
Hematopoietic Dysfunction during Graft-Versus-Host Disease: A Self-Destructive Process?


{kind=link}
{kind=link}
{kind=link}
Abstract
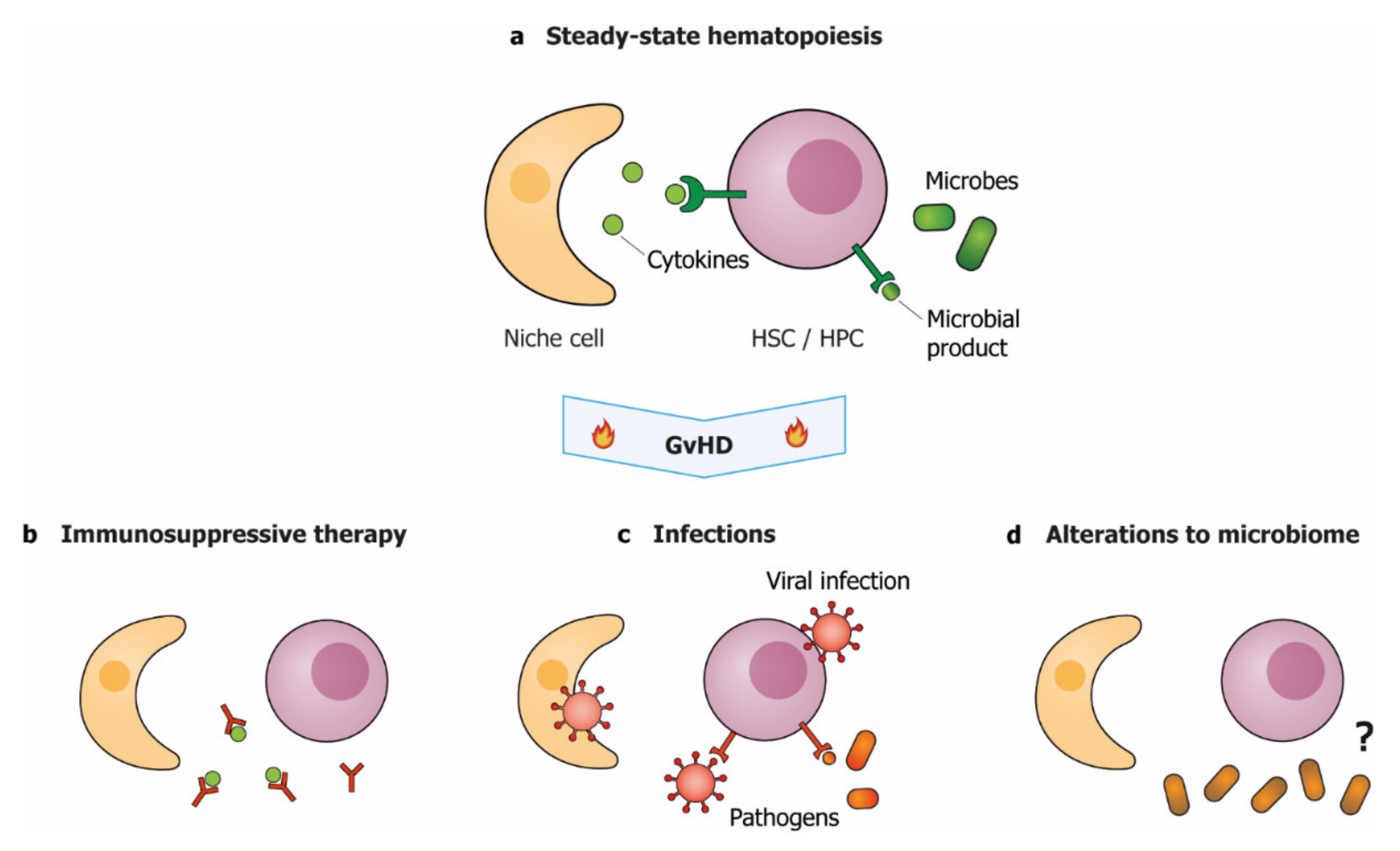
:1. Introduction
2. Bone Marrow Targets of GvHD: Hematopoietic Stem Cells and Their Niche
2.1. GvHD Reduces HSPC Number and Function
2.2. GvHD Damages BM Niche Components, Impairing Overall Niche Function
3. Mechanisms of GvHD-Related Hematopoietic Dysfunction
3.1. Fas-Mediated Cytotoxicity Is Important for GvHD-Mediated Hematopoietic Dysfunction
3.2. Cytokines Induce Cellular Destruction and Dysfunction of Niche and HSPCs
4. Clinical Factors Associated with GvHD Impair Hematopoietic Function
4.1. Immune Suppressive Therapy May Aggravate Hematopoietic Dysfunction
4.1.1. Corticosteroids
4.1.2. IL-2 Inhibitors
4.1.3. TNFα Inhibitors
4.1.4. IFN-γ Inhibitors
4.2. Infections and Viral Reactivations Exacerbate Hematopoietic Dysfunction
4.3. Antibiotic, Antifungal and Antiviral Therapy Can Directly Impair Hematopoiesis
4.4. Disruption of the Microbiome May Be Detrimental for Hematopoietic Function
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Passweg, J.R.; Baldomero, H.; Bader, P.; Bonini, C.; Cesaro, S.; Dreger, P.; Duarte, R.F.; Dufour, C.; Kuball, J.; Farge-Bancel, D.; et al. Hematopoietic stem cell transplantation in Europe 2014: More than 40 000 transplants annually. Bone Marrow Transplant. 2016, 51, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Lee, J.H.; Choi, S.J.; Kim, S.; Seol, M.; Lee, Y.S.; Kim, W.K. Failure of trilineage blood cell reconstitution after initial neutrophil engraftment in patients undergoing allogeneic hematopoietic cell transplantation–frequency and outcomes. Bone Marrow Transplant. 2004, 33, 729–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogonek, J.; Juric, M.K.; Ghimire, S.; Varanasi, P.R.; Holler, E.; Greinix, H.; Weissinger, E. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 2016, 7, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartelink, I.H.; Belitser, S.V.; Knibbe, C.A.J.; Danhof, M.; de Pagter, A.J.; Egberts, T.C.G.; Boelens, J.J. Immune reconstitution kinetics as an early predictor for mortality using various hematopoietic stem cell sources in children. Biol. Blood Marrow Transplant. 2013, 19, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Blazar, B.R.; Murphy, W.J.; Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 2012, 12, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlomchik, W.D. Graft-versus-host disease. Nat. Rev. Immunol. 2007, 7, 340–352. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Reddy, P.; Ferrara, J.L.M. Immunobiology of acute graft-versus-host disease. Blood Rev. 2003, 17, 187–194. [Google Scholar] [CrossRef]
- Wu, S.R.; Reddy, P. Tissue tolerance: A distinct concept to control acute GVHD severity. Blood 2017, 129, 1747–1752. [Google Scholar] [CrossRef]
- Zeiser, R.; Blazar, B.R. Pathophysiology of chronic graft-versus-host disease and therapeutic targets. N. Engl. J. Med. 2017, 377, 2565–2579. [Google Scholar] [CrossRef]
