
A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics

 , , ,
, , ,
Abstract
:1. Introduction
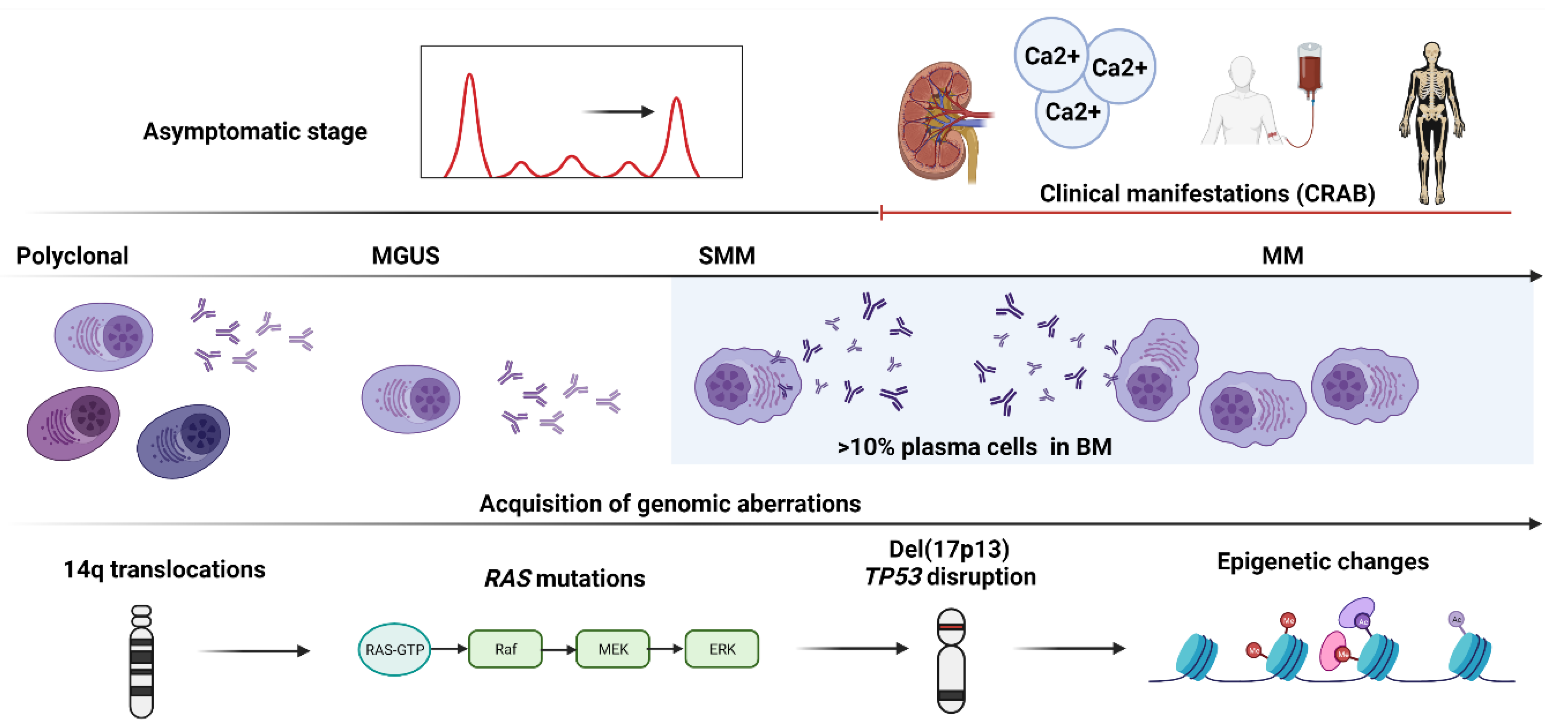
2. Genetic and Cytogenetic Abnormalities: A Long Way from MGUS to MM Progression
2.1. Primary Events Driving MGUS Progression to MM
2.2. Secondary Events Driving MGUS Progression to MM
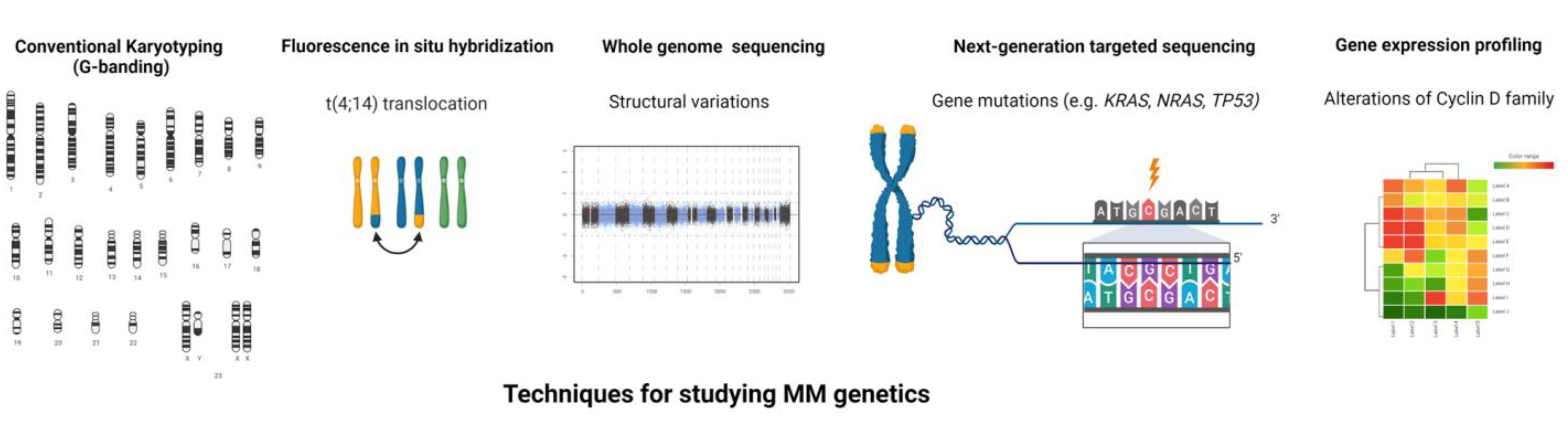
3. Fluorescent In-Situ Hybridization (FISH)
4. Current Data on Co-Existing Abnormalities
5. Gene Expression Profiling
Brief Summary of Genomic-Based Risk Stratification Studies
6. DNA Sequencing and Data on Mutations
7. Concept of Clonal and Subclonal Evolution
8. Evolving Therapeutic Implications
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- SEER Cancer Statistics Review. 1975–2017. SEER. Available online: https://seer.cancer.gov/csr/1975_2017/index.html (accessed on 7 May 2021).
- Goldschmidt, H.; Lokhorst, H.M.; Mai, E.K.; Van Der Holt, B.; Blau, I.W.; Zweegman, S.; Weisel, K.C.; Vellenga, E.; Pfreundschuh, M.; Kersten, M.J.; et al. Bortezomib before and after high-dose therapy in myeloma: Long-term results from the phase III HOVON-65/GMMG-HD4 trial. Leukemia 2018, 32, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.K.; Barlogie, B.; Van Rhee, F.; Zangari, M.; Walker, B.A.; Rosenthal, A.; Schinke, C.; Thanendrarajan, S.; Davies, F.; Hoering, A.; et al. Long-term outcomes after autologous stem cell transplantation for multiple myeloma. Blood Adv. 2020, 4, 422–431. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.C.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia 2017, 31, 1915–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalat, R.; Munshi, N.C. Genomic heterogeneity in multiple myeloma. Curr. Opin. Genet. Dev. 2015, 30, 56–65. [Google Scholar] [CrossRef]
- Yle, R.O.A.K.; Therneau, T.M.; Rajkumar, S.V.; Fford, J.A.R.O.; Arson, D.I.R.L.; Plevak, M.F.; Melton, L.J. A Long-Term Study of Prognosis in Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, S.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Keats, J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef]
- Walker, B.; Wardell, C.; Melchor, L.; Hulkki, S.; Potter, N.E.; Johnson, D.C.; Fenwick, K.; Kozarewa, I.; Gonzalez, D.; Lord, C.; et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 2012, 120, 1077–1086. [Google Scholar] [CrossRef]
- Egan, J.B.; Shi, C.-X.; Tembe, W.; Christoforides, A.; Kurdoglu, A.; Sinari, S.; Middha, S.; Asmann, Y.; Schmidt, J.; Braggio, E.; et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012, 120, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Hultcrantz, M.; Yellapantula, V.; Rustad, E.H. Genomic profiling of multiple myeloma: New insights and modern technologies. Best Pr. Res. Clin. Haematol. 2020, 33, 101153. [Google Scholar] [CrossRef]
