
Human Vascular Smooth Muscle Function and Oxidative Stress Induced by NADPH Oxidase with the Clinical Implications


Abstract
:1. Introduction
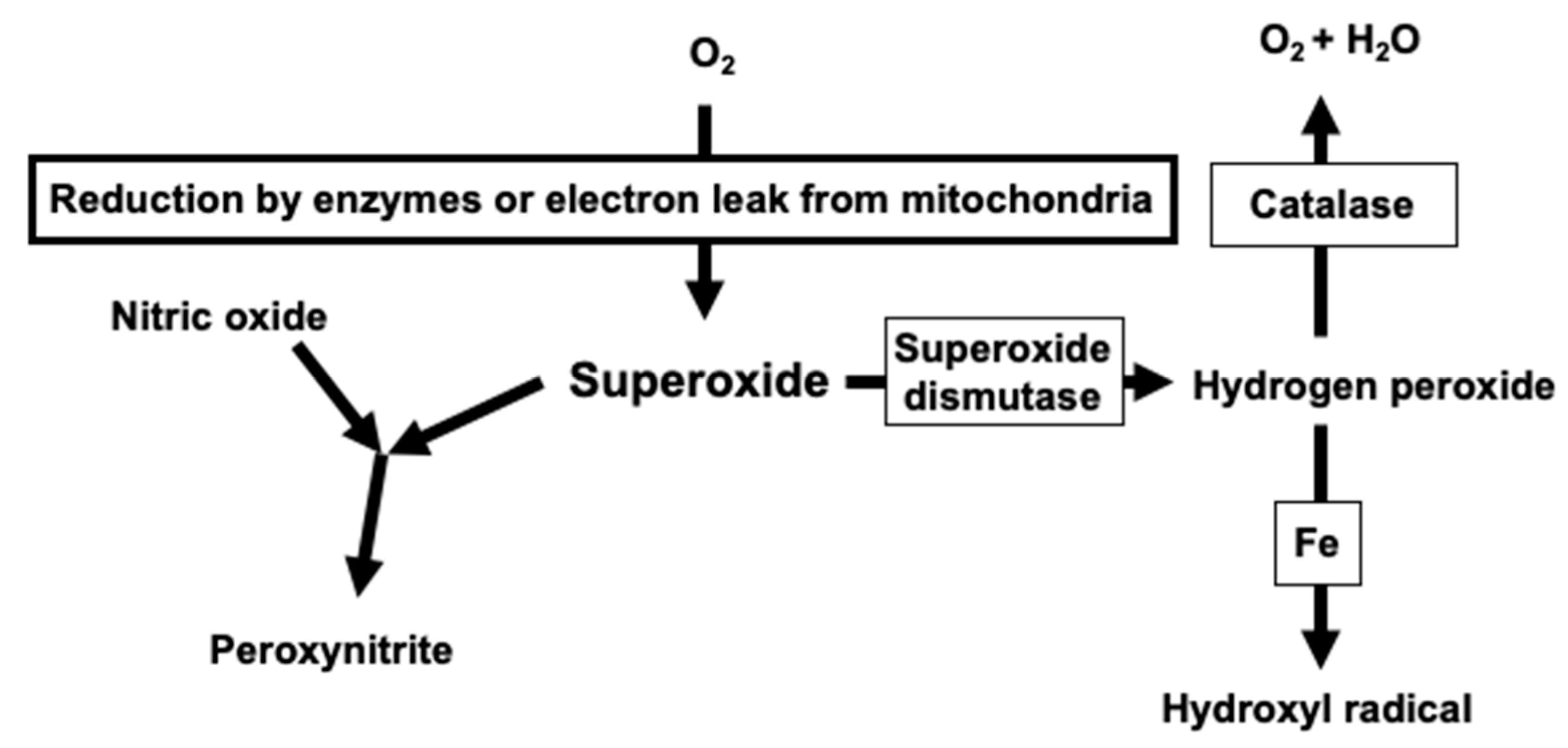
2. Critical Role of Superoxide in Vascular Oxidative Stress
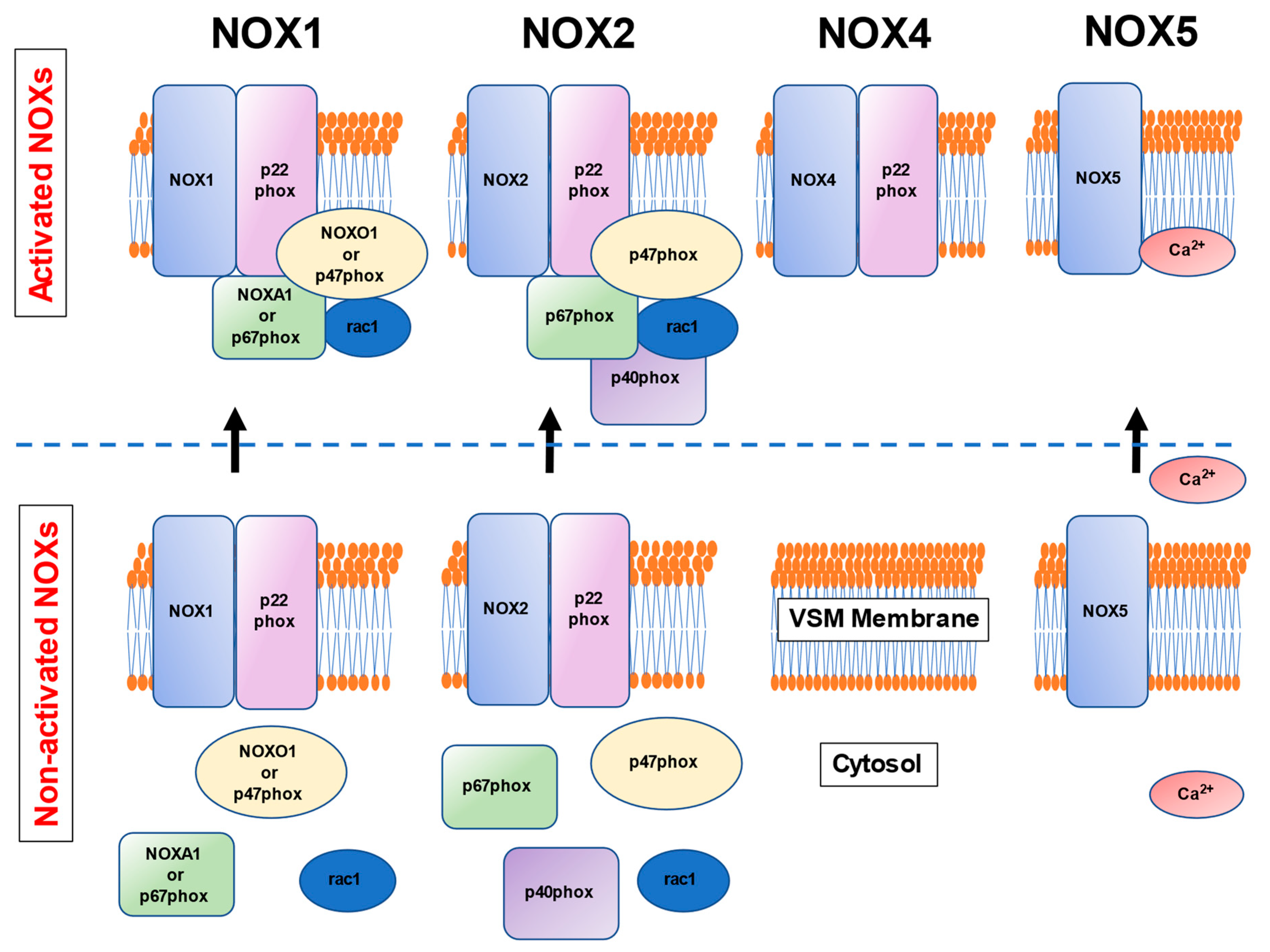
3. NOXs in the Human Vascular Smooth Muscle Cells
4. Pathophysiology Related to Oxidative Stress Induced by NOX in Human Vascular Smooth Muscle Cells
4.1. Hypertension
4.2. Hyperglycemia and Diabetes Mellitus
4.3. Inflammation
4.4. Arteriosclerosis
4.5. Heart Failure and Coronary Artery Disease
4.6. Marfan Syndrome
4.7. Major Depressive Disorder
4.8. Chronic Obstructive Pulmonary Disease
4.9. Chronic Kidney Disease
4.10. Varicose Veins
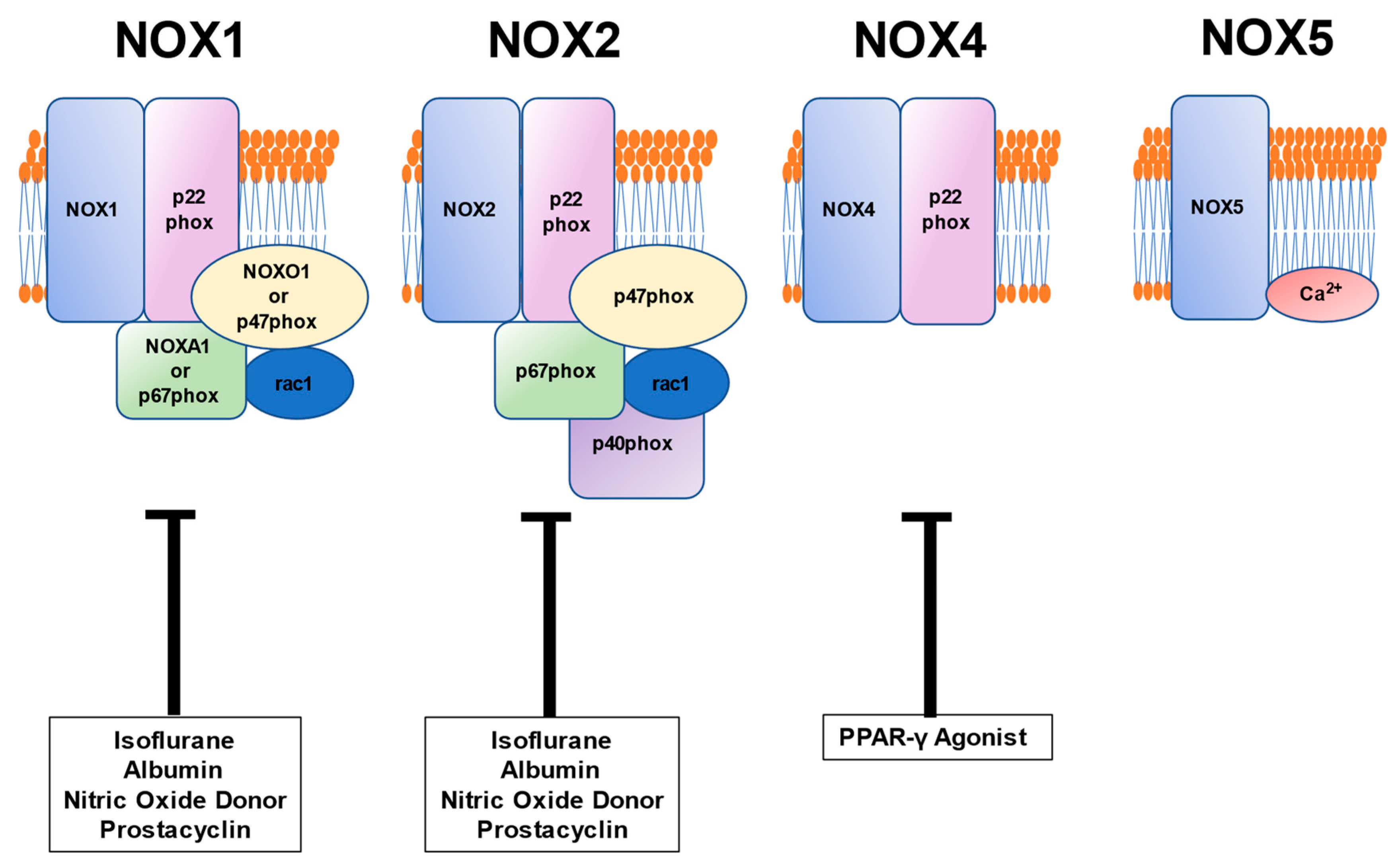
5. Relationship between Vascular Pathophysiology Induced by NOX and Pharmaceutical Formulation Used during the Perioperative Period
5.1. Anesthetic Isoflurane
5.2. Albumin
5.3. PPAR Agonist
5.4. Vasodilators: Nitric Oxide Donor and Prostacyclin
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, K.E.; Riches, K. The vascular smooth muscle cell: A therapeutic target in Type 2 diabetes? Clin. Sci. 2013, 125, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H. Effect of oxidative stress on vascular function, and the role of anesthetic. J. Anesth. 2012, 26, 141–142. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Matsuda, N.; Kaba, H.; Hatakeyama, N.; Azma, T.; Nakahata, K.; Kuroda, Y.; Tange, K.; Iranami, H.; Hatano, Y. Role of phosphatidylinositol 3-kinase-akt and NADPH oxidase in adenosine 5’-triphosphate-sensitive K+ channel function impaired by high glucose in the human artery. Hypertension 2008, 52, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef]
- Milstien, S.; Katusic, Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef] [PubMed]
- López-Acosta, O.; de Los Angeles Fortis-Barrera, M.; Barrios-Maya, M.A.; Ramírez, A.R.; Aguilar, F.J.A.; El-Hafidi, M. Reactive oxygen species from NADPH oxidase and mitochondria participate in the proliferation of aortic smooth muscle cells from a model of metabolic syndrome. Oxid. Med. Cell Longev. 2018, 2018, 5835072. [Google Scholar] [CrossRef] [Green Version]
