
Drosophila Models Rediscovered with Super-Resolution Microscopy


Abstract
:1. Introduction
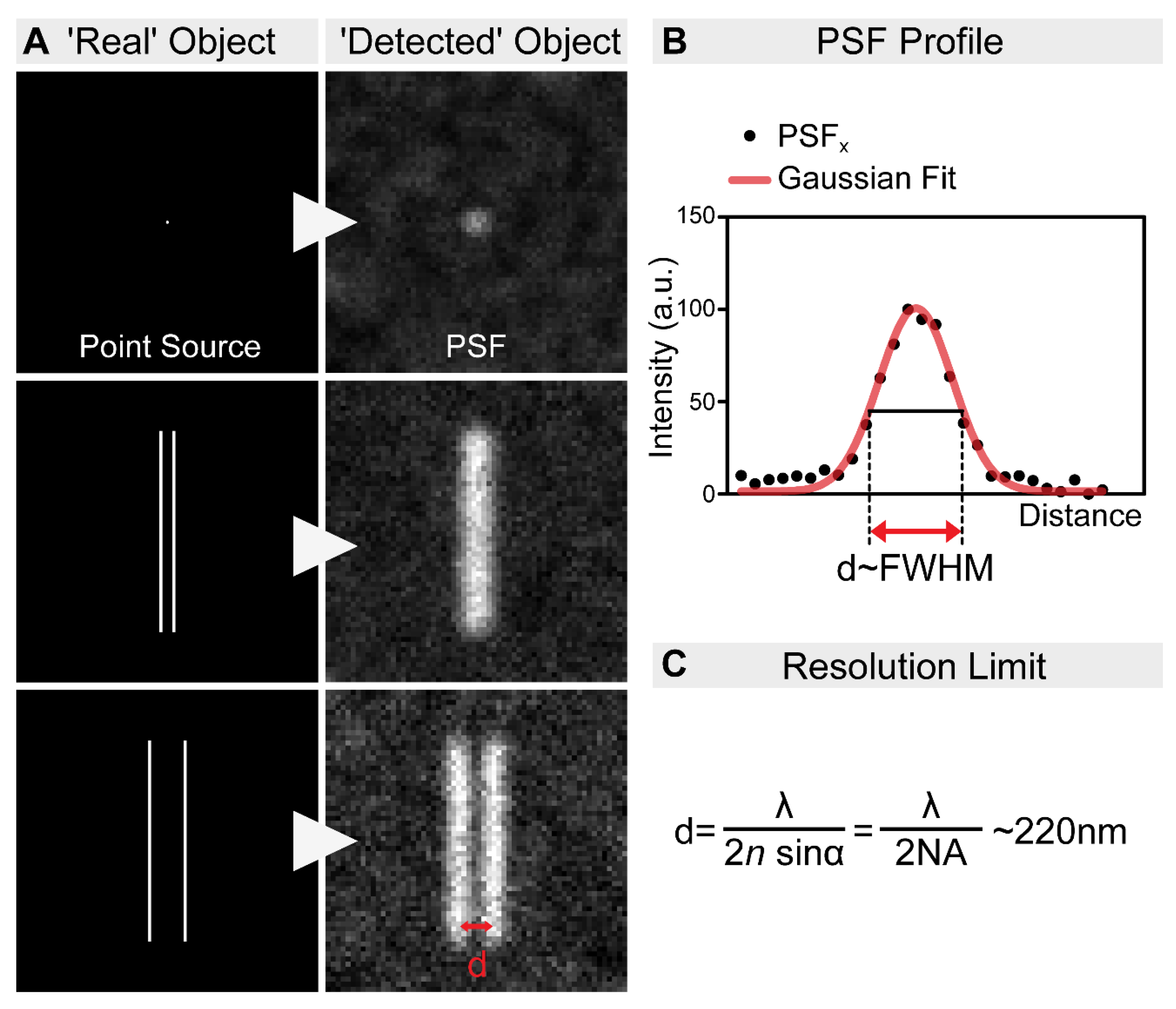
2. Overview of the Nanoscopic Methods
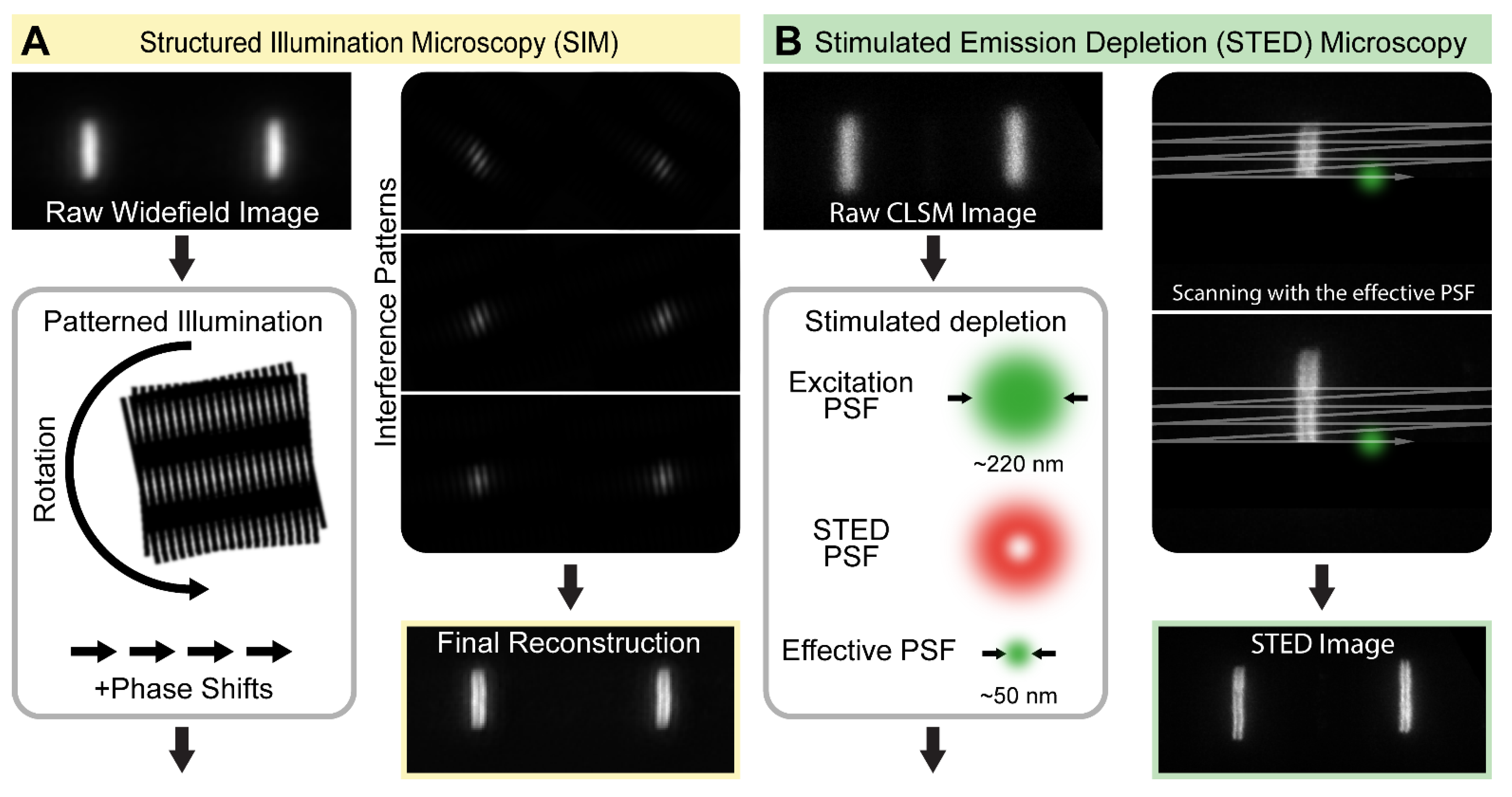
2.1. Structured Illumination Microscopy—SIM
2.2. Stimulated Emission Depletion—STED
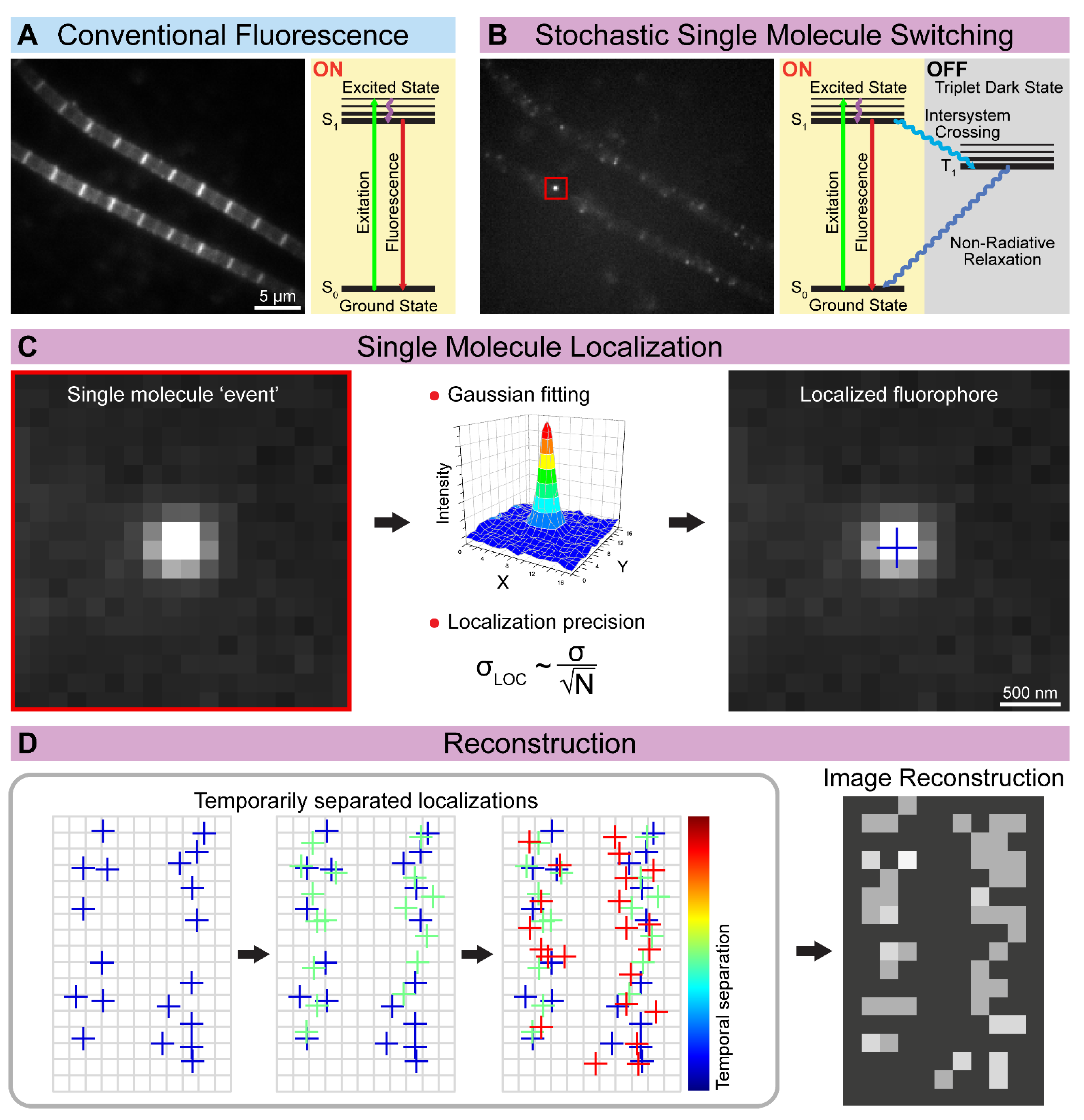
2.3. SMLM Methods—PALM, STORM, dSTORM, GSD, PAINT, MINFLUX
2.4. Expansion Microscopy
3. Biological Insights Offered by Nanoscopy
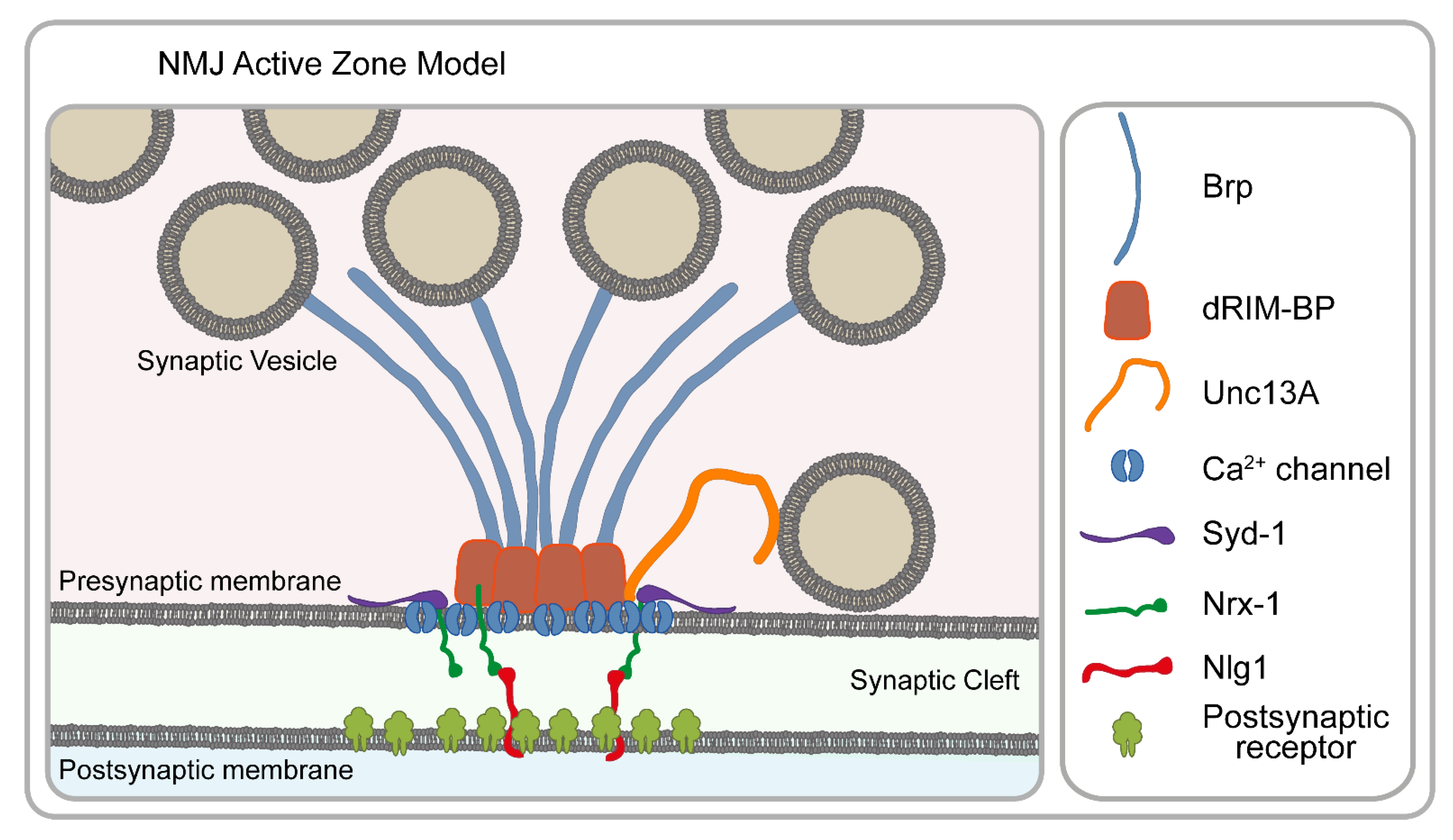
4. Chemical Synapses
5. Membrane-Associated Periodic Actin Skeleton
6. Nanoscopic Reconstruction of Protein Complexes with Structural Symmetry
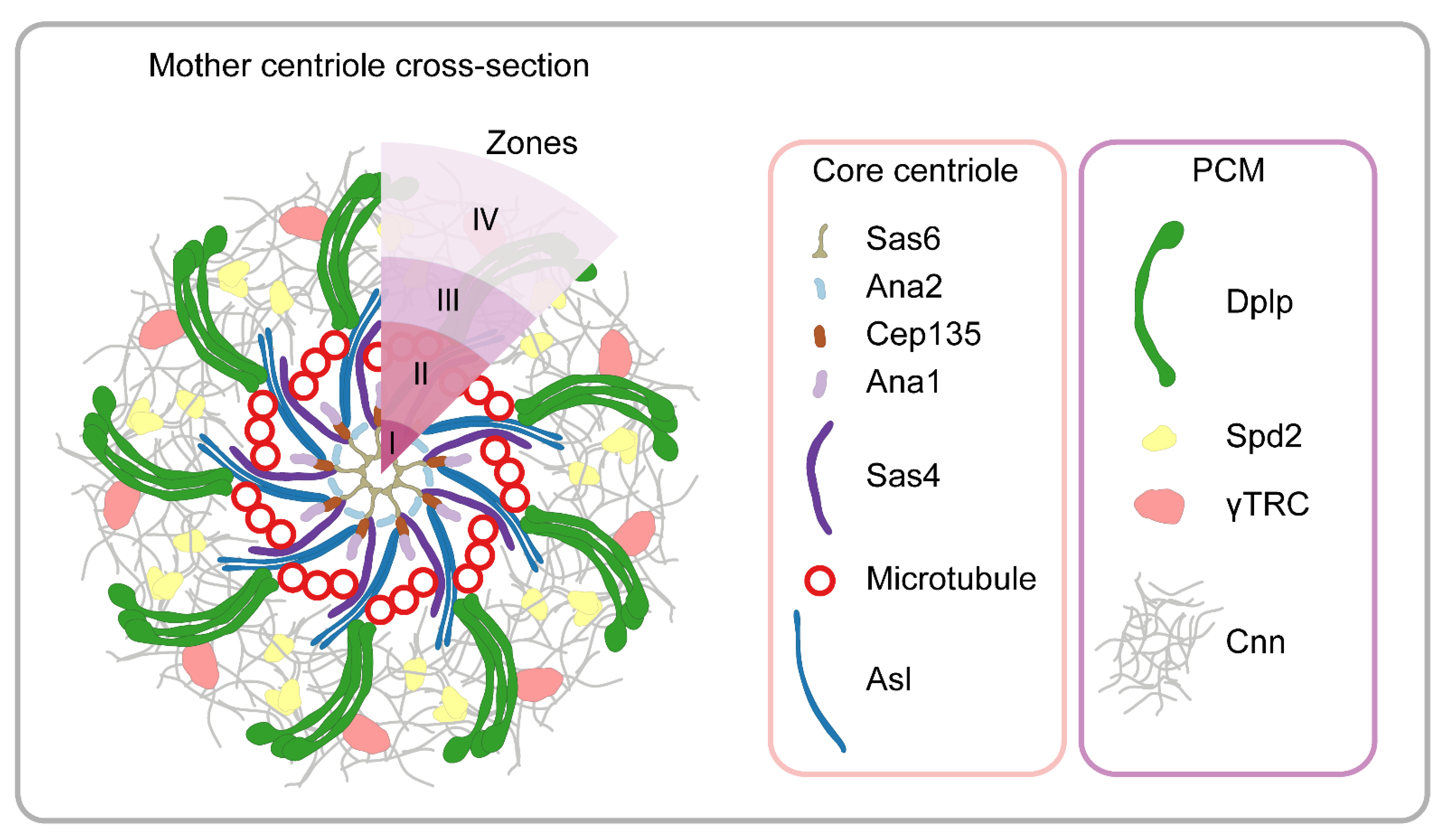
7. Centrosome
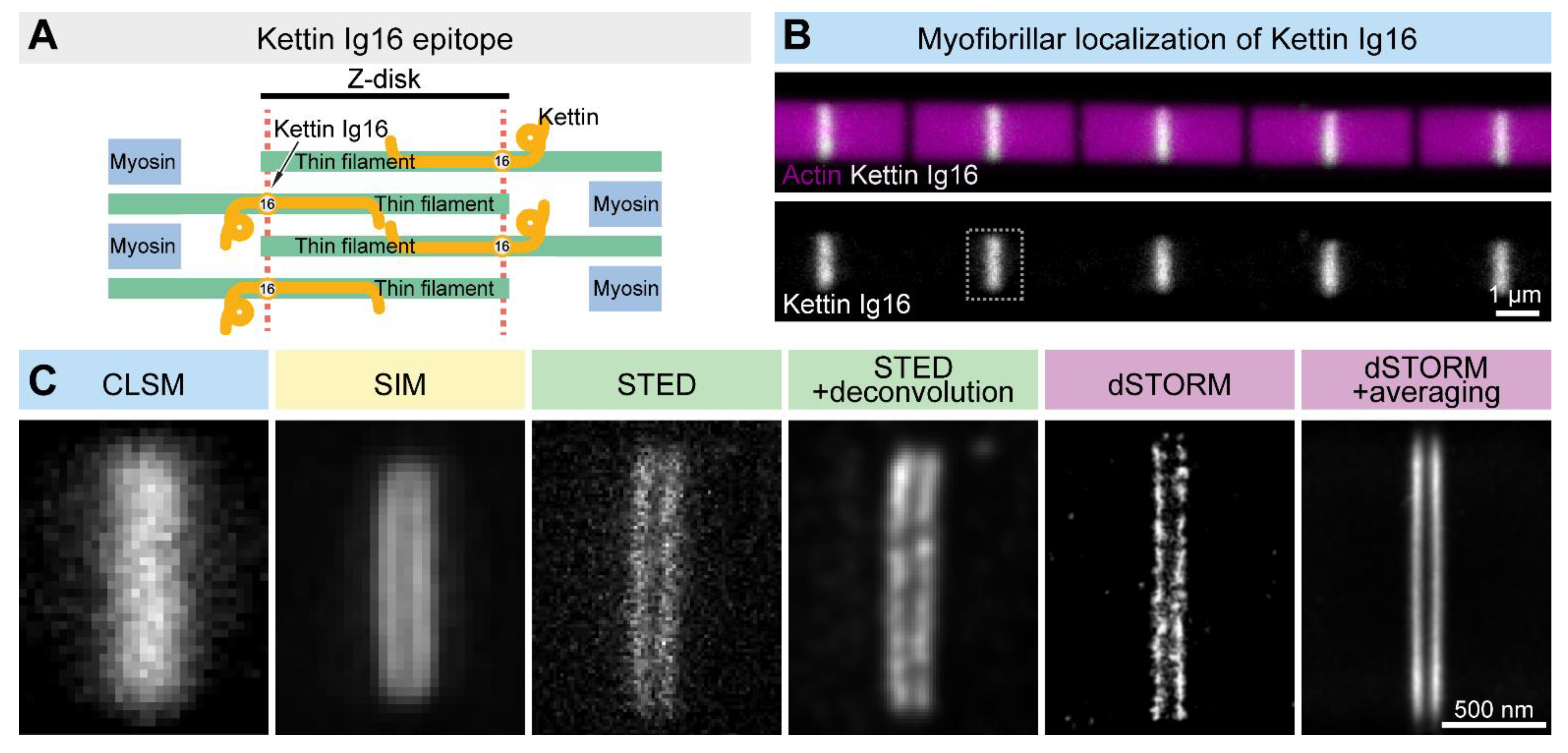
8. Sarcomeric Assemblies
9. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ana1 | Anastral spindle 1 |
| Ana2 | Anastral spindle 2 |
| Arp2/3 | Actin-related protein 2/3 |
| Asl | Asterless |
| AZ | Active zone |
| Brp | Bruchpilot |
| Cep97 | Centrosomal protein 97 kDa |
| Cep135 | Centrosomal protein 135 kDa |
| CLSM | Confocal laser scanning microscopy |
| Cnn | Centrosomin |
| CP110 | Centriolar coiled coil protein 110 kDa |
| DAAM | Disheveled Associated Activator of Morphogenesis |
| Dplp | Drosophila pericentrin-like protein |
| dRIM-BP | Drosophila Rab3-Interacting Molecule-Binding Protein |
| dSTORM | Direct stochastic optical reconstruction microscopy |
| EM | Electron microscopy |
| ExM | Expansion microscopy |
| F-actin | Filamentous actin |
| FHOS | Formin homology 2 domain containing |
| FWHM | Full width at half maximum |
| G-actin | Globular actin |
| gp210 | Glycoprotein 210 |
| GSD | Ground state depletion |
| γTRC | γ-tubulin ring complex |
| Hts | Hu-li tai shao |
| IFM | Indirect flight muscle |
| MINFLUX | Minimal photon flux |
| MPS | Membrane-associated periodic skeleton |
| MT | Microtubule |
| Nlg-1 | Neuroligin-1 |
| NMJ | Neuromuscular junction |
| NPC | Nuclear pore complex |
| Nrx-1 | Neurexin-1 |
| Nup | Nucleoporin |
| PAINT | Point Accumulation for Imaging in Nanoscale Topography |
| PALM | Photoactivated localization microscopy |
| PA NL-SIM | Patterned activation nonlinear-structured illumination microscopy |
| PCM | Pericentriolar material |
| PSF | Point spread function |
| SALS | Sarcomere length short |
| Sas4 | Spindle assembly abnormal 4 |
| Sas6 | Spindle assembly abnormal 6 |
| SIM | Structured illumination microscopy |
| SMLM | Single-molecule localization microscopy |
| Spd2 | Spindle defective 2 |
| SPIM | Single-plane illumination microscopy |
| Spn | Spinophilin |
| SRM | Super-resolution microscopy |
| STED | Stimulated emission depletion |
| STORM | Stochastic optical reconstruction microscopy |
| SV | Synaptic vesicle |
| Syd-1 | Synapse Defective 1 |
| TIRF | Total internal reflection fluorescence |
| Tmod | Tropomodulin |
| Unc13A | Uncoordinated 13A |
| Unc13B | Uncoordinated 13B |
References
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198 Pt 2, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.G.; Shao, L.; Carlton, P.M.; Wang, C.J.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., 3rd; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [Green Version]
- Markwirth, A.; Lachetta, M.; Monkemoller, V.; Heintzmann, R.; Hubner, W.; Huser, T.; Muller, M. Video-rate multi-color structured illumination microscopy with simultaneous real-time reconstruction. Nat. Commun. 2019, 10, 4315. [Google Scholar] [CrossRef]
