
cAMP Compartmentalization in Cerebrovascular Endothelial Cells: New Therapeutic Opportunities in Alzheimer’s Disease





{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Vascular Hypothesis of Alzheimer’s Disease (AD)
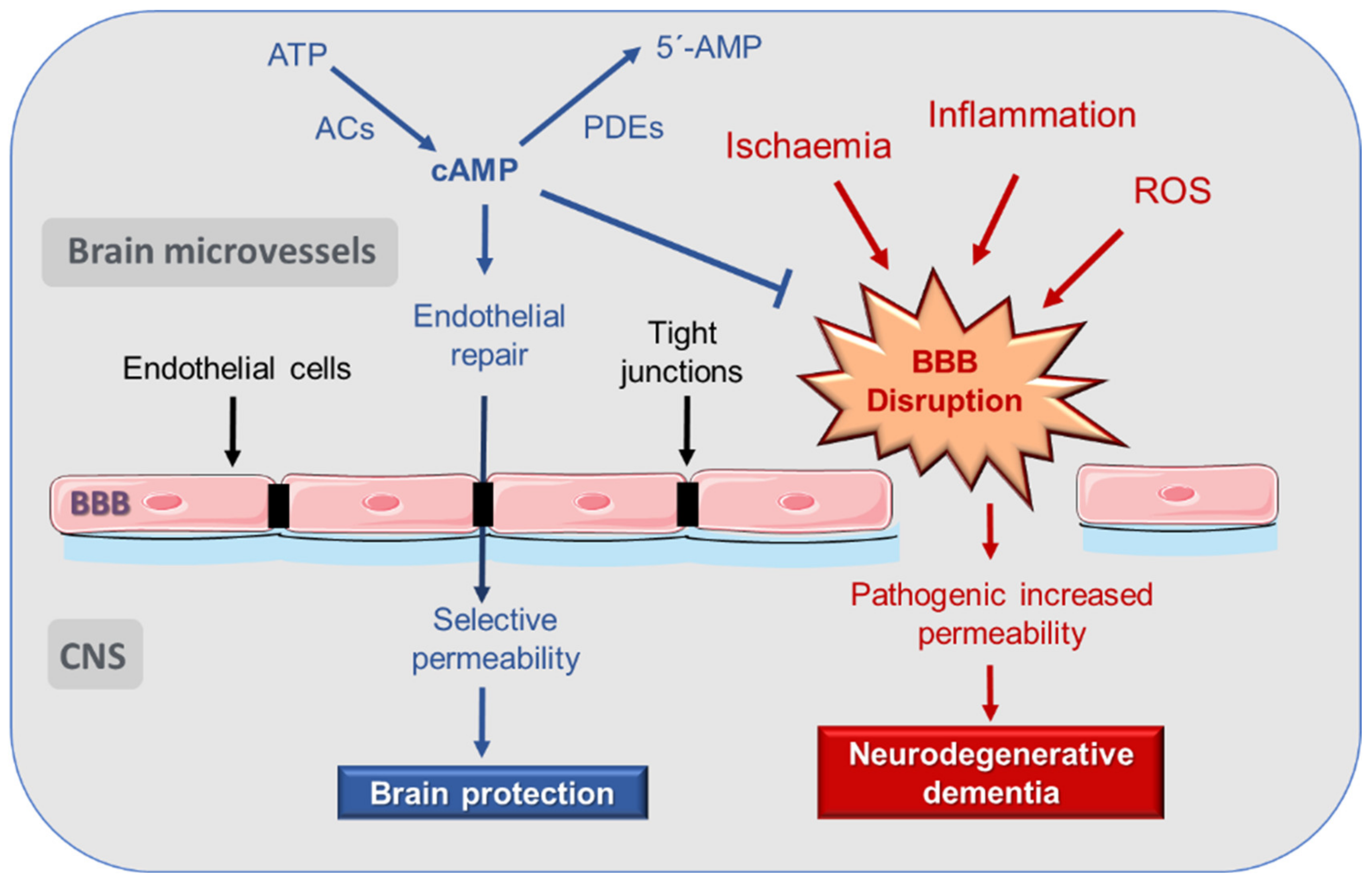
3. Disruption of Blood–Brain Barrier (BBB) in AD
4. cAMP Regulation of Endothelial and BBB Permeability
5. cAMP Compartmentalization
5.1. Methods for Studying the cAMP Compartmentalization
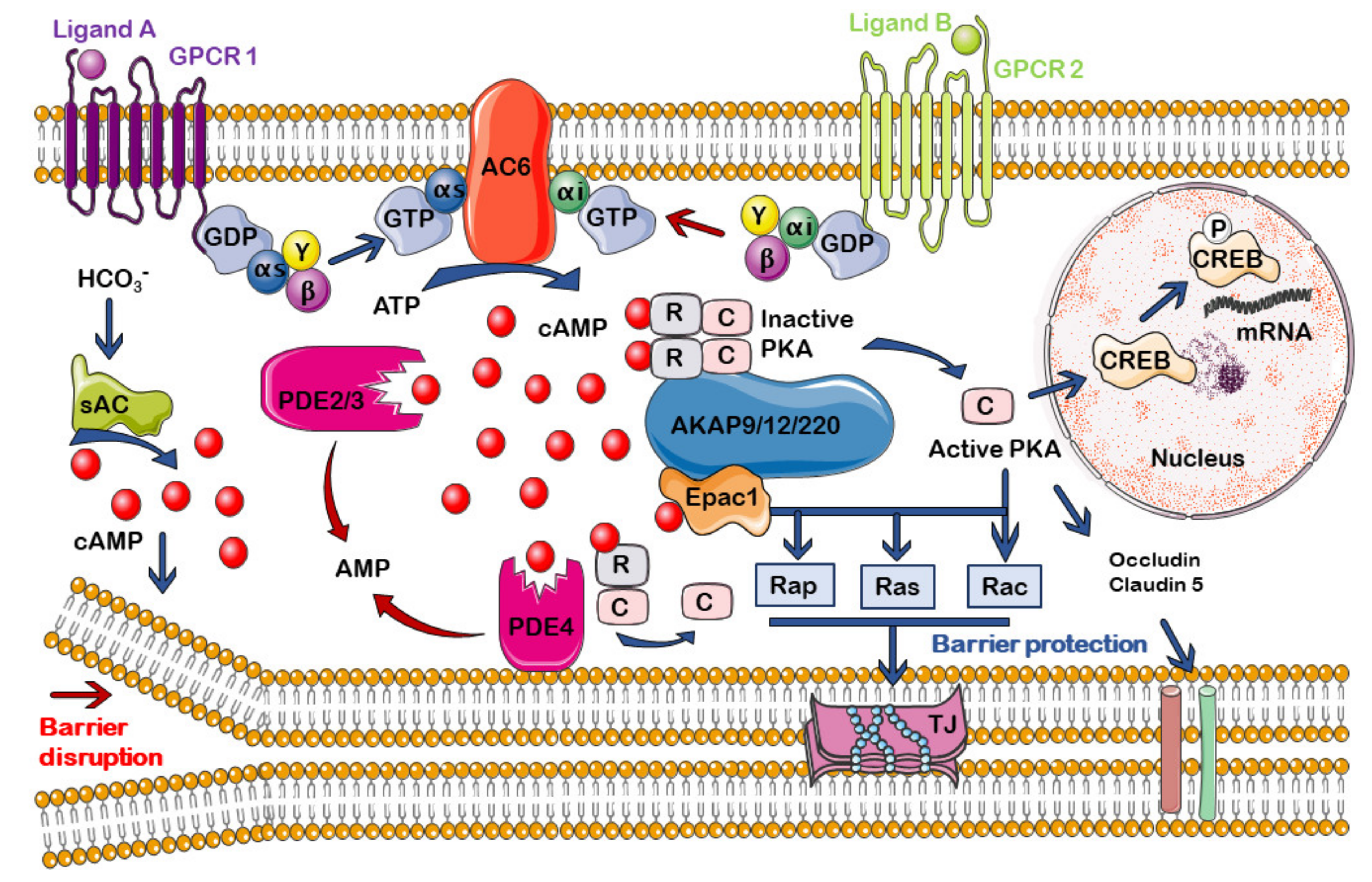
5.2. cAMP Signalosomes in Endothelium and BBB
6. Regulation of the cAMP Compartmentalization at the Endothelium and the BBB
6.1. cAMP Generation and Degradation: ACs and PDEs
6.1.1. Adenylyl Cyclases (ACs)
6.1.2. cAMP Phosphodiesterases (PDEs)
6.2. A-Kinase Anchoring Proteins (AKAPs)
6.3. cAMP Effectors: PKA and Epac
6.3.1. PKA
6.3.2. Exchange Proteins Directly Activated by cAMP (Epac)
7. cAMP Compartmentalization in the BBB and Aging
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Farotti, L.; Sepe, F.N.; Toja, A.; Rinaldi, R.; Parnetti, L. Differential diagnosis between Alzheimer’s disease and other dementias: Role of cerebrospinal fluid biomarkers. Clin. Biochem. 2019, 72, 24–29. [Google Scholar] [CrossRef]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 1, 108352. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, B.; Yin, W.; Han, Z.; Zheng, C.; Wang, P.; Ran, C. Differentiating Ab40 and Ab42 in amyloid plaques with a small molecule fluorescence probe. Chem. Sci. 2020, 11, 5238–5245. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Ismail, R.; Parbo, P.; Madsen, L.S.; Hansen, A.K.; Hansen, K.V.; Schaldemose, J.L.; Kjeldsen, P.L.; Stokholm, M.G.; Gottrup, H.; Eskildsen, S.F.; et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: A longitudinal PET study. J. Neuroinflamm. 2020, 17, 151. [Google Scholar] [CrossRef]
- Huang, X.; Hussain, B.; Chang, J. Peripheral inflammation and blood–brain barrier disruption: Effects and mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef]
- Rubin, L.L.; Hall, D.E.; Porter, S.; Barbu, K.; Cannon, C.; Horner, H.C.; Janatpour, M.; Liaw, C.W.; Manning, K.; Morales, J.; et al. A cell culture model of the blood-brain barrier. J. Cell Biol. 1991, 115, 1725–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef]
- De la Torre, J.C.; Mussivand, T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol. Res. 1993, 15, 146–153. [Google Scholar] [CrossRef]
- Hays, C.C.; Zlatar, Z.Z.; Wierenga, C.E. The utility of cerebral blood flow as a biomarker of preclinical Alzheimer’s disease. Cell. Mol. Neurobiol. 2016, 36, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Markus, H.S. Cerebrovascular abnormalities in Alzheimer’s dementia: A more tractable treatment target? Brain 2017, 140, 1822–1825. [Google Scholar] [CrossRef]
- Govindpani, K.; McNamara, L.G.; Smith, N.R.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Vascular dysfunction in Alzheimer’s disease: A prelude to the pathological process or a consequence of it? J. Clin. Med. 2019, 8, 651. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Braidy, N.; Poljak, A.; Chan, D.K.Y.; Sachdev, P. Cerebral small vessel disease and the risk of Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2018, 47, 41–48. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, S.E.; Raible, K.; Brettschneider, J.; Xie, S.X.; Grossman, M.; Monsell, S.E.; Kukull, W.A.; Trojanowski, J.Q. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013, 136, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Chiu, M.J.; Chen, Y.F.; Cheng, T.W.; Lai, Y.M.; Chen, T.F. The contribution of vascular risk factors in neurodegenerative disorders: From mild cognitive impairment to Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 91. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Kruyer, A.; Fernandez-Nued, I.; Marcos-Diaz, A.; Ceron, C.; Richards, A.T.; Jno-Charles, O.C.; Rodriguez, I.; Callejas, S.; Norris, E.H.; et al. Long-term dabigatran treatment delays Alzheimer’s disease pathogenesis in the TgCRND8 mouse model. J. Am. Coll. Cardiol. 2019, 7, 1910–1923. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reduction exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hou, D.; Lin, F.; Luo, J.; Wen Xie, J.; Wang, Y.; Tian, Y. The role of neurovascular unit damage in the occurrence and development of Alzheimer’s disease. Rev. Neurosci. 2019, 30, 477–484. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, function, and regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- Rodrigues, S.F.; Granger, D.N. Blood cells and endothelial barrier function. Tissue Barriers 2015, 3, e978720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. [Google Scholar] [CrossRef]
- Nava, E.; Llorens, S. The local regulation of vascular function: From an inside-outside to an outside-inside model. Front. Physiol. 2019, 10, 729. [Google Scholar] [CrossRef]
- Ashby, J.W.; Mack, J.J. Endothelial control of cerebral blood flow. Am. J. Pathol. 2021, in press. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular endothelial cell biology: An update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Takeda, Y.; Matoba, K.; Sekiguchi, K.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Endothelial dysfunction in diabetes. Biomedicines 2020, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Hang, W.; Chen, C.; Zhang, X.A.; Wang, D.W. Endothelial dysfunction in COVID-19 calls for immediate attention: The emerging roles of the endothelium in inflammation caused by SARS-CoV-2. Front. Med. 2021, 1–6. [Google Scholar] [CrossRef]
- McRae, M.; LaFratta, L.M.; Nguyen, B.M.; Paris, J.J.; Hauser, K.F.; Conway, D.E. Characterization of cell-cell junction changes associated with the formation of a strong endothelial barrier. Tissue Barriers 2018, 6, e1405774. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Wong, L.W.; Su, Y.; Huang, X.; Wang, N.; Chen, H.; Yi, C. Blood-brain barrier integrity in the pathogenesis of Alzheimer’s disease. Front. Neuroendocrinol. 2020, 59, 100857. [Google Scholar] [CrossRef]
- Armulik, A.; Mäe, M.; Betsholtz, C. Pericytes and the blood-brain barrier: Recent advances and implications for the delivery of CNS therapy. Ther. Deliv. 2011, 2, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Wan, C.Q.; Liu, Z. Astrocyte and Alzheimer’s disease. J. Neurol. 2017, 264, 2068–2074. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Ukidve, A.; Kim, J.; Mitragotri, S. Targeting strategies for tissue-specific drug delivery. Cell 2020, 181, 151–167. [Google Scholar] [CrossRef]