- Mistry, Y.; Poolman, T.; Williams, B.; Herbert, K.E. A role for mitochondrial oxidants in stress-induced premature senescence of human vascular smooth muscle cells. Redox Biol. 2013, 1, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Armstead, W.M. Endothelin-Induced cyclooxygenase-dependent superoxide generation contributes to K+ channel functional impairment after brain injury. J. Neurotrauma 2001, 18, 1039–1048. [Google Scholar] [CrossRef]
- Kinoshita, H.; Azma, T.; Iranami, H.; Nakahata, K.; Kimoto, Y.; Dojo, M.; Yuge, O.; Hatano, Y. Synthetic peroxisome proliferator-activated receptor-gamma agonists restore impaired vasorelaxation via ATP-sensitive K+ channels by high glucose. J. Pharmacol. Exp. Ther. 2006, 318, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Matesanz, N.; Lafuente, N.; Azcutia, V.; Martín, D.; Cuadrado, A.; Nevado, J.; Rodríguez-Mañas, L.; Sánchez-Ferrer, C.F.; Peiró, C. Xanthine oxidase-derived extracellular superoxide anions stimulate activator protein 1 activity and hypertrophy in human vascular smooth muscle via c-Jun N-terminal kinase and p38 mitogen-activated protein kinases. J. Hypertens. 2007, 25, 609–618. [Google Scholar] [CrossRef]
- Kinoshita, H.; Milstien, S.; Wambi, C.; Katusic, Z.S. Inhibition of tetrahydrobiopterin biosynthesis impairs endothelium-dependent relaxations in canine basilar artery. Am. J. Physiol. 1997, 273, H718–H724. [Google Scholar] [CrossRef]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Kreuzer, J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc. Res. 2005, 65, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Griendling, K.K.; Harrison, D.G. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 2003, 24, 471–478. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Rad. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Münzel, T. NOX5, a new radical player in human atherosclerosis? J. Am. Coll. Cardiol. 2008, 52, 1810–1812. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Babior, B.M. The respiratory burst oxidase. Hematol. Oncol. Clin. North Am. 1988, 2, 201–212. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. NOX4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Banfi, B.; Molnar, G.; Maturana, A.; Steger, K.; Hegedus, B.; Demaurex, N.; Krause, K.H. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef] [Green Version]
- Furmanik, M.; Chatrou, M.; van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive oxygen-forming Nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef]