- Rego, E.H.; Shao, L.; Macklin, J.J.; Winoto, L.; Johansson, G.A.; Kamps-Hughes, N.; Davidson, M.W.; Gustafsson, M.G. Nonlinear structured-illumination microscopy with a photoswitchable protein reveals cellular structures at 50-nm resolution. Proc. Natl. Acad. Sci. USA 2012, 109, E135–E143. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Shao, L.; Chen, B.-C.; Zhang, X.; Zhang, M.; Moses, B.; Milkie, D.E.; Beach, J.R.; Hammer, J.A., 3rd; Pasham, M.; et al. ADVANCED IMAGING. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 2015, 349, aab3500. [Google Scholar] [CrossRef] [Green Version]
- Mennella, V.; Keszthelyi, B.; McDonald, K.L.; Chhun, B.; Kan, F.; Rogers, G.C.; Huang, B.; Agard, D.A. Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat. Cell Biol. 2012, 14, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Glover, D.M. Structured illumination of the interface between centriole and peri-centriolar material. Open Biol. 2012, 2, 120104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, I.; Schöck, F. The nebulin repeat protein Lasp regulates I-band architecture and filament spacing in myofibrils. J. Cell Biol. 2014, 206, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shroff, H. Faster, sharper, and deeper: Structured illumination microscopy for biological imaging. Nat. Methods 2018, 15, 1011–1019. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [Green Version]
- Trifonov, A.S.; Jaskula, J.-C.; Teulon, C.; Glenn, D.R.; Bar-Gill, N.; Walsworth, R.L. Chapter 5—Limits to Resolution of CW STED Microscopy. In Advances In Atomic, Molecular, and Optical Physics; Arimondo, E., Berman, P.R., Lin, C.C., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 62, pp. 279–302. [Google Scholar]
- Wäldchen, S.; Lehmann, J.; Klein, T.; van de Linde, S.; Sauer, M. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 2015, 5, 15348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnert, G.; Keller, J.; Wurm, C.A.; Rizzoli, S.O.; Westphal, V.; Schönle, A.; Jahn, R.; Jakobs, S.; Eggeling, C.; Hell, S.W. Two-color far-field fluorescence nanoscopy. Biophys. J. 2007, 92, L67–L69. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Wurm, C.A.; Jakobs, S.; Engelhardt, J.; Egner, A.; Hell, S.W. Spherical nanosized focal spot unravels the interior of cells. Nat. Methods 2008, 5, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Harke, B.; Keller, J.; Ullal, C.K.; Westphal, V.; Schönle, A.; Hell, S.W. Resolution scaling in STED microscopy. Opt. Express 2008, 16, 4154–4162. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Wurm, C.A.; Punge, A.; Egner, A.; Jakobs, S.; Hell, S.W. Mitochondrial cristae revealed with focused light. Nano Lett. 2009, 9, 2508–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hell, S.W. Microscopy and its focal switch. Nat. Methods 2009, 6, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.; Gan, Q.; Ermolayev, V.; Harms, G.S. STED-SPIM: Stimulated emission depletion improves sheet illumination microscopy resolution. Biophys. J. 2011, 100, L43–L45. [Google Scholar] [CrossRef] [Green Version]