- Szalat, R.; Avet-Loiseau, H.; Munshi, N.C. Gene Expression Profiles in Myeloma: Ready for the Real World? Clin. Cancer Res. 2016, 22, 5434–5442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, N.; Rajkumar, S.V.; Greipp, P.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Baughn, L.B.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; et al. Cytogenetic abnormalities in multiple myeloma: Association with disease characteristics and treatment response. Blood Cancer J. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Prideaux, S.M.; O’Brien, E.C.; Chevassut, T.J. The Genetic Architecture of Multiple Myeloma. Adv. Hematol. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.; Haaber, J.; Dahl, I.M.; Knudsen, L.M.; Kerndrup, G.B.; Lodahl, M.; Johnsen, H.E.; Kuehl, M. Identification of translocation products but not K-RAS mutations in memory B cells from patients with multiple myeloma. Haematologica 2010, 95, 1730–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Ordoñez, R.; Kulis, M.; Russiñol, N.; Chapaprieta, V.; Carrasco-Leon, A.; García-Torre, B.; Charalampopoulou, S.; Clot, G.; Beekman, R.; Meydan, C.; et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020, 30, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, H.; Campo, C.; Weinhold, N.; Filho, M.I.D.S.; Pour, L.; Gregora, E.; Vodička, P.; Vodickova, L.; Hoffmann, P.; Nöthen, M.M.; et al. Genomewide association study on monoclonal gammopathy of unknown significance (MGUS). Eur. J. Haematol. 2017, 99, 70–79. [Google Scholar] [CrossRef]
- Broderick, P.; Chubb, D.; Johnson, D.C.; Weinhold, N.; Försti, A.; Lloyd, A.; Olver, B.; Ma, Y.P.; Dobbins, S.E.; Walker, B.; et al. Common variation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nat. Genet. 2011, 44, 58–61. [Google Scholar] [CrossRef] [Green Version]
- Beksac, M.; Gragert, L.; Fingerson, S.; Maiers, M.; Zhang, M.-J.; Albrecht, M.; Zhong, X.; Cozen, W.; Dispenzieri, A.; Lonial, S.; et al. HLA polymorphism and risk of multiple myeloma. Leukemia 2016, 30, 2260–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brousseau, M.; Leleu, X.; Gerard, J.; Gastinne, T.; Godon, A.; Genevieve, F.; Dib, M.; Lai, J.-L.; Facon, T.; Zandecki, M. Hyperdiploidy Is a Common Finding in Monoclonal Gammopathy of Undetermined Significance and Monosomy 13 Is Restricted to These Hyperdiploid Patients. Clin. Cancer Res. 2007, 13, 6026–6031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, W.J. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood 2005, 106, 2156–2161. [Google Scholar] [CrossRef] [Green Version]
- Magrangeas, F.; Lodé, L.; Wuillème, S.; Minvielle, S.; Avet-Loiseau, H. Genetic heterogeneity in multiple myeloma. Leukemia 2004, 19, 191–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Fonseca, R.; Ketterling, R.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood 2012, 119, 2100–2105. [Google Scholar] [CrossRef] [Green Version]
- Chng, W.J.; Glebov, O.; Bergsagel, P.; Kuehl, W.M. Genetic events in the pathogenesis of multiple myeloma. Best Pr. Res. Clin. Haematol. 2007, 20, 571–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef] [Green Version]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef]
- Bourguet, C.C.; Logue, E.E. Antigenic stimulation and multiple myeloma. A prospective study. Cancer 1993, 72, 2148–2154. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C. Inflammation and multiple myeloma: The Toll connection. Leukemia 2006, 20, 937–938. [Google Scholar] [CrossRef]
