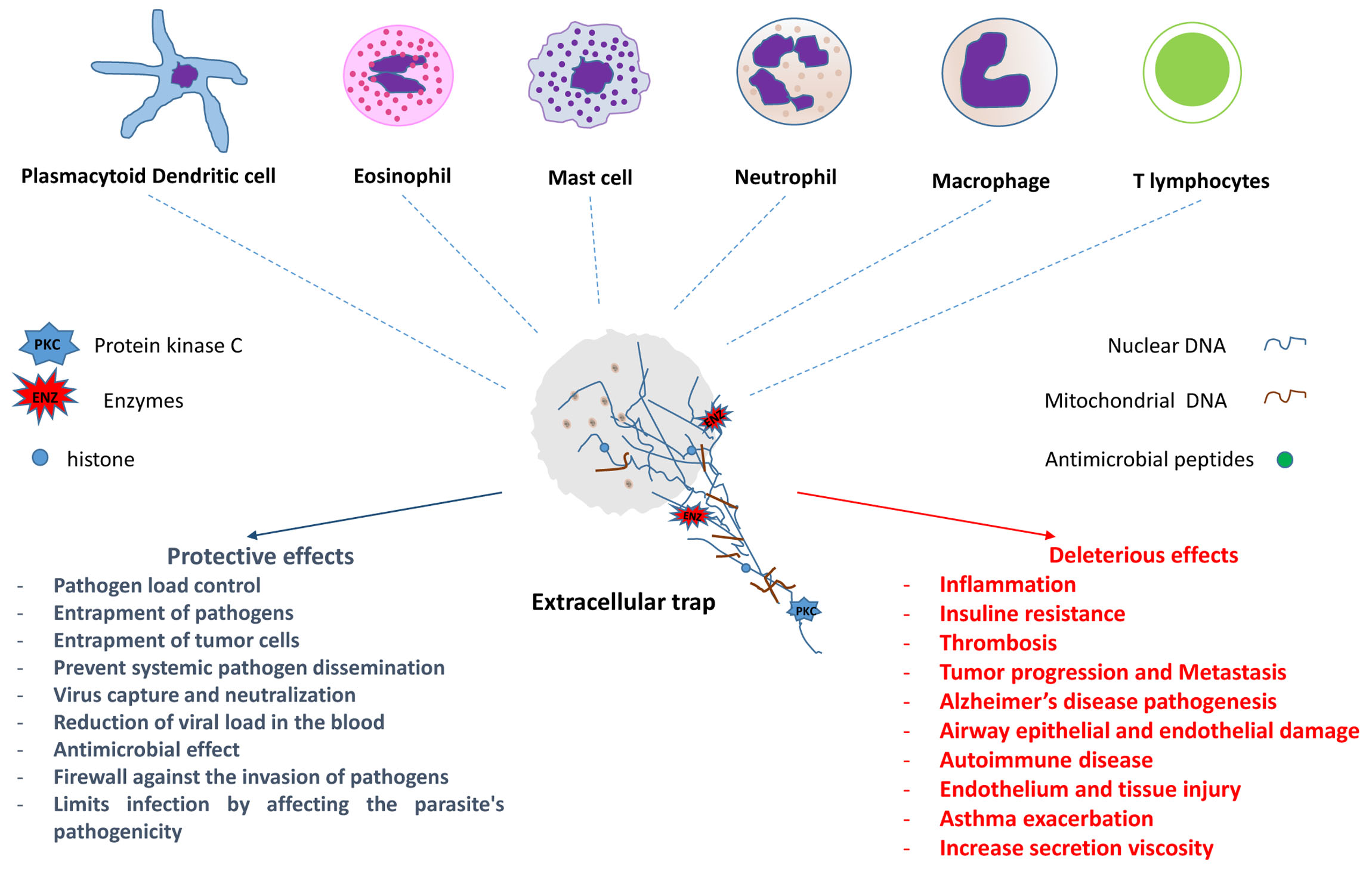{kind=link}
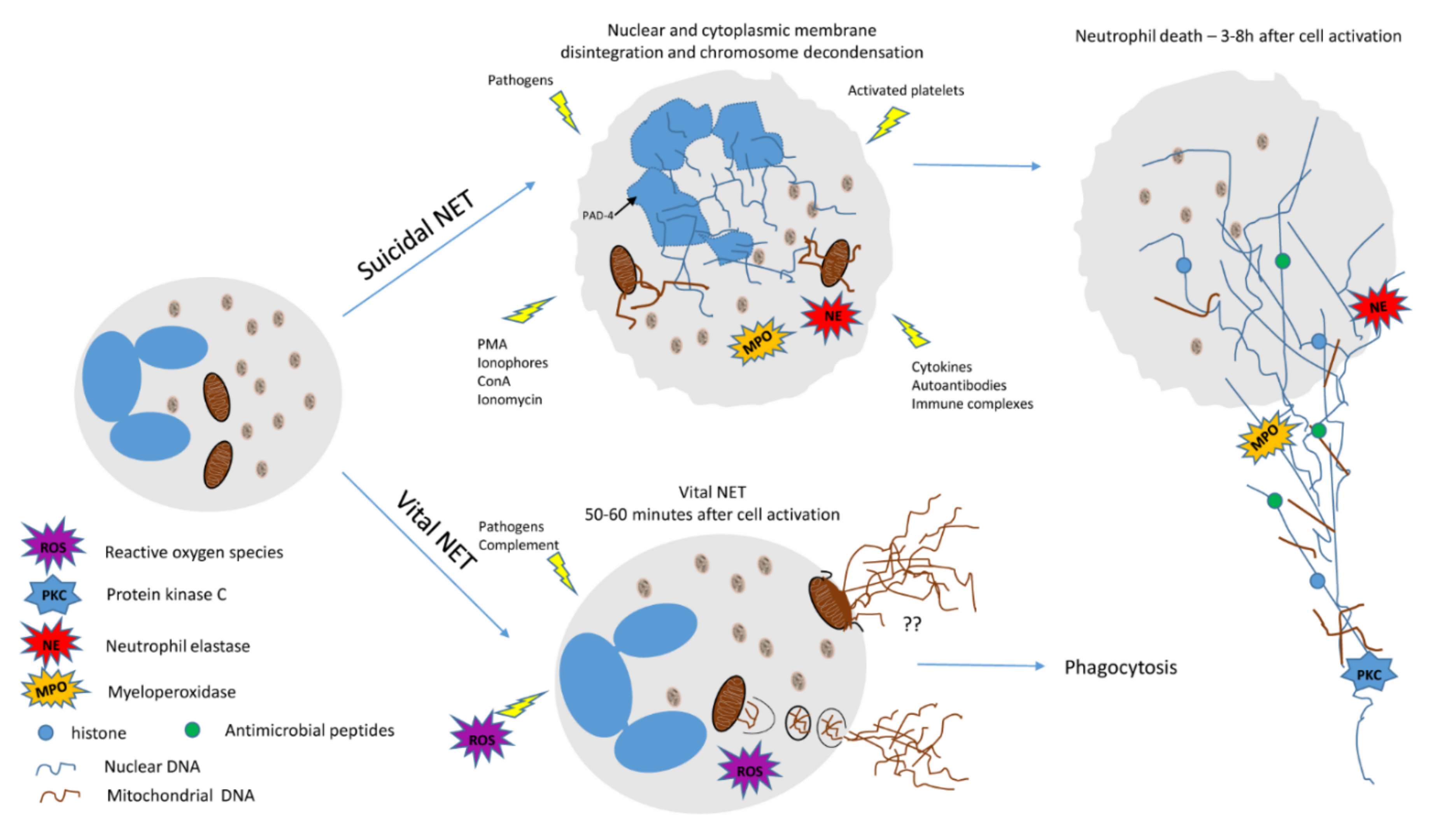{kind=link}
{kind=link}
| Cell | Mechanism of ETs Formation | Stimulus/Models | Biological Effect Protective Deleterious | |
|---|---|---|---|---|
| Neutrophil in Cancer | Suicidal (ROS-dependent) [91,107,108,109,110] Early/rapid ROS-independent (but may alternatively be dependent on autophagy) [111] Mitochondrial NETs [61] | In vivo Murine models of: breast cancer [91], lung carcinoma [107], metastatic colorectal cancer [109,110], lung carcinoma [108] Ex vivo Serum samples of patients with metastatic colorectal [109,110] and human tissue samples of breast cancer [91] In vitro Cancer cells [91], pancreatic cancer cells [111], anaplastic thyroid cancer cells [61] | Entrapment of tumor cells [107] | Association with an aggressive subtype of breast cancer [91] Tumor progression [61,110] Metastasis [91,107,108,109,110] Reduction in disease-free survival [109] Cancer-associated thrombosis [111] |
| Neutrophil in Central Nervous System Diseases | ROS-dependent [112] Nuclear DNA [113,114,115,116] | In vivo Murine model of Alzheimer’s disease, meningitis and [112,116] Piglet model of S. suis meningitis [113] In vitro Thrombi from patients with acute ischemic stroke [114,115]; paraffin sections of human cortex from Alzheimer’s disease brains [116] CSF of patients with S. pneumoniae meningitis [112] Modified human BCSFB model [113] | Entrapment of streptococci [113] | Alzheimer’s disease pathogenesis [116] Impairment of pneumococci clearance in meningitis [112] Poorer clinical outcomes and inflammation aggravation in patients with acute ischemic stroke [115]; Important constituents of cerebral thrombi [114] |
| Neutrophil in Pulmonary Diseases | Suicidal, ROS-dependent [117,118] ROS-dependent [119] Nuclear DNA [120,121,122] | In vivo Murine and human model of rhinovirus-induced allergic asthma exacerbation [122], murine model of S. pneumoniae induced pneumonia [119], and PTB [121] Ex vivo Human lung samples [121] In vitro Sputum samples of asthma patients/human airway epithelial cells [117] Sputum samples of COPD patients [118,120] | Asthma severity and exacerbation [117,122] Airway epithelial and endothelial damage [117] Severity of S. pneumoniae induced pneumonia [119] COPD severity and airway flow limitation [118,120] PTB pathogenesis and severity [121] | |
| Neutrophil in Autoimmune Diseases | ROS-dependent [123] Mitochondrial NETs (mtDNA, mtROS) [25] Not described [15] | In vitro Immune complexes (Anti-LL-37, anti-HNP, PR3 and MPO, ANCAs) [123] Healthy and lupus neutrophils (PMA and immune complexes) [25] Healthy and rheumatoid arthritis neutrophils (PMA and A23187) [15] | Autoimmune diseases (systemic lupus erythematosus, psoriasis, vasculitis, rheumatoid arthritis) [15,25,123,124] | |