- Montezano, A.C.; De Lucca Camargo, L.; Persson, P.; Rios, F.J.; Harvey, A.P.; Anagnostopoulou, A.; Palacios, R.; Gandara, A.C.P.; Alves-Lopes, R.; Neves, K.B.; et al. NADPH oxidase 5 is a pro-contractile nox isoform and a point of cross-talk for calcium and redox signaling-implications in vascular function. J. Am. Heart Assoc. 2018, 7, e009388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagnostopoulou, A.; Camargo, L.L.; Rodrigues, D.; Montezano, A.C.; Touyz, R.M. Importance of cholesterol-rich microdomains in the regulation of Nox isoforms and redox signaling in human vascular smooth muscle cells. Sci. Rep. 2020, 10, 17818. [Google Scholar] [CrossRef]
- Touyz, R.M.; Chen, X.; Yao, G.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of functional active gp91phox-containing neutrophiltype NAD(P)H oxidase in smooth muscle cells from human resistance arteries. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touyz, R.M.; Yao, G.; Schiffrin, E.L. c-Src induces phosphorylation and translocation of p47phox: Role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touyz, R.M.; Yao, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: Role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, P.; Miao, S.B.; Shu, Y.N.; Dong, L.H.; Liu, G.; Xie, X.L.; Gao, M.; Wang, Y.C.; Yin, Y.J.; Wang, X.J.; et al. Phosphorylation of smooth muscle 22α facilitates angiotensin ii–induced ROS production via activation of the PKCδ-p47phox axis through release of PKCδ and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ. Res. 2012, 111, 697–707. [Google Scholar]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M.; Simionescu, M. AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: Role of p22phox subunit. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 878–885. [Google Scholar] [CrossRef] [Green Version]
- Muzaffar, S.; Shukla, N.; Massey, Y.; Angelini, G.D.; Jeremy, J.Y. NADPH oxidase 1 mediates upregulation of thromboxane A2 synthase in human vascular smooth muscle cells: Inhibition with iloprost. Eur. J. Pharmacol. 2011, 658, 187–192. [Google Scholar] [CrossRef]