- Jagasia, M.H.; Greinix, H.T.; Arora, M.; Williams, K.M.; Wolff, D.; Cowen, E.W.; Palmer, J.; Weisdorf, D.; Treister, N.S.; Cheng, G.-S.; et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 diagnosis and staging working group report. Biol. Blood Marrow Transplant. 2015, 21, 389–401.e1. [Google Scholar] [CrossRef] [Green Version]
- Von Bonin, M.; Bornhäuser, M.; Bornhaüser, B. Concise review: The bone marrow niche as a target of graft versus host disease. Stem Cells 2014, 32, 1420–1428. [Google Scholar] [CrossRef]
- Szyska, M.; Na, I.K. Bone marrow GvHD after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 2016, 7, 118. [Google Scholar] [CrossRef] [Green Version]
- Dominietto, A.; Raiola, A.M.; Van Lint, M.T.; Lamparelli, T.; Gualandi, F.; Berisso, G.; Bregante, S.; Frassoni, F.; Casarino, L.; Verdiani, S.; et al. Factors influencing haematological recovery after allogeneic haemopoietic stem cell transplants: Graft-versus-host disease, donor type, cytomegalovirus infections and cell dose. Br. J. Haematol. 2001, 112, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Peralvo, J.; Bacigalupo, A.; Pittaluga, P.A.; Occhini, D.; Van Lint, M.T.; Frassoni, F.; Nardelli, E.; Transino, A.; Pantarotto, M.; Marmout, A.M. Poor graft function associated with graft-versus-host disease after allogeneic marrow transplantation. Bone Marrow Transplant. 1987, 2, 279–285. [Google Scholar]
- Bruno, B.; Gooley, T.; Sullivan, K.M.; Davis, C.; Bensinger, W.I.; Storb, R.; Nash, R.A. Secondary failure of platelet recovery after hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2001, 7, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Hu, X.; Cheng, H.; Pang, Y.; Wang, L.; Zou, L.; Xu, S.; Zhuang, X.; Jiang, C.; Yuan, W.; et al. Graft-versus-host disease causes broad suppression of hematopoietic primitive cells and blocks megakaryocyte differentiation in a murine model. Biol. Blood Marrow Transplant. 2014, 20, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Mensen, A.; Jöhrens, K.; Anagnostopoulos, I.; Demski, S.; Oey, M.; Stroux, A.; Hemmati, P.; Westermann, J.; Blau, O.; Wittenbecher, F.; et al. Bone marrow T-cell infiltration during acute GVHD is associated with delayed B-cell recovery and function after HSCT. Blood 2014, 124, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Storek, J.; Wells, D.; Dawson, M.A.; Storer, B.; Maloney, D.G. Factors influencing B lymphopoiesis after allogeneic hematopoietic cell transplantation. Blood 2001, 98, 489–491. [Google Scholar] [CrossRef] [Green Version]
- Krenger, W.; Holländer, G.A. The immunopathology of thymic GVHD. Semin. Immunopathol. 2008, 30, 439–456. [Google Scholar] [CrossRef]
- Kuzmina, Z.; Eder, S.; Bohm, A.; Pernicka, E.; Vormittag, L.; Kalhs, P.; Petkov, V.; Stary, G.; Nepp, J.; Knobler, R.; et al. Significantly worse survival of patients with NIH-defined chronic graft-versus-host disease and thrombocytopenia or progressive onset type: Results of a prospective study. Leukemia 2011, 26, 746–756. [Google Scholar] [CrossRef]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiser, R.; Blazar, B.R. Acute graft-versus-host disease—Biologic process, prevention, and therapy. N. Engl. J. Med. 2017, 377, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Cheshier, S.H.; Prohaska, S.S.; Weissman, I.L. The effect of bleeding on hematopoietic stem cell cycling and self-renewal. Stem Cells Dev. 2007, 16, 707–718. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Mori, T.; Nishimura, T.; Ikeda, Y.; Hotta, T.; Yagita, H.; Ando, K. Involvement of FAS-mediated apoptosis in the hematopoietic progenitor cells of graft-versus-host reaction-associated myelosuppression. Blood 1998, 92, 101–107. [Google Scholar] [CrossRef]