| Neutrophil in Thrombosis/Cardiovascular Disorders | Nuclear DNA [125] ROS-dependent [126] | In vitro Blood neutrophils and platelets [125] In vivo Deep vein thrombosis model (Baboons) [125] In vivo Murine model (cholesterol crystals) [126] | Thrombosis [125] Atherosclerosis [126] | |
| Neutrophil and Virus | ROS-dependent [127,128] Suicidal, ROS-dependent [92] PAD-4 dependent [94] Suicidal, presence of Cit-H3 and MPO-DNA complexes [94,95,96,97,98,99] | In vivo Murine model of influenza A virus H1N1pneumonia [127] and Chikungunya virus infection [128] In vitro Neutrophils + influenza virus–primed epithelial cells [127] Serum samples and/or nasal swab specimens from COVID-19 patients [92,93,94,95,96,97,98,99] Neutrophils + SARS-CoV-2 [92,94] Neutrophils + Chikungunya virus [128] Ex vivo BALF and lung autopsies from COVID-19 patients [94,95,96] | Virus capture, Neutralization and reduction of viral load in the blood. [128] | Lung injury [127] Thrombosis formation in COVID-19 [92,96,99] COVID-19 Pneumonia [97] COVID-19 severity and vascular damage [94,95,98,99] |
| Neutrophil and Fungi | Suicidal, ROS-dependent [66,129,130] Vital NETs, ROS-independent [65] Not described [30] | In vivo Murine model of A. fumigatus [66] Murine model of C. albicans infection [129] In vitro A fumigatus conidia [130] C. albicans (β-glucan) [65] Ex vivo Active sporotrichosis lesion [30] | Entrapment of conidia, the only fungistatic effect [66,130] Capture and kill C. albicans yeast and hyphal forms [65,129] Antimicrobial effect [30] | |
| Neutrophil and Protozoa | Early/rapid, ROS-independent, and late ROS-dependent [68] Suicidal, ROS-dependent [64] ROS-dependent [28,73,131] ROS-independent [132] Not described [27,29,67] | In vivo Murine model of T. cruzi [131] Murine model of Malaria with P. berghei [132] and P. chabaudi [73] Murine model of T. gondii [28] Ex vivo ATL active cutaneous lesions [29] In vitro Leishmania spp.—amastigotes, promastigote/lipophosphoglycan [64,67,68] T. cruzi [131] Blood samples from patients infected with P. falciparum [73,132] | Containment of promastigotes at the inoculation site and Leishmania killing [64,68] Limits infection by affecting the parasite’s pathogenicity [131] Antimicrobial effect [29,73,132] Interferes with the parasite’s ability to invade cells [28] | Activation of emergency granulopoiesis via GM-CSF production, and induction of the endothelial cytoadhesion receptor ICAM-1 [73] Stimulus of ANA production, which may lead to autoimmunity [27] |