- Erdener, Ş.E.; Dalkara, T. Small vessels are a big problem in neurodegeneration and neuroprotection. Front. Neurol. 2019, 10, 889. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Miners, J.S.; Schulz, I.; Love, S. Differing associations between Aβ accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2018, 38, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Bowman, G.L.; Dayon, L.; Kirkland, R.; Wojcik, J.; Peyratout, G.; Severin, I.C.; Henry, H.; Oikonomidi, A.; Migliavacca, E.; Bacher, M.; et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimers Dement. 2018, 14, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Yousef, H.; Czupalla, C.J.; Lee, D.; Chen, M.B.; Burke, A.N.; Zera, K.A.; Zandstra, J.; Berber, E.; Lehallier, B.; Mathur, V.; et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 2019, 25, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Novak, V.; Zhao, P.; Manor, B.; Sejdic, E.; Alsop, D.; Abduljalil, A.; Roberson, P.K.; Munshi, M.; Novak, P. Adhesion molecules, altered vasoreactivity, and brain atrophy in type 2 diabetes. Diabetes Care 2011, 34, 2438–2441. [Google Scholar] [CrossRef] [Green Version]
- Takemori, K.; Ito, H.; Suzuki, T. Effects of the AT1 receptor antagonist on adhesion molecule expression in leukocytes and brain microvessels of stroke-prone spontaneously hypertensive rats. Am. J. Hypertens. 2000, 13, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Iannucci, J.; Renehan, W.; Grammas, P. Thrombin, a mediator of coagulation, inflammation, and neurotoxicity at the neurovascular interface: Implications for Alzheimer’s disease. Front. Neurosci. 2020, 14, 762. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harb. Perspect. Biol. 2014, 7, a016345. [Google Scholar] [CrossRef]
- Chrissobolis, S.; Faraci, F.M. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol. Med. 2008, 14, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Huang, C.; Wu, Y.; Chen, B.; Zhang, W.; Zhang, J. Autophagy protects the Blood-Brain Barrier through regulating the dynamic of claudin-5 in short-term starvation. Front. Physiol. 2019, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Cai, Y.B.; Liu, Z. Activation of AMPK improves lipopolysaccharide-induced dysfunction of the blood-brain barrier in mice. Brain Inj. 2015, 29, 777–784. [Google Scholar] [CrossRef]
- Seok, S.M.; Kim, J.M.; Park, T.Y.; Baik, E.J.; Lee, S.H. Fructose-1,6-bisphosphate ameliorates lipopolysaccharide-induced dysfunction of blood-brain barrier. Arch. Pharm. Res. 2013, 36, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S.; Manneville, J.B.; Adamson, P.; Wilbourn, B.; Greenwood, J.; Couraud, P.O. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J. Immunol. 2000, 165, 3375–3383. [Google Scholar] [CrossRef] [Green Version]
- Mamelak, M. Energy and the Alzheimer brain. Neurosci. Biobehav. Rev. 2017, 75, 297–313. [Google Scholar] [CrossRef]
- Halls, M.L.; Cooper, D.M.F. Adenylyl cyclase signalling complexes–Pharmacological challenges and opportunities. Pharmacol. Ther. 2017, 172, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Iannucci, L.F.; Surdo, N.C.; Zanin, S.; Conca, F.; Grisan, F.; Gerbino, A.; Lefkimmiatis, K. Compartmentalized signaling in aging and neurodegeneration. Cells 2021, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, C.; Foster, K.; Corley, J.E.; Dimri, M.; Brady, M.F. Biochemistry, cAMP. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK535431/ (accessed on 30 July 2021).