- Westphal, V.; Rizzoli, S.O.; Lauterbach, M.A.; Kamin, D.; Jahn, R.; Hell, S.W. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science 2008, 320, 246–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.; Zahn, J.; Maglione, M.; Sigrist, S.J.; Marquard, J.; Chojnacki, J.; Kräusslich, H.G.; Sahl, S.J.; Engelhardt, J.; Hell, S.W. Ultrafast, temporally stochastic STED nanoscopy of millisecond dynamics. Nat. Methods 2015, 12, 827–830. [Google Scholar] [CrossRef]
- Fouquet, W.; Owald, D.; Wichmann, C.; Mertel, S.; Depner, H.; Dyba, M.; Hallermann, S.; Kittel, R.J.; Eimer, S.; Sigrist, S.J. Maturation of active zone assembly by Drosophila Bruchpilot. J. Cell Biol. 2009, 186, 129–145. [Google Scholar] [CrossRef] [Green Version]
- Kittel, R.J.; Wichmann, C.; Rasse, T.M.; Fouquet, W.; Schmidt, M.; Schmid, A.; Wagh, D.A.; Pawlu, C.; Kellner, R.R.; Willig, K.I.; et al. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 2006, 312, 1051–1054. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. STED super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Weisenburger, S.; Boening, D.; Schomburg, B.; Giller, K.; Becker, S.; Griesinger, C.; Sandoghdar, V. Cryogenic optical localization provides 3D protein structure data with Angstrom resolution. Nat. Methods 2017, 14, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moerner, W.E.; Kador, L. Optical detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett. 1989, 62, 2535–2538. [Google Scholar] [CrossRef] [Green Version]
- Yildiz, A.; Tomishige, M.; Vale, R.D.; Selvin, P.R. Kinesin Walks Hand-Over-Hand. Science 2004, 303, 676. [Google Scholar] [CrossRef] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilemann, M.; van de Linde, S.; Schüttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008, 47, 6172–6176. [Google Scholar] [CrossRef]
- Bretschneider, S.; Eggeling, C.; Hell, S.W. Breaking the diffraction barrier in fluorescence microscopy by optical shelving. Phys. Rev. Lett. 2007, 98, 218103. [Google Scholar] [CrossRef] [Green Version]
- Fölling, J.; Bossi, M.; Bock, H.; Medda, R.; Wurm, C.A.; Hein, B.; Jakobs, S.; Eggeling, C.; Hell, S.W. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 2008, 5, 943–945. [Google Scholar] [CrossRef]
- Sharonov, A.; Hochstrasser, R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA 2006, 103, 18911–18916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606. [Google Scholar] [CrossRef] [Green Version]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [Green Version]
- van de Linde, S.; Löschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991–1009. [Google Scholar] [CrossRef] [PubMed]
- Gwosch, K.C.; Pape, J.K.; Balzarotti, F.; Hoess, P.; Ellenberg, J.; Ries, J.; Hell, S.W. MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat. Methods 2020, 17, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jones, S.A.; Brandenburg, B.; Zhuang, X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat. Methods 2008, 5, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Kao, H.P.; Verkman, A.S. Tracking of single fluorescent particles in three dimensions: Use of cylindrical optics to encode particle position. Biophys. J. 1994, 67, 1291–1300. [Google Scholar] [CrossRef] [Green Version]
- Shtengel, G.; Galbraith, J.A.; Galbraith, C.G.; Lippincott-Schwartz, J.; Gillette, J.M.; Manley, S.; Sougrat, R.; Waterman, C.M.; Kanchanawong, P.; Davidson, M.W.; et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Gajdos, T.; Cserteg, Z.; Szikora, S.; Novák, T.; Kovács, B.B.H.; Szabó, G.; Mihály, J.; Erdélyi, M. mmSTORM: Multimodal localization based super-resolution microscopy. Sci. Rep. 2019, 9, 798. [Google Scholar] [CrossRef]