- Kinoshita, H.; Azma, T.; Nakahata, K.; Iranami, H.; Kimoto, Y.; Dojo, M.; Yuge, O.; Hatano, Y. Inhibitory Effect of high concentration of glucose on relaxations to activation of ATP-sensitive K+ channels in human omental artery. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2290–2295. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Watanabe, K.; Azma, T.; Feng, G.G.; Akahori, T.; Hayashi, H.; Sato, M.; Fujiwara, Y.; Wakatsuki, A. Human serum albumin and oxidative stress in preeclamptic women and the mechanism of albumin for stress reduction. Heliyon 2017, 3, e00369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaishi, K.; Kinoshita, H.; Feng, G.G.; Azma, T.; Kawahito, S.; Kitahata, H. Cytoskeleton-disrupting agent cytochalasin B reduces oxidative stress caused by high glucose in the human arterial smooth muscle. J. Pharmacol. Sci. 2020, 144, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Margaritis, M.; Coutinho, P.; Shirodaria, C.; Psarros, C.; Herdman, L.; Sanna, F.; De Silva, R.; Petrou, M.; Sayeed, R.; et al. Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: The regulatory role of perivascular adipose tissue. Diabetes 2015, 64, 2207–2219. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [Green Version]
- Moe, K.T.; Aulis, S.; Jiang, F.; Chua, Y.L.; Koh, T.H.; Wong, M.C.; Dusting, G.J. Differential upregulation of Nox homologues of NADPH oxidase by tumor necrosis factor-alpha in human aortic smooth muscle and embryonic kidney cells. J. Cell Mol. Med. 2006, 10, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Saleh, M.A.; Kirabo, A.; Itani, H.A.; Montaniel, K.R.; Xiao, L.; Chen, W.; Mernaugh, R.L.; Cai, H.; Bernstein, K.E.; et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J. Clin. Investig. 2016, 126, 50–67. [Google Scholar] [CrossRef] [Green Version]
- Przybylska, D.; Janiszewska, D.; Goździk, A.; Bielak-Zmijewska, A.; Sunderland, P.; Sikora, E.; Mosieniak, G. NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget 2016, 7, 66429–66443. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworakowski, R.; Walker, S.; Momin, A.; Desai, J.; El-Gamel, A.; Wendler, O.; Kearney, M.T.; Shah, A.M. Reduced nicotinamide adenine dinucleotide phosphate oxidase-derived superoxide and vascular endothelial dysfunction in human heart failure. J. Am. Coll. Cardiol. 2008, 51, 1349–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Altayó, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; del Blanco, D.G.; López-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barberà, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef]