- Moore, T.M.; Chetham, P.M.; Kelly, J.J.; Stevens, T. Signal transduction and regulation of lung endothelial cell permeability. Interaction between calcium and cAMP. Am. J. Physiol. 1998, 275, L203–L222. [Google Scholar] [CrossRef]
- Adamson, R.H.; Liu, B.; Fry, G.N.; Rubin, L.L.; Curry, F.E. Microvascular permeability and number of tight junctions are modulated by cAMP. Am. J. Physiol. 1998, 274, H1885–H1894. [Google Scholar] [CrossRef]
- Feinstein, W.P.; Zhu, B.; Leavesley, S.J.; Sayner, S.L.; Rich, T.C. Assessment of cellular mechanisms contributing to cAMP compartmentalization in pulmonary microvascular endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 302, C839–C852. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of endothelial junctional permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef]
- Curry, F.R.; Adamson, R.H. Tonic regulation of vascular permeability. Acta Physiol. 2013, 207, 628–649. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, N.; Waschke, J. cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier—A signaling pathway compromised in inflammation. Cell Tissue Res. 2014, 355, 587–596. [Google Scholar] [CrossRef]
- Easton, A.S.; Sarker, M.H.; Fraser, P.A. Two components of blood-brain barrier disruption in the rat. J. Physiol. 1997, 503, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zeng, M.; Fu, B.M. Temporal effects of vascular endothelial growth factor and 3,5-cyclic monophosphate on blood-brain barrier solute permeability in vivo. J. Neurosci. Res. 2014, 92, 1678–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, A.; Bintig, W.; Begandt, D.; Klett, A.; Siller, I.G.; Gregor, C.; Schaarschmidt, F.; Weksler, B.; Romero, I.; Couraud, P.-O.; et al. Adenosine receptors regulate gap junction coupling of the human cerebral microvascular endothelial cells hCMEC/D3 by Ca2+ influx through cyclic nucleotide-gated channels. J. Physiol. 2017, 595, 2497–2517. [Google Scholar] [CrossRef] [Green Version]
- Perrot, C.Y.; Sawada, J.; Komatsu, M. Prolonged activation of cAMP signaling leads to endothelial barrier disruption via transcriptional repression of RRAS. FASEB J. 2018, 32, 5793–5812. [Google Scholar] [CrossRef] [Green Version]
- Sayner, S.L. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L667–L678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musheshe, N.; Schmidt, M.; Zaccolo, M. cAMP: From long-range second messenger to nanodomain signalling. Trends Pharmacol. Sci. 2018, 39, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Cattani-Cavalieri, I.; Nuñez, F.J.; Ostrom, R.S. Phosphodiesterase isoforms and cAMP compartments in the development of new therapies for obstructive pulmonary diseases. Curr. Opin. Pharmacol. 2020, 51, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef]
- Zaccolo, M.; De Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2000, 2, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [Green Version]
- Annamdevula, N.S.; Sweat, R.; Griswold, J.R.; Trinh, K.; Hoffman, C.; West, S.; Deal, J.; Britain, A.L.; Jalink, K.; Rich, T.C.; et al. Spectral imaging of FRET-based sensors reveals sustained cAMP gradients in three spatial dimensions. Cytometry A 2018, 93, 1029–1038. [Google Scholar] [CrossRef] [Green Version]
- Annamdevula, N.S.; Sweat, R.; Gunn, H.; Griswold, J.R.; Britain, A.L.; Rich, T.C.; Leavesley, S.J. Measurement of 3-dimensional cAMP distributions in living cells using 4-dimensional (x, y, z, and λ) hyperspectral FRET imaging and analysis. J. Vis. Exp. 2020, 164, e61720. [Google Scholar] [CrossRef]
- Kim, N.; Shin, S.; Bae, S.W. cAMP Biosensors based on genetically encoded fluorescent/luminescent proteins. Biosensors 2021, 11, 39. [Google Scholar] [CrossRef]
- Ghigo, A.; Mika, D. cAMP/PKA signaling compartmentalization in cardiomyocytes: Lessons from FRET-based biosensors. J. Mol. Cell Cardiol. 2019, 131, 112–121. [Google Scholar] [CrossRef]
- Judina, A.; Gorelik, J.; Wright, P.T. Studying signal compartmentation in adult cardio-myocytes. Biochem. Soc. Trans. 2020, 48, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.C.; Surdo, N.C.; Pantano, S.; Zaccolo, M. Imaging cAMP nanodomains in the heart. Biochem. Soc. Trans. 2019, 47, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannabiran, S.A.; Gosejacob, D.; Niemann, B.; Nikolaev, V.O.; Pfeifer, A. Real-time monitoring of cAMP in brown adipocytes reveals differential compartmentation of β1 and β3-adrenoceptor signalling. Mol. Metab. 2020, 37, 100986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bouadjel, K.; Manoury, B.; Vandecasteele, G.; Fischmeister, R.; Leblais, V. Cyclic nucleotide signalling compartmentation by PDEs in cultured vascular smooth muscle cells. Br. J. Pharmacol. 2019, 176, 1780–1792. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Cameron, E.G.; Yu, W.; Xia, X.; Shah, S.H.; Castillo Chabeco, B.; Galvao, J.; Nahmou, M.; Li, J.; Thakur, H.; et al. Regulation of neuronal survival and axon growth by a perinuclear cAMP compartment. J. Neurosci. 2019, 39, 5466–5480. [Google Scholar] [CrossRef] [Green Version]