- Juette, M.F.; Gould, T.J.; Lessard, M.D.; Mlodzianoski, M.J.; Nagpure, B.S.; Bennett, B.T.; Hess, S.T.; Bewersdorf, J. Three-dimensional sub–100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 2008, 5, 527–529. [Google Scholar] [CrossRef]
- Pavani, S.R.P.; Thompson, M.A.; Biteen, J.S.; Lord, S.J.; Liu, N.; Twieg, R.J.; Piestun, R.; Moerner, W.E. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc. Natl. Acad. Sci. USA 2009, 106, 2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Vaughan, J.C.; Zhuang, X. Isotropic three-dimensional super-resolution imaging with a self-bending point spread function. Nat. Photonics 2014, 8, 302–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; Betzig, E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 2008, 5, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Hartwich, T.M.P.; Rivera-Molina, F.E.; Lin, Y.; Duim, W.C.; Long, J.J.; Uchil, P.D.; Myers, J.R.; Baird, M.A.; Mothes, W.; et al. Video-rate nanoscopy using sCMOS camera–specific single-molecule localization algorithms. Nat. Methods 2013, 10, 653–658. [Google Scholar] [CrossRef]
- Szikora, S.; Gajdos, T.; Novák, T.; Farkas, D.; Földi, I.; Lenart, P.; Erdélyi, M.; Mihály, J. Nanoscopy reveals the layered organization of the sarcomeric H-zone and I-band complexes. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffié, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat. Rev. Methods Primers 2021, 1, 39. [Google Scholar] [CrossRef]
- Chen, F.; Tillberg, P.W.; Boyden, E.S. Expansion microscopy. Science 2015, 347, 543. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yu, Z.; Cahoon, C.K.; Parmely, T.; Thomas, N.; Unruh, J.R.; Slaughter, B.D.; Hawley, R.S. Combined expansion microscopy with structured illumination microscopy for analyzing protein complexes. Nat. Protoc. 2018, 13, 1869–1895. [Google Scholar] [CrossRef]
- Chang, J.-B.; Chen, F.; Yoon, Y.-G.; Jung, E.E.; Babcock, H.; Kang, J.S.; Asano, S.; Suk, H.-J.; Pak, N.; Tillberg, P.W.; et al. Iterative expansion microscopy. Nat. Methods 2017, 14, 593–599. [Google Scholar] [CrossRef]
- Wassie, A.T.; Zhao, Y.; Boyden, E.S. Expansion microscopy: Principles and uses in biological research. Nat. Methods 2019, 16, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Burkart, C.; Qiu, F.; Brendel, S.; Benes, V.; Hååg, P.; Labeit, S.; Leonard, K.; Bullard, B. Modular proteins from the Drosophila sallimus (sls) gene and their expression in muscles with different extensibility. J. Mol. Biol. 2007, 367, 953–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhme, M.A.; Beis, C.; Reddy-Alla, S.; Reynolds, E.; Mampell, M.M.; Grasskamp, A.T.; Lützkendorf, J.; Bergeron, D.D.; Driller, J.H.; Babikir, H.; et al. Active zone scaffolds differentially accumulate Unc13 isoforms to tune Ca(2+) channel-vesicle coupling. Nat. Neurosci. 2016, 19, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Böhme, M.A.; McCarthy, A.W.; Grasskamp, A.T.; Beuschel, C.B.; Goel, P.; Jusyte, M.; Laber, D.; Huang, S.; Rey, U.; Petzoldt, A.G.; et al. Rapid active zone remodeling consolidates presynaptic potentiation. Nat. Commun. 2019, 10, 1085. [Google Scholar] [CrossRef] [Green Version]