- Brubaker, D.B. Immunopathogenic mechanisms of posttransfusion graft-vs-host disease. Proc. Soc. Exp. Biol. Med. 1993, 202, 122–147. [Google Scholar] [CrossRef] [PubMed]
- Bloom, M.L.; Wolk, A.G.; Simon-Stoos, K.L.; Bard, J.S.; Chen, J.; Young, N.S. A mouse model of lymphocyte infusion-induced bone marrow failure. Exp. Hematol. 2004, 32, 1163–1172. [Google Scholar] [CrossRef]
- Iwasaki, T.; Fujiwara, H.; Iwasaki, T.; Shearer, G.M. Loss of proliferative capacity and T cell immune development potential by bone marrow from mice undergoing a graft-vs-host reaction. J. Immunol. 1986, 137. [Google Scholar]
- Chen, J.; Brandt, J.S.; Ellison, F.M.; Calado, R.T.; Young, N.S. Defective stromal cell function in a mouse model of infusion-induced bone marrow failure. Exp. Hematol. 2005, 33, 901–908. [Google Scholar] [CrossRef]
- Ferrara, J.L.M. Mouse models of graft-versus-host disease. StemBook 2009. [Google Scholar] [CrossRef] [PubMed]
- Van Dijken, P.J.; Wimperis, J.; Crawford, J.M.; Ferrara, J.L.M. Effect of graft-versus-host disease on hematopoiesis after bone marrow transplantation in mice. Blood 1991, 78, 2773–2779. [Google Scholar] [CrossRef] [Green Version]
- Garvy, B.A.; Elia, J.M.; Hamilton, B.L.; Riley, R.L. Suppression of B-cell development as a result of selective expansion of donor T cells during the minor H antigen graft-versus-host reaction. Blood 1993, 82, 2758–2766. [Google Scholar] [CrossRef] [Green Version]
- Shono, Y.; Ueha, S.; Wang, Y.; Abe, J.; Kurachi, M.; Matsuno, Y.; Sugiyama, T.; Nagasawa, T.; Imamura, M.; Matsushima, K. Bone marrow graft-versus-host disease: Early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood 2010, 115, 5401–5411. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jaramillo, G.; Gómez-Morales, E.; Sánchez-Valle, E.; Mayani, H. Severe hematopoietic alterations in vitro, in bone marrow transplant recipients who develop graft-versus-host disease. J. Hematother. Stem Cell Res. 2001, 10, 347–354. [Google Scholar] [CrossRef]
- Rieger, K.; Marinets, O.; Fietz, T.; Körper, S.; Sommer, D.; Mücke, C.; Reufi, B.; Blau, W.I.; Thiel, E.; Knauf, W.U. Mesenchymal stem cells remain of host origin even a long time after allogeneic peripheral blood stem cell or bone marrow transplantation. Exp. Hematol. 2005, 33, 605–611. [Google Scholar] [CrossRef]
- Yao, Y.; Song, X.; Cheng, H.; Tang, G.; Hu, X.; Zhou, H.; Wang, J. Dysfunction of bone marrow vascular niche in acute graft-versus-host disease after MHC-haploidentical bone marrow transplantation. PLoS ONE 2014, 9, e104607. [Google Scholar] [CrossRef]