- Reuschlein, A.K.; Jakobsen, E.; Mertz, C.; Bak, L.K. Aspects of astrocytic cAMP signaling with an emphasis on the putative power of compartmentalized signals in health and disease. Glia 2019, 67, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Elíes, J.; Cuíñas, A.; MacDougall, D.; Leiro, J.; Campos-Toimil, M. Trans-resveratrol down-regulates caveolin-1, up-regulates endothelial NO synthase and reduces their interaction in vascular smooth muscle and endothelial cells. Food Biosci. 2013, 1, 31–38. [Google Scholar] [CrossRef]
- Sampson, L.J.; Hayabuchi, Y.; Standen, N.B.; Dart, C. Caveolae localize protein kinase A signaling to arterial ATP-sensitive potassium channels. Circ. Res. 2004, 95, 1012–1018. [Google Scholar] [CrossRef] [Green Version]
- MacKeil, J.L.; Brzezinska, P.; Burke-Kleinman, J.; Theilmann, A.L.; Nicol, C.J.B.; Ormiston, M.L.; Maurice, D.H. Phosphodiesterase 3B (PDE3B) antagonizes the anti-angiogenic actions of PKA in human and murine endothelial cells. Cell Signal. 2019, 62, 109342. [Google Scholar] [CrossRef]
- Oldenburger, A.; Maarsingh, H.; Schmidt, M. Multiple facets of cAMP signalling and physiological impact: cAMP compartmentalization in the lung. Pharmaceuticals 2012, 5, 1291–1331. [Google Scholar] [CrossRef] [Green Version]
- Murray, F. The interplay of multiple molecular and cellular components is necessary for compartmentalization of cAMP. Focus on “Assessment of cellular mechanisms contributing to cAMP compartmentalization in pulmonary microvascular endothelial cells”. Am. J. Physiol. Cell Physiol. 2012, 302, C837–C838. [Google Scholar] [CrossRef]
- Tresguerres, M.; Levin, L.R.; Buck, J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011, 79, 1277–1288. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell. Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, L.K.; Scott, J.D. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell. Biol. 2015, 16, 232–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioffi, D.L.; Moore, T.M.; Schaack, J.; Creighton, J.R.; Cooper, D.M.; Stevens, T. Dominant regulation of interendothelial cell gap formation by calcium-inhibited type 6 adenylyl cyclase. J. Cell Biol. 2002, 157, 1267–1678. [Google Scholar] [CrossRef] [Green Version]
- Sayner, S.L.; Frank, D.W.; King, J.; Chen, H.; VandeWaa, J.; Stevens, T. Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ. Res. 2004, 95, 196–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, C.D.; Alexeyev, M.; Pastukh, V.; Balczon, R.; Stevens, T. Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial τ-phosphorylation and permeability. J. Biol. Chem. 2012, 287, 25407–25418. [Google Scholar] [CrossRef] [Green Version]
- Balczon, R.; Prasain, N.; Ochoa, C.; Prater, J.; Zhu, B.; Alexeyev, M.; Sayner, S.; Frank, D.W.; Stevens, T. Pseudomonas aeruginosa exotoxin Y-mediated tau hyperphosphorylation impairs microtubule assembly in pulmonary microvascular endothelial cells. PLoS ONE 2013, 8, e74343. [Google Scholar] [CrossRef]
- Nickols, J.; Obiako, B.; Ramila, K.C.; Putinta, K.; Schilling, S.; Sayner, S.L. Lipopolysaccharide-induced pulmonary endothelial barrier disruption and lung edema: Critical role for bicarbonate stimulation of AC10. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1430–L1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, K.A.; Frank, D.W.; Balczon, R.; Stevens, T. The Pseudomonas aeruginosa exoenzyme Y: A promiscuous nucleotidyl cyclase edema factor and virulence determinant. Handb. Exp. Pharmacol. 2017, 238, 67–85. [Google Scholar] [CrossRef]
- Sayner, S.; Stevens, T. Soluble adenylate cyclase reveals the significance of compartmentalized cAMP on endothelial cell barrier function. Biochem. Soc. Trans. 2006, 34, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayner, S.L.; Alexeyev, M.; Dessauer, C.W.; Stevens, T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ. Res. 2006, 98, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayner, S.L.; Choi, C.S.; Maulucci, M.E.; Ramila, K.C.; Zhou, C.; Scruggs, A.K.; Yarbrough, T.; Blair, L.A.; King, J.A.; Seifert, R.; et al. Extracellular vesicles: Another compartment for the second messenger, cyclic adenosine monophosphate. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L691–L700. [Google Scholar] [CrossRef]
- Khannpnavar, B.; Mehta, V.; Qi, C.; Korkhov, V. Structure and function of adenylyl cyclases, key enzymes in cellular signaling. Curr. Opin. Struct. Biol. 2020, 63, 34–41. [Google Scholar] [CrossRef]