- Reddy-Alla, S.; Böhme, M.A.; Reynolds, E.; Beis, C.; Grasskamp, A.T.; Mampell, M.M.; Maglione, M.; Jusyte, M.; Rey, U.; Babikir, H.; et al. Stable Positioning of Unc13 Restricts Synaptic Vesicle Fusion to Defined Release Sites to Promote Synchronous Neurotransmission. Neuron 2017, 95, 1350–1364.e1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owald, D.; Khorramshahi, O.; Gupta, V.K.; Banovic, D.; Depner, H.; Fouquet, W.; Wichmann, C.; Mertel, S.; Eimer, S.; Reynolds, E.; et al. Cooperation of Syd-1 with Neurexin synchronizes pre- with postsynaptic assembly. Nat. Neurosci. 2012, 15, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ashley, J.; Budnik, V.; Bhat, M.A. Crucial role of Drosophila neurexin in proper active zone apposition to postsynaptic densities, synaptic growth, and synaptic transmission. Neuron 2007, 55, 741–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banovic, D.; Khorramshahi, O.; Owald, D.; Wichmann, C.; Riedt, T.; Fouquet, W.; Tian, R.; Sigrist, S.J.; Aberle, H. Drosophila neuroligin 1 promotes growth and postsynaptic differentiation at glutamatergic neuromuscular junctions. Neuron 2010, 66, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Wagh, D.A.; Rasse, T.M.; Asan, E.; Hofbauer, A.; Schwenkert, I.; Dürrbeck, H.; Buchner, S.; Dabauvalle, M.C.; Schmidt, M.; Qin, G.; et al. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 2006, 49, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallermann, S.; Kittel, R.J.; Wichmann, C.; Weyhersmüller, A.; Fouquet, W.; Mertel, S.; Owald, D.; Eimer, S.; Depner, H.; Schwärzel, M.; et al. Naked dense bodies provoke depression. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 14340–14345. [Google Scholar] [CrossRef] [Green Version]
- Lepicard, S.; Franco, B.; de Bock, F.; Parmentier, M.L. A presynaptic role of microtubule-associated protein 1/Futsch in Drosophila: Regulation of active zone number and neurotransmitter release. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 6759–6771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migh, E.; Götz, T.; Földi, I.; Szikora, S.; Gombos, R.; Darula, Z.; Medzihradszky, K.F.; Maléth, J.; Hegyi, P.; Sigrist, S.; et al. Microtubule organization in presynaptic boutons relies on the formin DAAM. Development 2018, 145, dev158519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.S.; Siebert, M.; Mertel, S.; Knoche, E.; Wegener, S.; Wichmann, C.; Matkovic, T.; Muhammad, K.; Depner, H.; Mettke, C.; et al. RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 2011, 334, 1565–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owald, D.; Fouquet, W.; Schmidt, M.; Wichmann, C.; Mertel, S.; Depner, H.; Christiansen, F.; Zube, C.; Quentin, C.; Körner, J.; et al. A Syd-1 homologue regulates pre- and postsynaptic maturation in Drosophila. J. Cell Biol. 2010, 188, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Ehmann, N.; van de Linde, S.; Alon, A.; Ljaschenko, D.; Keung, X.Z.; Holm, T.; Rings, A.; DiAntonio, A.; Hallermann, S.; Ashery, U.; et al. Quantitative super-resolution imaging of Bruchpilot distinguishes active zone states. Nat. Commun. 2014, 5, 4650. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [Green Version]
- D’Este, E.; Kamin, D.; Göttfert, F.; El-Hady, A.; Hell, S.W. STED Nanoscopy Reveals the Ubiquity of Subcortical Cytoskeleton Periodicity in Living Neurons. Cell Rep. 2015, 10, 1246–1251. [Google Scholar] [CrossRef] [Green Version]