| Cell | Mechanism of ETs Formation | Stimulus/Models | Biological Effect | |
|---|---|---|---|---|
| Protective | Deleterious | |||
| Monocytes/Macrophages and Fungi | Not described [137] ROS and NADPH oxidase-independent manner, mtDNA only or mtDNA and nuclear DNA [136] | In vitro C. albicans [136,137] | C albicans load control in vitro [137] Entrapment of C. albicans [136,137] | |
| Monocytes/Macrophages and Bacteria | mtDNA only or mtDNA and nuclear DNA, ROS, and NADPH oxidase-independent manner [136] Not described [144] Elastase activity and M. tuberculosis ESX-1 [144] | In vitro E. coli [136,142] M tuberculosis [144] IFN-γ [144] Ex vivo U. urealyticum and C. trachomatis [142] | E. coli load control in vitro [136] Entrapment of E. coli and M. tuberculosis [136,142,144] | |
| Monocytes/Macrophages and Protozoa | MPO, ROS, and NADPH oxidase-dependent manner [139,140] | In vitro E. ninakohlyakimovae [140] B. besnoiti/E. bovis [139] | Entrapment of E. ninakohlyakimova, B besnoiti/E bovis [139,140] | |
| Monocytes/Macrophages in Diabetes and Obesity | PAD2/PAD4 mediated histone hypercitrulination [138] Not described [143] | In vitro TNF [138] Not described [143] | Induction of inflammation and insulin resistance [143] Acceleration of inflammation associated with obesity [138] | |
| Monocytes/Macrophages in Thrombosis | Not described [141,147] | Not described [141,147] | Arteriosclerotic plaques and coronary thrombosis formation [141,147] Thrombus instability [147] | |
| Cell | Mechanism of ETs Formation | Stimulus/Models | Biological Effect | |
|---|---|---|---|---|
| Protective | Deleterious | |||
| Mast cell and Bacteria | ROS-dependent [33,167,169,177] Suicidal MCETs [33,166] Not described, probably suicidal because DNA released was linked to dead cell staining or nuclear changes were observed [165,167,177] Not described [33,169,170,171,172] Suicidal and vital MCETs, ROS-independent [168] | In vitro HMC1 and BMMC lines + S. pyogenes/S. aureus/P. aeruginosa [33] HMC1 + GAS/Purified M1 GAS protein/L. lactis [170] HMC1 and BMMC lines + S.aureus [165,167] HMC-1 and BMMC lines + E. faecalis [172] HMC1 line + L. monocytogenes [177] HMC-1 and BMMC lines + Mtb (viable and HK-Mtb)/S.aureus [169] HMC-1 and BMMC lines + GAS/L.lactis/S.aureus [171] HMC-1 + S.aureus [166] HMC-1 + L.monocytogenes/E. coli/S.aureus/S. pneumoniae [168] | Antimicrobial effect [33,165,167,168,171,172,177] | M1 GAS protein contributes to GAS survival—invasive forms of infection [170] Mtb inhibit MCET formation—bacteria survival [169] Capture, phagocytosis, maintenance of infection [166] |