- Chewning, J.H.; Zhang, W.; Randolph, D.A.; Swindle, C.S.; Schoeb, T.R.; Weaver, C.T. Allogeneic TH1 cells home to host bone marrow and spleen and mediate IFNγ-dependent aplasia. Biol. Blood Marrow Transplant. 2013, 19, 876–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medinger, M.; Krenger, W.; Jakab, A.; Halter, J.; Buser, A.; Bucher, C.; Passweg, J.; Tzankov, A. Numerical impairment of nestin+ bone marrow niches in acute GvHD after allogeneic hematopoietic stem cell transplantation for AML. Bone Marrow Transplant. 2015, 50, 1453–1458. [Google Scholar] [CrossRef] [Green Version]
- Shono, Y.; Shiratori, S.; Kosugi-Kanaya, M.; Ueha, S.; Sugita, J.; Shigematsu, A.; Kondo, T.; Hashimoto, D.; Fujimoto, K.; Endo, T.; et al. Bone marrow graft-versus-host disease: Evaluation of its clinical impact on disrupted hematopoiesis after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2014, 20, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, A.; Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. T cell-mediated cytotoxicity. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Graubert, T.A.; DiPersio, J.F.; Russell, J.H.; Ley, T.J. Perforin/granzyme-dependent and independent mechanisms are both important for the development of graft-versus-host disease after murine bone marrow transplantation. J. Clin. Investig. 1997, 100, 904–911. [Google Scholar] [CrossRef]
- Baker, M.B.; Altman, N.H.; Podack, E.R.; Levy, R.B. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J. Exp. Med. 1996, 183, 2645–2656. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.Y.; Lowin, B.; French, L.; Acha-Orbea, H.; Tschopp, J. Cytotoxic T cells deficient in both functional fas ligand and perforin show residual cytolytic activity yet lose their capacity to induce lethal acute graft-versus-host disease. J. Exp. Med. 1996, 183, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Marks, L.; Altman, N.H.; Podack, E.R.; Levy, R.B. Donor T cells lacking Fas ligand and perforin retain the capacity to induce severe GvHD in minor histocompatibility antigen mismatched bone-marrow transplantation recipients. Transplantation 2004, 77, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.B.; Riley, R.L.; Podack, E.R.; Levy, R.B. Graft-versus-host-disease-associated lymphoid hypoplasia and B cell dysfunction is dependent upon donor T cell-mediated Fas-ligand function, but not perforin function. Proc. Natl. Acad. Sci. USA 1997, 94, 1366–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarcon, A.K.; Desierto, M.J.; Zhou, W.; Visconte, V.; Gibellini, F.; Chen, J.; Young, N.S. Role of perforin-mediated cell apoptosis in murine models of infusion-induced bone marrow failure. Exp. Hematol. 2009, 37, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewski, J.; Selleri, C.; Anderson, S.; Young, N.S. Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood 1995, 85, 3183–3190. [Google Scholar] [CrossRef]