- Bagratuni, T.; Sklirou, A.D.; Kastritis, E.; Liacos, C.I.; Spilioti, C.; Eleutherakis-Papaiakovou, E.; Kanellias, N.; Gavriatopoulou, M.; Terpos, E.; Trougakos, I.P.; et al. Toll-Like Receptor 4 Activation Promotes Multiple Myeloma Cell Growth and Survival Via Suppression of The Endoplasmic Reticulum Stress Factor Chop. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Fenton, J.A.; Ashcroft, J.; English, A.; Jones, R.A.; Richards, S.J.; Pratt, G.; Owen, R.; Davies, F.E.; Child, J.A.; et al. The interleukin-6 receptor alpha-chain (CD126) is expressed by neoplastic but not normal plasma cells. Blood 2000, 96, 3880–3886. [Google Scholar] [CrossRef]
- Puthier, D.; Derenne, S.; Barille-Nion, S.; Moreau, P.; Harousseau, J.-L.; Bataille, R.; Amiot, M. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br. J. Haematol. 1999, 107, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. The screening imperative for multiple myeloma. Nat. Cell Biol. 2020, 587, S63. [Google Scholar] [CrossRef]
- Pang, L.; Rajkumar, S.V.; Kapoor, P.; Buadi, F.; Dispenzieri, A.; Gertz, M.; Lacy, M.; Kyle, R.; Kumar, S. Prognosis of young patients with monoclonal gammopathy of undetermined significance (MGUS). Blood Cancer J. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Landgren, O.; Hofmann, J.N.; McShane, C.M.; Santo, L.; Hultcrantz, M.; Korde, N.; Mailankody, S.; Kazandjian, D.; Murata, K.; Thoren, K.; et al. Association of Immune Marker Changes with Progression of Monoclonal Gammopathy of Undetermined Significance to Multiple Myeloma. JAMA Oncol. 2019, 5, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Dib, A.; Gabrea, A.; Glebov, O.K.; Bergsagel, P.L.; Kuehl, W.M. Characterization of MYC Translocations in Multiple Myeloma Cell Lines. J. Natl. Cancer Inst. Monogr. 2008, 2008, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Misund, K.; Network, M.C.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020, 34, 322–326. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Gerson, F.; Magrangeas, F.; Minvielle, S.; Harousseau, J.-L.; Bataille, R. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001, 98, 3082–3086. [Google Scholar] [CrossRef] [Green Version]
- Gabrea, A.; Martelli, M.L.; Qi, Y.; Roschke, A.; Barlogie, B.; Shaughnessy, J.D., Jr.; Sawyer, J.R.; Kuehl, W.M. Secondary genomic rearrangements involving immunoglobulin or MYC loci show similar prevalences in hyperdiploid and nonhyperdiploid myeloma tumors. Genes Chromosom. Cancer 2008, 47, 573–590. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, N.; Baughn, L.B.; Rajkumar, S.V.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; et al. Implications of MYC Rearrangements in Newly Diagnosed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 6581–6588. [Google Scholar] [CrossRef]