| Mast cell and Fungi | Not described, probably suicidal, but dead MC numbers were higher than MCETs observed [173] | In vitro HMC1 + C. albicans [173] | Physical restraint only [173] | |
| Mast cell and Protozoa | Suicidal MCETs ROS-dependent [174] | In vitro RBL MC line + L. donovani/L. tropica [174] | Antimicrobial effect [174] | |
| Mast cell and Psoriasis | Not described, probably suicidal because it was observed that MCs were not intact in lesions [176] | Ex vivo MCs from psoriasis lesions [176] | IL-17 release, leading to pathogenic effect [176] | |
| Mast cell And Atherothrombosis | Not described [141] | Ex vivo MCs from coronary plaques and thrombus [141] | Thrombus progression and maturation [141] | |
| Cell | Mechanism of ETs Formation | Stimulus/Models | Biological Effect | |
|---|---|---|---|---|
| Protective | Deleterious | |||
| Eosinophil in Intestinal (Colon) Diseases | Vital (mtDNA) ROS-dependent [183] | Ex vivo Colon Biopsies from Crohn’s disease, schistosomiasis, and intestinal spirochetosis patients | Entrapment of bacteria [183] | |
| Eosinophil In vitro (Human PBMC) | Vital (mtDNA) ROS-dependent [183] NADPH oxidase-dependent [184,188] Suicidal (Nuclear DNA) dependent of histone citrullination, CD11b, and the Syk tyrosine kinase pathway [185,187,189] Suicidal-independent of PAD4 histone citrullination and depends on the Src family, Akt, Ca, and p38 MAPK signaling pathways [197] | LPS, C5a, cotaxin/CCL11 [183] Opsonized E. coli [183] A. fumigatus [189,197] Thymic stromal lymphopoietin [184] Immobilized immunoglobulins (IgG, IgA), cytokines with PAF, Ca ionophore, or PMA [185,187] IL-5 and LPS [188] | Bactericidal activity [183] Entrapment of fungi [197] | Airway inflammation and obstruction in Asthma [188] |
| Eosinophils in Eosinophilic Diseases | Suicidal (Nuclear DNA) [187,194] Not described [186,190,195,202] | Ex vivo Secretions and tissue slides ECRS patients [187,190,195] Secretions from EOM patients Tissue slides [187,194] Biopsies from EOE patients [186] Skin biopsy tissues of 25 different eosinophilic skin diseases [202] | Firewall against the invasion of pathogens [186,195] | Increase in secretion viscosity [187,194] Inflammation [202] |
| Eosinophils in Allergic Bronchopulmonary Diseases | Suicidal (Nuclear DNA) [189,196] Dependent of histone citrullination, CD11b, and the Syk tyrosine kinase pathway [189] Not described [193] | Ex vivo BALF [196] Bronchial mucus plugs [189] In vivo Murine animal model of Asthma [193] | Increase in secretion viscosity [189,196] Asthma exacerbation [193] | |
| Eosinophils in Atherothrombosis | Suicidal (Nuclear DNA) [141,198] | In vivo Murine model [198] Ex vivo Human autopsy [141] | Thrombus formation [141,198] | |