- Bassler, J.; Schultz, J.E.; Lupas, A.N. Adenylate cyclases: Receivers, transducers, and generators of signals. Cell. Signal. 2018, 46, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.B.; Agarwal, S.R.; Harvey, R.D.; Ostrom, R.S. cAMP Signaling compartmentation: Adenylyl cyclases as anchors of dynamic signaling complexes. Mol. Pharmacol. 2018, 93, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Spindler, V.; Waschke, J. Beta-adrenergic stimulation contributes to maintenance of endothelial barrier functions under baseline conditions. Microcirculation 2011, 18, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Zagranichnaya, T.; Fu, P.; Alekseeva, E.; Chen, W.; Jacobson, J.R.; Birukov, K.G. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp. Cell Res. 2007, 313, 2504–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Meng, F.; Tian, Y.; Meliton, A.; Sarich, N.; Quilliam, L.A.; Birukov, K.G. Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochim. Biophys. Acta 2015, 1852, 778–791. [Google Scholar] [CrossRef] [Green Version]
- Callesen, K.T.; Yuste-Montalvo, A.; Poulsen, L.K.; Jensen, B.M.; Esteban, V. In vitro investigation of vascular permeability in endothelial cells from human artery, vein and lung microvessels at steady-state and anaphylactic conditions. Biomedicines 2021, 9, 439. [Google Scholar] [CrossRef]
- Sayner, S.L.; Balczon, R.; Frank, D.W.; Cooper, D.M.; Stevens, T. Filamin A is a phosphorylation target of membrane but not cytosolic adenylyl cyclase activity. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L117–L124. [Google Scholar] [CrossRef] [Green Version]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Scalzotto, E.; Mongillo, M.; Pozzan, T. Mitochondrial Ca²⁺ uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 2013, 17, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steegborn, C. Structure, mechanism, and regulation of soluble Adenylyl Cyclases–Similarities and differences to transmembrane Adenylyl Cyclases. Biochim. Biophys. Acta 2014, 1842, 2535–2547. [Google Scholar] [CrossRef] [Green Version]
- Buck, J.; Sinclair, M.L.; Schapal, L.; Cann, M.J.; Levin, L.R. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc. Natl. Acad. Sci. USA 1999, 96, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kostin, S.; Flacke, J.P.; Reusch, H.P.; Ladilov, Y. Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells. J. Biol. Chem. 2009, 284, 14760–14768. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, B.; Brand, S.M.; Brand, E. Aldosterone signaling and soluble adenylyl cyclase—A nexus for the kidney and vascular endothelium. Biochim. Biophys. Acta 2014, 1842, 2601–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mewes, M.; Lenders, M.; Stappers, F.; Scharnetzki, D.; Nedele, J.; Fels, J.; Wedlich-Söldner, R.; Brand, S.M.; Schmitz, B.; Brand, E. Soluble adenylyl cyclase (sAC) regulates calcium signaling in the vascular endothelium. FASEB J. 2019, 33, 13762–13774. [Google Scholar] [CrossRef] [Green Version]
- Khanppnavar, B.; Datta, S. Crystal structure and substrate specificity of ExoY, a unique T3SS mediated secreted nucleotidyl cyclase toxin from Pseudomonas aeruginosa. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2090–2103. [Google Scholar] [CrossRef]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [Green Version]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Lugnier, C.; Meyer, A.; Talha, S.; Geny, B. Cyclic nucleotide phosphodiesterases: New targets in the metabolic syndrome? Pharmacol. Ther. 2020, 208, 107475. [Google Scholar] [CrossRef]
- Lohse, C.; Bock, A.; Maiellaro, I.; Hannawacker, A.; Schad, L.R.; Lohse, M.J.; Bauer, W.R. Experimental and mathematical analysis of cAMP nanodomains. PLoS ONE 2017, 12, e0174856. [Google Scholar] [CrossRef] [Green Version]
- Surapisitchat, J.; Beavo, J.A. Regulation of endothelial barrier function by cyclic nucleo-tides: The role of phosphodiesterases. Handb. Exp. Pharmacol. 2011, 204, 193–210. [Google Scholar] [CrossRef] [Green Version]
- Fertig, B.A.; Baillie, G.S. PDE4-Mediated cAMP signalling. J. Cardiovasc. Dev. Dis. 2018, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Mokra, D.; Mokry, J. Phosphodiesterase inhibitors in acute lung injury: What are the perspectives? Int. J. Mol. Sci. 2021, 22, 1929. [Google Scholar] [CrossRef]
- Creighton, J.; Zhu, B.; Alexeyev, M.; Stevens, T. Spectrin-anchored phosphodiesterase 4D4 restricts cAMP from disrupting microtubules and inducing endothelial cell gap formation. J. Cell Sci. 2008, 121, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Rampersad, S.N.; Ovens, J.D.; Huston, E.; Umana, M.B.; Wilson, L.S.; Netherton, S.J.; Lynch, M.J.; Baillie, G.S.; Houslay, M.D.; Maurice, D.H. Cyclic AMP phosphodiesterase 4D (PDE4D) Tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J. Biol. Chem. 2010, 285, 33614–33622. [Google Scholar] [CrossRef] [Green Version]
- Flemming, S.; Schlegel, N.; Wunder, C.; Meir, M.; Baar, W.; Wollborn, J.; Roewer, N.; Germer, C.-T.; Schick, M.A. Phosphodiesterase 4 inhibition dose dependently stabilizes microvascular barrier functions and microcirculation in a rodent model of polymicrobial sepsis. Shock 2014, 41, 537–545. [Google Scholar] [CrossRef]