- Lukinavičius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Göttfert, F.; Ta, H.; Güther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef]
- Qu, Y.; Hahn, I.; Webb, S.E.; Pearce, S.P.; Prokop, A. Periodic actin structures in neuronal axons are required to maintain microtubules. Mol. Biol. Cell 2017, 28, 296–308. [Google Scholar] [CrossRef]
- Han, B.; Zhou, R.; Xia, C.; Zhuang, X. Structural organization of the actin-spectrin–based membrane skeleton in dendrites and soma of neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E6678. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhou, R.; Wu, Z.; Carrasco, M.A.; Kurshan, P.T.; Farley, J.E.; Simon, D.J.; Wang, G.; Han, B.; Hao, J.; et al. Prevalent presence of periodic actin–spectrin-based membrane skeleton in a broad range of neuronal cell types and animal species. Proc. Natl. Acad. Sci. USA 2016, 113, 6029. [Google Scholar] [CrossRef] [Green Version]
- D’Este, E.; Kamin, D.; Velte, C.; Göttfert, F.; Simons, M.; Hell, S.W. Subcortical cytoskeleton periodicity throughout the nervous system. Sci. Rep. 2016, 6, 22741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokop, A.; Beaven, R.; Qu, Y.; Sánchez-Soriano, N. Using fly genetics to dissect the cytoskeletal machinery of neurons during axonal growth and maintenance. J. Cell Sci. 2013, 126, 2331–2341. [Google Scholar] [CrossRef] [Green Version]
- Matusek, T.; Djiane, A.; Jankovics, F.; Brunner, D.; Mlodzik, M.; Mihály, J. The Drosophila formin DAAM regulates the tracheal cuticle pattern through organizing the actin cytoskeleton. Development 2006, 133, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löschberger, A.; van de Linde, S.; Dabauvalle, M.C.; Rieger, B.; Heilemann, M.; Krohne, G.; Sauer, M. Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. J. Cell Sci. 2012, 125, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Szymborska, A.; de Marco, A.; Daigle, N.; Cordes, V.C.; Briggs, J.A.G.; Ellenberg, J. Nuclear Pore Scaffold Structure Analyzed by Super-Resolution Microscopy and Particle Averaging. Science 2013, 341, 655. [Google Scholar] [CrossRef] [Green Version]
- Cahoon, C.K.; Yu, Z.; Wang, Y.; Guo, F.; Unruh, J.R.; Slaughter, B.D.; Hawley, R.S. Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proc. Natl. Acad. Sci. USA 2017, 114, E6857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mund, M.; van der Beek, J.A.; Deschamps, J.; Dmitrieff, S.; Hoess, P.; Monster, J.L.; Picco, A.; Nédélec, F.; Kaksonen, M.; Ries, J. Systematic Nanoscale Analysis of Endocytosis Links Efficient Vesicle Formation to Patterned Actin Nucleation. Cell 2018, 174, 884–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattao, R.; Kovács, L.; Glover, D.M. The Centrioles, Centrosomes, Basal Bodies, and Cilia of Drosophila melanogaster. Genetics 2017, 206, 33–53. [Google Scholar] [CrossRef] [Green Version]
- Dobbelaere, J.; Josué, F.; Suijkerbuijk, S.; Baum, B.; Tapon, N.; Raff, J. A Genome-Wide RNAi Screen to Dissect Centriole Duplication and Centrosome Maturation in Drosophila. PLoS Biol. 2008, 6, e224. [Google Scholar] [CrossRef]