| Cell | Mechanism of ETs Formation | Stimulus/Models | Biological Effect | |
|---|---|---|---|---|
| Protective | Deleterious | |||
| B lymphocytes | Not described, probably suicidal, since membrane damage is described [202] Vital [14] | In vitro PMA, ionomycin,, anti-IgM,, LPS, SLE serum [202] CPG motifs [14] | Probably autoimmune diseases, SLE, cryoglobulemic vasculitis, and Sjögren syndrome [202] Autoimmune diseases [14] | |
| CD4 T lymphocytes | In vitro antiCD3/antiCD28 [203] antiCD4/antiCD28 [32] In vivo Experimental model of encephalomyelitis [203] | Autoimmune diseases [203] American Tegumentary Leishmaniasis [32] | ||
| CD8 T lymphocytes | Suicidal [32] | In vitro antiCD3/antiCD28 [32] Ex vivo American Tegumentary Leishmaniasis lesions [32] | American Tegumentary Leishmaniasis [32] | |
| Cell | Mechanism of ETs Formation | Stimulus/Models | Biological Effect | |
|---|---|---|---|---|
| Protective | Deleterious | |||
| Basophils | Vital (mtDNA), NADPH oxidase independent [204] Not described [205,206] | In vitro (human blood) Monosodium urate [205] Staphylococcus aureus [206] In vitro (human blood and murine Hoxb8-immortalized myeloid progenitors derived basophils) IL-3 priming and subsequent activation of the C5a receptor or FcεRI [204] | Bactericidal activity [206] | |
| Plasmacytoid dendritic cells | Suicidal (Nuclear DNA) Citrullinated histone H3 Dectin-2-dependent [207] | In vitro (Human PBMC) Aspergillus fumigatus [207] | Antifungal activity [207] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conceição-Silva, F.; Reis, C.S.M.; De Luca, P.M.; Leite-Silva, J.; Santiago, M.A.; Morrot, A.; Morgado, F.N. The Immune System Throws Its Traps: Cells and Their Extracellular Traps in Disease and Protection. Cells 2021, 10, 1891. https://doi.org/10.3390/cells10081891
Conceição-Silva F, Reis CSM, De Luca PM, Leite-Silva J, Santiago MA, Morrot A, Morgado FN. The Immune System Throws Its Traps: Cells and Their Extracellular Traps in Disease and Protection. Cells. 2021; 10(8):1891. https://doi.org/10.3390/cells10081891
Chicago/Turabian StyleConceição-Silva, Fátima, Clarissa S. M. Reis, Paula Mello De Luca, Jessica Leite-Silva, Marta A. Santiago, Alexandre Morrot, and Fernanda N. Morgado. 2021. "The Immune System Throws Its Traps: Cells and Their Extracellular Traps in Disease and Protection" Cells 10, no. 8: 1891. https://doi.org/10.3390/cells10081891
APA StyleConceição-Silva, F., Reis, C. S. M., De Luca, P. M., Leite-Silva, J., Santiago, M. A., Morrot, A., & Morgado, F. N. (2021). The Immune System Throws Its Traps: Cells and Their Extracellular Traps in Disease and Protection. Cells, 10(8), 1891. https://doi.org/10.3390/cells10081891