- Greaney, J.L.; Saunders, E.F.H.; Santhanam, L.; Alexander, L.M. Oxidative stress contributes to microvascular endothelial dysfunction in men and women with major depressive disorder. Circ. Res. 2019, 124, 564–574. [Google Scholar] [CrossRef]
- Oswald-Mammosser, M.; Weitzenblum, E.; Quoix, E.; Moser, G.; Chaouat, A.; Charpentier, C.; Kessler, R. Prognostic factors in COPD patients receiving longterm oxygen therapy. Importance of pulmonary artery pressure. Chest 1995, 107, 1193–1998. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Fan, Y.; Cui, J.; Hao, B.; Zhu, L.; Sun, X.; He, J.; Yang, J.; Dong, J.; Wang, Y.; et al. NOX4 expression and distal arteriolar remodeling correlate with pulmonary hypertension in COPD. BMC Pulm. Med. 2018, 18, 111. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22phox-containing NADPH oxidase. Circ. Res. 2001, 89, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Carrizzo, A.; Carrizzo, A.; Vecchione, C.; Damato, A.; di Nonno, F.; Ambrosio, M.; Pompeo, F.; Cappello, E.; Capocci, L.; Peruzzi, M.; et al. Rac1 Pharmacological inhibition rescues human endothelial dysfunction. J. Am. Heart Assoc. 2017, 6, e004746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, H.; Matsuda, N.; Iranami, H.; Ogawa, K.; Hatakeyama, H.; Azma, T.; Kawahito, S.; Yamazaki, M. Isoflurane pretreatment preserves adenosine triphosphate–sensitive K+ channel function in the human artery exposed to oxidative stress caused by high glucose levels. Anesth. Analg. 2012, 115, 54–61. [Google Scholar] [CrossRef]
- Bijli, K.M.; Kleinhenz, J.M.; Murphy, T.C.; Kang, B.J.; Adesina, S.E.; Sutiff, R.L.; Hart, C.M. Peroxisome proliferator-activated receptor gamma depletion stimulates NOX4 expression and human pulmonary artery smooth muscle cell. Free. Radic. Biol. Med. 2015, 80, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Muzaffar, S.; Shukla, N.; Bond, M.; Sala-Newby, G.; Angelini, G.D.; Newby, A.C.; Jeremy, J.Y. Acute inhibition of superoxide formation and Rac1 activation by nitric oxide and iloprost in human vascular smooth muscle cells in response to the thromboxane A2 analogue, U46619. Prostaglandins Leukot. Essent. Fat. Acids 2008, 78, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Saito, T.; Feng, G.