- Folcik, V.A.; Smith, T.; O’Bryant, S.; Kawczak, J.A.; Zhu, B.; Sakurai, H.; Kajiwara, A.; Staddon, J.M.; Glabinski, A.; Chernosky, A.L.; et al. Treatment with BBB022A or rolipram stabilizes the blood-brain barrier in experimental autoimmune encephalomyelitis: An additional mechanism for the therapeutic effect of type IV phosphodiesterase inhibitors. J. Neuroimmunol. 1999, 97, 119–128. [Google Scholar] [CrossRef]
- Kraft, P.; Schwarz, T.; Göb, E.; Heydenreich, N.; Brede, M.; Meuth, S.G.; Kleinschnitz, C. The phosphodiesterase-4 inhibitor rolipram protects from ischemic stroke in mice by reducing blood-brain-barrier damage, inflammation and thrombosis. Exp. Neurol. 2013, 247, 80–90. [Google Scholar] [CrossRef]
- Witzenrath, M.; Gutbier, B.; Schmeck, B.; Tenor, H.; Seybold, J.; Kuelzer, R.; Grentzmann, G.; Hatzelmann, A.; van Laak, V.; Tschernig, T.; et al. Phosphodiesterase 2 inhibition diminished acute lung injury in murine pneumococcal pneumonia. Crit. Care Med. 2009, 37, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Horai, S.; Nakagawa, S.; Tanaka, K.; Morofuji, Y.; Couraud, P.O.; Deli, M.A.; Ozawa, M.; Niwa, M. Cilostazol strengthens barrier integrity in brain endothelial cells. Cell. Mol. Neurobiol. 2013, 33, 291–307. [Google Scholar] [CrossRef] [Green Version]
- Poppinga, W.J.; Muñoz-Llancao, P.; González-Billault, C.; Schmidt, M. A-kinase anchoring proteins: cAMP compartmentalization in neurodegenerative and obstructive pulmonary diseases. Br. J. Pharmacol. 2014, 171, 5603–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omar, M.H.; Scott, J.D. AKAP Signaling islands: Venues for precision pharmacology. Trends Pharmacol. Sci. 2020, 41, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Deák, V.A.; Klussmann, E. Pharmacological interference with protein-protein interactions of Akinase anchoring proteins as a strategy for the treatment of disease. Curr. Drug Targets 2016, 17, 1147–1171. [Google Scholar] [CrossRef] [Green Version]
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef]
- Ercu, M.; Klussmann, E. Roles of A-Kinase anchoring proteins and phosphodiesterases in the cardiovascular system. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Sehrawat, S.; Ernandez, T.; Cullere, X.; Takahashi, M.; Ono, Y.; Komarova, Y.; Mayadas, T.N. AKAP9 regulation of microtubule dynamics promotes Epac1-induced endothelial barrier properties. Blood 2011, 117, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.-J.; Kim, K.-W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, J.H.; Kim, W.J.; Lee, H.Y.; Park, J.A.; Lee, S.-W.; Yoon, D.-K.; Kim, H.H.; Chung, H.; Yu, Y.S.; et al. AKAP12 regulates human blood-retinal barrier formation by downregulation of hypoxia-inducible factor-1alpha. J. Neurosci. 2007, 27, 4472–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radeva, M.Y.; Kugelmann, D.; Spindler, V.; Waschke, J. PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS ONE 2014, 9, e106733. [Google Scholar] [CrossRef] [Green Version]
- Colombe, A.S.; Pidoux, G. Cardiac cAMP-PKA signaling compartmentalization in myocardial infarction. Cells 2021, 10, 922. [Google Scholar] [CrossRef]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Bloyd, M.; Stratakis, C.A. PKA functions in metabolism and resistance to obesity: Lessons from mouse and human studies. J. Endocrinol. 2020, 246, R51–R64. [Google Scholar] [CrossRef]
- Taylor, S.S.; Buechler, J.A.; Yonemoto, W. cAMP-dependent protein kinase: Framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem. 1990, 59, 971–1005. [Google Scholar] [CrossRef]
- Haushalter, K.J.; Casteel, D.E.; Raffeiner, A.; Stefan, E.; Patel, H.H.; Taylor, S.S. Phosphorylation of protein kinase A (PKA) regulatory subunit RIα by protein kinase G (PKG) primes PKA for catalytic activity in cells. J. Biol. Chem. 2018, 293, 4411–4421. [Google Scholar] [CrossRef] [Green Version]
- Cong, X.; Kong, W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell. Signal. 2020, 66, 109485. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, L.; Creighton, J.; Alexeyev, M.; Strada, S.J.; Stevens, T. Protein kinase A phosphorylation of tau-serine 214 reorganizes microtubules and disrupts the endothelial cell barrier. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L493–L501. [Google Scholar] [CrossRef] [Green Version]
- Roscioni, S.S.; Elzinga, C.R.; Schmidt, M. Epac: Effectors and biological functions. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 377, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [Green Version]
- Robichaux, W.G., 3rd; Cheng, X. Intracellular cAMP sensor EPAC: Physiology, pathophysiology, and therapeutics development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef]
- García-Morales, V.; Cuíñas, A.; Elíes, J.; Campos-Toimil, M. PKA and Epac activation mediates cAMP-induced vasorelaxation by increasing endothelial NO production. Vascul. Pharmacol. 2014, 60, 95–101. [Google Scholar] [CrossRef]