- Xiong, W.; Wu, X.; Starnes, S.; Johnson, S.K.; Haessler, J.; Wang, S.; Chen, L.; Barlogie, B.; Shaughnessy, J.J.D.; Zhan, F. An analysis of the clinical and biologic significance of TP53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood 2008, 112, 4235–4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, R.; Barlogie, B.; Bataille, R.; Bastard, C.; Bergsagel, P.L.; Chesi, M.; Davies, F.; Drach, J.; Greipp, P.R.; Kirsch, I.R.; et al. Genetics and Cytogenetics of Multiple Myeloma. Cancer Res. 2004, 64, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Sloan, S.; Li, D.; Stewart, A.K. Multiple myeloma involving central nervous system: High frequency of chromosome 17p13.1 (p53) deletions. Br. J. Haematol. 2004, 127, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, A.; Ortiz, M.; Blecua, P.; Towfic, F.; Corre, J.; Serbina, N.V.; Flynt, E.; Yu, Z.; Yang, Z.; Palumbo, A.; et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood 2019, 133, 1217–1221. [Google Scholar] [CrossRef] [Green Version]
- Kalakonda, N.; Rothwell, D.; Scarffe, J.H.; Norton, J.D. Detection of N-Ras codon 61 mutations in subpopulations of tumor cells in multiple myeloma at presentation. Blood 2001, 98, 1555–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, W.J.; Gonzalez-Paz, N.; Price-Troska, T.; Jacobus, S.; Rajkumar, S.V.; Oken, M.M.; Kyle, R.A.; Henderson, K.J.; Van Wier, S.; Greipp, P.; et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia 2008, 22, 2280–2284. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, G.; Lichter, D.I.; Di Bacco, A.; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent Engagement of the Classical and Alternative NF-κB Pathways by Diverse Genetic Abnormalities in Multiple Myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demchenko, Y.N.; Kuehl, W.M. A critical role for the NFkB pathway in multiple myeloma. Oncotarget 2010, 1, 59–68. [Google Scholar] [CrossRef]
- Keats, J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous Mutations Activate the Noncanonical NF-κB Pathway in Multiple Myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, M.; Daggett, J.; Bender, T.; Kuehl, W.; Bergsagel, P.; Williams, M. Frequent inactivation of the cyclin-dependent kinase inhibitor p18 by homozygous deletion in multiple myeloma cell lines: Ectopic p18 expression inhibits growth and induces apoptosis. Leukemia 2002, 16, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, A.; Tu, Y.; Fady, C.; Vescio, R.; Berenson, J. Interleukin-6 Inhibits Apoptosis of Malignant Plasma Cells. Cell. Immunol. 1995, 162, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Ong, F.; Van Nieuwkoop, J.A.; De Groot-Swings, G.M.; Hermans, J.; Harvey, M.S.; Kluin, P.M.; Kluin-Nelemans, J.C. Bcl-2 protein expression is not related to short survival in multiple myeloma. Leukemia 1995, 9, 1282–1284. [Google Scholar] [PubMed]
- Landowski, T.H.; Qu, N.; Buyuksal, I.; Painter, J.S.; Dalton, W.S. Mutations in the Fas antigen in patients with multiple myeloma. Blood 1997, 90, 4266–4270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.; Chung, Y.; Lo, K.W.; Wickham, N.; Lee, J.; Huang, D. Frequent Hypermethylation of p16 and p15 Genes in Multiple Myeloma. Blood 1997, 89, 2500–2506. [Google Scholar] [CrossRef]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Muddasani, R.; Orlowski, R.; Abruzzo, L.V.; Qazilbash, M.H.; You, M.J.; Wang, Y.; Zhao, M.; Chen, S.; Glitza, I.C.; et al. Plasma Cell Enrichment Enhances Detection of High-Risk Cytogenomic Abnormalities by Fluorescence In Situ Hybridization and Improves Risk Stratification of Patients with Plasma Cell Neoplasms. Arch. Pathol. Lab. Med. 2013, 137, 625–631. [Google Scholar] [CrossRef]