- Goshima, G.; Wollman, R.; Goodwin, S.S.; Zhang, N.; Scholey, J.M.; Vale, R.D.; Stuurman, N. Genes Required for Mitotic Spindle Assembly in Drosophila S2 Cells. Science 2007, 316, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Zhang, C. Super-resolution microscopy: Successful applications in centrosome study and beyond. Biophys. Rep. 2019, 5, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Dzhindzhev, N.S.; Tzolovsky, G.; Lipinszki, Z.; Schneider, S.; Lattao, R.; Fu, J.; Debski, J.; Dadlez, M.; Glover, D.M. Plk4 phosphorylates Ana2 to trigger Sas6 recruitment and procentriole formation. Curr. Biol. CB 2014, 24, 2526–2532. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Lipinszki, Z.; Rangone, H.; Min, M.; Mykura, C.; Chao-Chu, J.; Schneider, S.; Dzhindzhev, N.S.; Gottardo, M.; Riparbelli, M.G.; et al. Conserved molecular interactions in centriole-to-centrosome conversion. Nat. Cell Biol. 2016, 18, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roque, H.; Wainman, A.; Richens, J.; Kozyrska, K.; Franz, A.; Raff, J.W. Drosophila Cep135/Bld10 maintains proper centriole structure but is dispensable for cartwheel formation. J. Cell Sci. 2012, 125 Pt 23, 5881–5886. [Google Scholar] [CrossRef] [Green Version]
- Conduit, P.T.; Richens, J.H.; Wainman, A.; Holder, J.; Vicente, C.C.; Pratt, M.B.; Dix, C.I.; Novak, Z.A.; Dobbie, I.M.; Schermelleh, L.; et al. A molecular mechanism of mitotic centrosome assembly in Drosophila. eLife 2014, 3, e03399. [Google Scholar] [CrossRef] [PubMed]
- Conduit, P.T.; Wainman, A.; Novak, Z.A.; Weil, T.T.; Raff, J.W. Re-examining the role of Drosophila Sas-4 in centrosome assembly using two-colour-3D-SIM FRAP. eLife 2015, 4, e08483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, A.; Roque, H.; Saurya, S.; Dobbelaere, J.; Raff, J.W. CP110 exhibits novel regulatory activities during centriole assembly in Drosophila. J. Cell Biol. 2013, 203, 785–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbelaere, J.; Cernohorska, M.S.; Huranova, M.; Slade, D.; Dammermann, A. Cep97 Is Required for Centriole Structural Integrity and Cilia Formation in Drosophila. Curr. Biol. CB 2020, 30, 3045–3056.e3047. [Google Scholar] [CrossRef] [PubMed]
- Lawo, S.; Hasegan, M.; Gupta, G.D.; Pelletier, L. Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nat. Cell Biol. 2012, 14, 1148–1158. [Google Scholar] [CrossRef]
- Sonnen, K.F.; Schermelleh, L.; Leonhardt, H.; Nigg, E.A. 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol. Open 2012, 1, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.T.; Chong, W.M.; Wang, W.-J.; Mazo, G.; Tanos, B.; Chen, Z.; Tran, T.M.N.; Chen, Y.-D.; Weng, R.R.; Huang, C.-E.; et al. Super-resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat. Commun. 2018, 9, 2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Garcia, G.; Van De Weghe, J.C.; McGorty, R.; Pazour, G.J.; Doherty, D.; Huang, B.; Reiter, J.F. Super-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat. Cell Biol. 2017, 19, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.; Banterle, N.; Douglass, K.M.; Gönczy, P.; Manley, S. Multicolor single-particle reconstruction of protein complexes. Nat. Methods 2018, 15, 777–780. [Google Scholar] [CrossRef]
- von der Ecken, J.; Müller, M.; Lehman, W.; Manstein, D.J.; Penczek, P.A.; Raunser, S. Structure of the F-actin–tropomyosin complex. Nature 2015, 519, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Taylor, D.W.; Reedy, M.K.; Edwards, R.J.; Taylor, K.A. Structure of myosin filaments from relaxed Lethocerus flight muscle by cryo-EM at 6 Å resolution. Sci. Adv. 2016, 2, e1600058. [Google Scholar] [CrossRef] [Green Version]