- García-Morales, V.; Luaces-Regueira, M.; Campos-Toimil, M. The cAMP effectors PKA and Epac activate endothelial NO synthase through PI3K/Akt pathway in human endothelial cells. Biochem. Pharmacol. 2017, 145, 94–101. [Google Scholar] [CrossRef]
- Cuíñas, A.; García-Morales, V.; Viña, D.; Gil-Longo, J.; Campos-Toimil, M. Activation of PKA and Epac proteins by cyclic AMP depletes intracellular calcium stores and reduces calcium availability for vasoconstriction. Life Sci. 2016, 155, 102–109. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Luchowska-Stańska, U.; Morgan, D.; Yarwood, S.J.; Barker, G. Selective small-molecule EPAC activators. Biochem. Soc. Trans. 2019, 47, 1415–1427. [Google Scholar] [CrossRef] [Green Version]
- Parnell, E.; Smith, B.O.; Palmer, T.M.; Terrin, A.; Zaccolo, M.; Yarwood, S.J. Regulation of the inflammatory response of vascular endothelial cells by EPAC1. Br. J. Pharmacol. 2012, 166, 434–446. [Google Scholar] [CrossRef] [Green Version]
- García-Ponce, A.; Schuster, K.; Døskeland, S.O.; Reed, R.K.; Curry, F.E.; Waschke, J.; Radeva, M.Y. Epac1 is crucial for maintenance of endothelial barrier function through A mechanism partly independent of Rac1. Cells 2020, 9, 2170. [Google Scholar] [CrossRef]
- Garcia-Morales, V.; Friedrich, J.; Jorna, L.M.; Campos-Toimil, M.; Hammes, H.P.; Schmidt, M.; Krenning, G. The microRNA-7-mediated reduction in EPAC-1 contributes to vascular endothelial permeability and eNOS uncoupling in murine experimental retinopathy. Acta Diabetol. 2017, 54, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Kopperud, R.K.; Brekke Rygh, C.; Karlsen, T.V.; Krakstad, C.; Kleppe, R.; Hoivikl, E.A. Increased microvascular permeability in mice lacking Epac1 (Rapgef3). Acta Physiol. 2017, 219, 441–452. [Google Scholar] [CrossRef]
- Wang, X.; Song, S.; Hu, Z.; Zhang, Z.; Li, Y.; Yan, C. Activation of Epac alleviates inflammation and vascular leakage in LPS-induced acute murine lung injury. Biomed. Pharmacother. 2017, 96, 1127–1136. [Google Scholar] [CrossRef]
- Kakogiannos, N.; Ferrari, L.; Giampietro, C.; Scalise, A.A.; Maderna, C.; Ravà, M.; Taddei, A.; Lampugnani, M.G.; Pisati, F.; Malinverno, M.; et al. JAM-A Acts via C/EBP-α to promote claudin-5 expression and enhance endothelial barrier function. Circ. Res. 2020, 127, 1056–1073. [Google Scholar] [CrossRef]
- Ramos, C.J.; Antonetti, D.A. The role of small GTPases and EPAC-Rap signaling in the regulation of the blood-brain and blood-retinal barriers. Tissue Barriers 2017, 5, e1339768. [Google Scholar] [CrossRef] [Green Version]
- Parnell, E.; Yarwood, S.J. Interactions between Epac1 and ezrin in the control of endothelial barrier function. Biochem. Soc. Trans. 2014, 42, 274–278. [Google Scholar] [CrossRef]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.E.; Adderley, S.P.; Breslin, J.W. Activation of RhoA, but not Rac1, mediates early stages of S1P-induced endothelial barrier enhancement. PLoS ONE 2016, 11, e0155490. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.J.; Lin, C.; Liu, X.; Antonetti, D.A. The EPAC-Rap1 pathway prevents and reverses cytokine-induced retinal vascular permeability. J. Biol. Chem. 2018, 293, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 2018, 42, 281–291. [Google Scholar] [CrossRef]
- Yanai, S.; Toyohara, J.; Ishiwata, K.; Ito, H.; Endo, S. Long-term cilostazol administration ameliorates memory decline in senescence-accelerated mouse prone 8 (SAMP8) through a dual effect on cAMP and blood-brain barrier. Neuropharmacology 2017, 116, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Welter, J.; Pfitzer, G.; Grisk, O.; Hescheler, J.; Lubomirov, L.T. Epac-mediated relaxation in murine basilar arteries depends on membrane permeability of cyclic nucleotide analogues and endothelial aging. Gen. Physiol. Biophys. 2020, 39, 157–168. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viña, D.; Seoane, N.; Vasquez, E.C.; Campos-Toimil, M. cAMP Compartmentalization in Cerebrovascular Endothelial Cells: New Therapeutic Opportunities in Alzheimer’s Disease. Cells 2021, 10, 1951. https://doi.org/10.3390/cells10081951
Viña D, Seoane N, Vasquez EC, Campos-Toimil M. cAMP Compartmentalization in Cerebrovascular Endothelial Cells: New Therapeutic Opportunities in Alzheimer’s Disease. Cells. 2021; 10(8):1951. https://doi.org/10.3390/cells10081951
Chicago/Turabian StyleViña, Dolores, Nuria Seoane, Elisardo C. Vasquez, and Manuel Campos-Toimil. 2021. "cAMP Compartmentalization in Cerebrovascular Endothelial Cells: New Therapeutic Opportunities in Alzheimer’s Disease" Cells 10, no. 8: 1951. https://doi.org/10.3390/cells10081951
APA StyleViña, D., Seoane, N., Vasquez, E. C., & Campos-Toimil, M. (2021). cAMP Compartmentalization in Cerebrovascular Endothelial Cells: New Therapeutic Opportunities in Alzheimer’s Disease. Cells, 10(8), 1951. https://doi.org/10.3390/cells10081951