- Pozdnyakova, O.; Crowley-Larsen, P.; Zota, V.; Wang, S.A.; Miron, P.M. Interphase FISH in plasma cell dyscrasia: Increase in abnormality detection with plasma cell enrichment. Cancer Genet. Cytogenet. 2009, 189, 112–117. [Google Scholar] [CrossRef]
- Ma, E.S.K.; Wang, C.L.N.; Wong, A.T.C.; Choy, G.; Chan, T.L. Target fluorescence in-situ hybridization (Target FISH) for plasma cell enrichment in myeloma. Mol. Cytogenet. 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [Green Version]
- Perrot, A.; Lauwers-Cances, V.; Tournay, E.; Hulin, C.; Chretien, M.-L.; Royer, B.; Dib, M.; Decaux, O.; Jaccard, A.; Belhadj, K.; et al. Development and Validation of a Cytogenetic Prognostic Index Predicting Survival in Multiple Myeloma. J. Clin. Oncol. 2019, 37, 1657–1665. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.; Ashby, C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Baysal, M.; Demirci, U.; Umit, E.; Kirkizlar, H.O.; Atli, E.I.; Gurkan, H.; Gulsaran, S.K.; Bas, V.; Mail, C.; Demir, A.M. Concepts of Double Hit and Triple Hit Disease in Multiple Myeloma, Entity and Prognostic Significance. Sci. Rep. 2020, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J.J. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Lida, S.; Hanamura, I.; Kato, M.; Banno, S.; Ishida, T.; Kusumoto, S.; Takeuchi, G.; Miwa, H.; Nitta, M.; et al. Frequent occurrence of CCND1 deregulation in patients with early stages of plasma cell dyscrasia. Cancer Sci. 2003, 94, 350–354. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Molecular Pathogenesis and a Consequent Classification of Multiple Myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broyl, A.; Hose, D.; Lokhorst, H.; De Knegt, Y.; Peeters, J.; Jauch, A.; Bertsch, U.; Buijs, A.; Stevens-Kroef, M.; Beverloo, H.B.; et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 2010, 116, 2543–2553. [Google Scholar] [CrossRef] [Green Version]
- Shaughnessy, J.D.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2006, 109, 2276–2284. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, R.; Broyl, A.; De Knegt, Y.; Van Vliet, M.H.; Van Beers, E.H.; Van Der Holt, B.; El Jarari, L.; Mulligan, G.; Gregory, W.; Morgan, G.; et al. A gene expression signature for high-risk multiple myeloma. Leukemia 2012, 26, 2406–2413. [Google Scholar] [CrossRef] [Green Version]
- Decaux, O.; Lodé, L.; Magrangeas, F.; Charbonnel, C.; Gouraud, W.; Jézéquel, P.; Attal, M.; Harousseau, J.-L.; Moreau, P.; Bataille, R.; et al. Prediction of Survival in Multiple Myeloma Based on Gene Expression Profiles Reveals Cell Cycle and Chromosomal Instability Signatures in High-Risk Patients and Hyperdiploid Signatures in Low-Risk Patients: A Study of the Intergroupe Francophone du Myélome. J. Clin. Oncol. 2008, 26, 4798–4805. [Google Scholar] [CrossRef] [PubMed]