- Szikora, S.; Novák, T.; Gajdos, T.; Erdélyi, M.; Mihály, J. Superresolution Microscopy of Drosophila Indirect Flight Muscle Sarcomeres. Bio-protocol 2020, 10, e3654. [Google Scholar] [CrossRef]
- González-Morales, N.; Schöck, F. Commentary: Nanoscopy reveals the layered organization of the sarcomeric H-zone and I-band complexes. Front. Cell Dev. Biol. 2020, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Deschout, H.; Zanacchi, F.C.; Mlodzianoski, M.; Diaspro, A.; Bewersdorf, J.; Hess, S.T.; Braeckmans, K. Precisely and accurately localizing single emitters in fluorescence microscopy. Nat. Methods 2014, 11, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Sarov, M.; Barz, C.; Jambor, H.; Hein, M.Y.; Schmied, C.; Suchold, D.; Stender, B.; Janosch, S.; Kj, V.V.; Krishnan, R.T.; et al. A genome-wide resource for the analysis of protein localisation in Drosophila. eLife 2016, 5, e12068. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, J.; Kaplan, C.; Platonova, E.; Eghlidi, H.; Ewers, H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 2012, 9, 582–584. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SIM | STED | STORM/PALM | ExM | |
|---|---|---|---|---|
| Concept to overcome the resolution limit | High-frequency information containing interference generated by patterned illumination | Stimulated de-excitation is used to produce a narrower emission zone | Fluorophores are modulated in the time scale to separate and localize them one by one | Isotropic sample expansion is used to increase the distance between the molecules |
| Microscopy type | Widefield | Laser scanning confocal | Widefield | Widefield/laser scanning confocal/spinning disk confocal |
| Lateral resolution | ~100 nm (linear) ~50 nm (nonlinear) | ~30–50 nm | ~20 nm | ~70 nm ~25 nm (with SIM) |
| Axial resolution | ~300 nm (linear) ~120 nm (nonlinear) | ~30 nm | ~50 nm | ~200 nm ~60 nm (with SIM) |
| Fluorophore type | Conventional fluorescent proteins and dyes | Photostable dyes and fluorescent proteins | STORM: photoswitchable dyes PALM: photoswitchable fluorescent proteins/dyes | Special labeling probe (able to survive the homogenization) |
| Phototoxicity | */** | **/*** | ** | - |
| Photobleaching | **/*** | **/*** | * | * |
| Live imaging | Well suited | Moderately suited | Limitedly suited | No |
| Post-image processing | Yes | No | Yes | Yes |
| Maximum number of simultaneous colors | 4 | 2 | 2–3 | 4 |
| Concerns | Out-of-focus signals | Photobleaching | Over/under-labeling artifacts | Time-consuming optimization |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szikora, S.; Görög, P.; Kozma, C.; Mihály, J. Drosophila Models Rediscovered with Super-Resolution Microscopy. Cells 2021, 10, 1924. https://doi.org/10.3390/cells10081924
Szikora S, Görög P, Kozma C, Mihály J. Drosophila Models Rediscovered with Super-Resolution Microscopy. Cells. 2021; 10(8):1924. https://doi.org/10.3390/cells10081924
Chicago/Turabian StyleSzikora, Szilárd, Péter Görög, Csaba Kozma, and József Mihály. 2021. "Drosophila Models Rediscovered with Super-Resolution Microscopy" Cells 10, no. 8: 1924. https://doi.org/10.3390/cells10081924
APA StyleSzikora, S., Görög, P., Kozma, C., & Mihály, J. (2021). Drosophila Models Rediscovered with Super-Resolution Microscopy. Cells, 10(8), 1924. https://doi.org/10.3390/cells10081924