-G.; Li, J.; Yasuda, Y.; Kazaoka, Y.; Fujiwara, Y.; Kinoshita, H. Intermittent local periodontal inflammation causes endothelial dysfunction of the systemic artery via increased levels of hydrogen peroxide concomitantly with overexpression of superoxide dismutase. Int. J. Cardiol. 2016, 222, 901–907. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Pathology | NOX1 | NOX2 | NOX4 | NOX5 | Reference Number |
|---|---|---|---|---|---|
| Hypertension | Peripheral artery | Peripheral artery | [28,29,30] | ||
| Umbilical artery | Umbilical artery | [31] | |||
| Aorta | Aorta | [32] | |||
| Hyperglycemia | Omental artery | Omental artery | [6] | ||
| Coronary artery | Coronary artery | [36] | |||
| Diabetes Mellitus | Mammary artery | Mammary artery | [37,38] | ||
| Saphenous vein | [38] | ||||
| Inflammation | Aorta | Aorta | Aorta | [32,39,40] | |
| Arteriosclerosis | Aorta | [43] | |||
| Aorta, Coronary artery | [5,25] | ||||
| Heart Failure | Saphenous vein | Saphenous vein | [45] | ||
| Coronary artery disease | Coronary artery | Coronary artery | [44] | ||
| Marfan Syndrome | Aorta | [46] | |||
| Depressive disorder | Cutaneous vessel | Cutaneous vessel | [47] | ||
| COPD | Pulmonary artery | [48] | |||
| CKD | Renal artery | Renal artery | Renal artery | [50] | |
| Varicose Veins | Saphenous vein | Saphenous vein | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takaishi, K.; Kinoshita, H.; Kawashima, S.; Kawahito, S. Human Vascular Smooth Muscle Function and Oxidative Stress Induced by NADPH Oxidase with the Clinical Implications. Cells 2021, 10, 1947. https://doi.org/10.3390/cells10081947
Takaishi K, Kinoshita H, Kawashima S, Kawahito S. Human Vascular Smooth Muscle Function and Oxidative Stress Induced by NADPH Oxidase with the Clinical Implications. Cells. 2021; 10(8):1947. https://doi.org/10.3390/cells10081947
Chicago/Turabian StyleTakaishi, Kazumi, Hiroyuki Kinoshita, Shingo Kawashima, and Shinji Kawahito. 2021. "Human Vascular Smooth Muscle Function and Oxidative Stress Induced by NADPH Oxidase with the Clinical Implications" Cells 10, no. 8: 1947. https://doi.org/10.3390/cells10081947
APA StyleTakaishi, K., Kinoshita, H., Kawashima, S., & Kawahito, S. (2021). Human Vascular Smooth Muscle Function and Oxidative Stress Induced by NADPH Oxidase with the Clinical Implications. Cells, 10(8), 1947. https://doi.org/10.3390/cells10081947