- Dickens, N.; Walker, B.A.; Leone, P.E.; Johnson, D.C.; Brito, J.L.; Zeisig, A.; Jenner, M.; Boyd, K.; Gonzalez, D.; Gregory, W.M.; et al. Homozygous Deletion Mapping in Myeloma Samples Identifies Genes and an Expression Signature Relevant to Pathogenesis and Outcome. Clin. Cancer Res. 2010, 16, 1856–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2006, 109, 3177–3188. [Google Scholar] [CrossRef] [PubMed]
- Hose, D.; Rème, T.; Hielscher, T.; Moreaux, J.; Messner, T.; Seckinger, A.; Benner, A.; Shaughnessy, J.D.; Barlogie, B.; Zhou, Y.; et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica 2010, 96, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V. Sequencing of myeloma therapy: Finding the right path among many standards. Hematol. Oncol. 2021, 39, 68–72. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nat. Cell Biol. 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Biancon, G.; Moarii, M.; Gimondi, S.; Li, Y.; De Philippis, C.; Maura, F.; Sathiaseelan, V.; Tai, Y.-T.; Mudie, L.; et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 2018, 32, 2604–2616. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.; Boyle, E.M.; Wardell, C.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Hoang, P.H.; Dobbins, S.E.; Cornish, A.J.; Chubb, D.; Law, P.; Kaiser, M.; Houlston, R.S. Whole-genome sequencing of multiple myeloma reveals oncogenic pathways are targeted somatically through multiple mechanisms. Leukemia 2018, 32, 2459–2470. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Smadbeck, J.B.; Abdallah, N.; Zepeda-Mendoza, C.; Binder, M.; Pearce, K.E.; Asmann, Y.W.; Peterson, J.F.; Ketterling, R.P.; Greipp, P.T.; et al. The prognostic role of MYC structural variants identified by NGS and FISH in multiple myeloma. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.; Van Loo, P.; Alexandrov, L.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Rustad, E.H.; Yellapantula, V.; Łuksza, M.; Hoyos, D.; MacLachlan, K.H.; Diamond, B.T.; Greenbaum, B.D.; Morgan, G.; Lesokhin, A.; et al. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia 2020, 34, 1476–1480. [Google Scholar] [CrossRef]
- Jänne, P.A.; Shaw, A.T.; Pereira, J.R.; Jeannin, G.; Vansteenkiste, J.; Barrios, C.; Franke, F.A.; Grinsted, L.; Zazulina, V.; Smith, P.; et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013, 14, 38–47. [Google Scholar] [CrossRef]
- Adjei, A.A.; Cohen, R.B.; Franklin, W.; Morris, C.; Wilson, D.; Molina, J.R.; Hanson, L.J.; Gore, L.; Chow, L.; Leong, S.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the Oral, Small-Molecule Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor AZD6244 (ARRY-142886) in Patients with Advanced Cancers. J. Clin. Oncol. 2008, 26, 2139–2146. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Klimek, V.M.; Gaskell, A.A.; Viale, A.; Cheng, D.; Kim, E.; Rampal, R.; Bluth, M.; Harding, J.J.; Callahan, M.K.; et al. Efficacy of Intermittent Combined RAF and MEK Inhibition in a Patient with Concurrent BRAF- and NRAS-Mutant Malignancies. Cancer Discov. 2014, 4, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Baughn, L.B.; Di Liberto, M.; Wu, K.; Toogood, P.L.; Louie, T.; Gottschalk, R.; Niesvizky, R.; Cho, H.; Ely, S.; Moore, M.A.; et al. A Novel Orally Active Small Molecule Potently Induces G1 Arrest in Primary Myeloma Cells and Prevents Tumor Growth by Specific Inhibition of Cyclin-Dependent Kinase 4/6. Cancer Res. 2006, 66, 7661–7667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Pont, S.R.; Cleynen, A.; Fontan, C.; Attal, M.; Munshi, N.; Corre, J.; Avet-Loiseau, H. Genomics of Multiple Myeloma. J. Clin. Oncol. 2017, 35, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Bahlis, N.J. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012, 120, 927–928. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, Y.; Kikuchi, J. Molecular basis of clonal evolution in multiple myeloma. Int. J. Hematol. 2020, 111, 496–511. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Cardona-Benavides, I.J.; de Ramón, C.; Gutiérrez, N.C. Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells 2021, 10, 336. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Nakamura, R.; Lear, S.; Wilson, D.; Koeppen, H.; Vaze, A.; Trudel, M.S.; Spencer, A.; Harrison, M.M.; Cohen, A.D.; Fine, B.M.; et al. Early Pharmacodynamic Changes in T-Cell Activation, Proliferation, and Cytokine Production Confirm the Mode of Action of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 14–15. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, M.M.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A.; Berdeja, J.G.; Laubach, J.P.; Li, M.; Choeurng, V.; et al. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Kumar, S.; Rajkumar, S.V. BiTEing the Tumor. J. Clin. Oncol. 2020, 38, 2077–2079. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| I | Serum β2 microglobulin < 3.5 mg/L |
| Serum albumin ≥ 3.5 g/dL | |
| Normal serum lactate dehydrogenase | |
| No high-risk cytogenetics * | |
| II | Not meeting criteria for Stages I or III |
| III | Serum β2 microglobulin > 5.5 mg/L and at least one of the following: |
| -Elevated serum lactate dehydrogenase | |
| -High-risk cytogenetics * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awada, H.; Thapa, B.; Awada, H.; Dong, J.; Gurnari, C.; Hari, P.; Dhakal, B. A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics. Cells 2021, 10, 1961. https://doi.org/10.3390/cells10081961
Awada H, Thapa B, Awada H, Dong J, Gurnari C, Hari P, Dhakal B. A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics. Cells. 2021; 10(8):1961. https://doi.org/10.3390/cells10081961
Chicago/Turabian StyleAwada, Hassan, Bicky Thapa, Hussein Awada, Jing Dong, Carmelo Gurnari, Parameswaran Hari, and Binod Dhakal. 2021. "A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics" Cells 10, no. 8: 1961. https://doi.org/10.3390/cells10081961
APA StyleAwada, H., Thapa, B., Awada, H., Dong, J., Gurnari, C., Hari, P., & Dhakal, B. (2021). A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics. Cells, 10(8), 1961. https://doi.org/10.3390/cells10081961