- Nagafuji, K.; Shibuya, T.; Harada, M.; Mizuno, S.; Takenaka, K.; Miyamoto, T.; Okamura, T.; Gondo, H.; Niho, Y. Functional expression of Fas antigen (CD95) on hematopoietic progenitor cells. Blood 1995, 86, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, T.; Hamano, T.; Saheki, K.; Kuroiwa, T.; Kataoka, Y.; Takemoto, Y.; Ogata, A.; Sugihara, A.; Terada, N.; Fujimoto, J.; et al. Effect of graft-versus-host disease (GVHD) On host hematopoietic progenitor cells is mediated by FAS–FAS ligand interactions but this does not explain the effect of GVHD on donor cells. Cell. Immunol. 1999, 197, 30–38. [Google Scholar] [CrossRef]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Celso, C.L.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Teshima, T.; Ordemann, R.; Reddy, P.; Gagin, S.; Liu, C.; Cooke, K.R.; Ferrara, J.L.M. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat. Med. 2002, 8, 575–581. [Google Scholar] [CrossRef]
- Faßlrinner, F.; Wobus, M.; Duryagina, R.; Müller, K.; Stopp, S.; Wehner, R.; Rauner, M.; Hofbauer, L.C.; Schmitz, M.; Bornhäuser, M. Differential effects of mixed lymphocyte reaction supernatant on human mesenchymal stromal cells. Exp. Hematol. 2012, 40, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Lepri, E.; Delfino, D.V.; Migliorati, G.; Moraca, R.; Ayroldi, E.; Riccardi, C. Functional expression of Fas on mouse bone marrow stromal cells: Upregulation by tumor necrosis factor-α and interferon-γ. Exp. Hematol. 1998, 26, 1202–1208. [Google Scholar] [PubMed]
- Schürch, C.; Riether, C.; Ochsenbein, A.F. Cytotoxic CD8+ t cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 2014, 14, 460–472. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, M.; Kuck, A.; Van Essen, M.; Haas, S.; Blaszkiewicz, S.; Essers, M.A.G. IFNα-mediated remodeling of endothelial cells in the bone marrow niche. Haematology 2016, 102, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mohammadpour, H.; Cao, X. Targeting cytokines in GVHD therapy. J. Immunol. Res. Ther. 2017, 2, 90–99. [Google Scholar] [PubMed]
- Takashima, S.; Martin, M.L.; Jansen, S.A.; Fu, Y.; Bos, J.; Chandra, D.; O’Connor, M.H.; Mertelsmann, A.M.; Vinci, P.; Kuttiyara, J.; et al. T cell–derived interferon-γ programs stem cell death in immune-mediated intestinal damage. Sci. Immunol. 2019, 4, eaay8556. [Google Scholar] [CrossRef]
- Na, I.-K.; Lu, S.X.; Yim, N.L.; Goldberg, G.L.; Tsai, J.; Rao, U.; Smith, O.M.; King, C.G.; Suh, D.; Hirschhorn-Cymerman, D.; et al. The cytolytic molecules Fas ligand and TRAIL are required for murine thymic graft-versus-host disease. J. Clin. Investig. 2010, 120, 343–356. [Google Scholar] [CrossRef]
- Piguet, P.F.; Grau, G.; Allet, B.; Vassalli, P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions of the acute phase of graft-vs.-host disease. J. Exp. Med. 1987, 166, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wang, W.; Ni, J.; Mao, X.; Song, D.; Liu, T.; Wei, J.; Zhou, H. Role of autophagy in tumor necrosis factor-α-induced apoptosis of osteoblast cells. J. Investig. Med. 2017, 65, 1014–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Shang, B.; Li, Y.-N.; Shi, Y.; Shao, C. IFNγ and TNFα synergistically induce apoptosis of mesenchymal stem/stromal cells via the induction of nitric oxide. Stem Cell Res. Ther. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Baldridge, M.T.; King, K.Y.; Goodell, M.A. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011, 32, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Morales-Mantilla, D.E.; King, K.Y. The role of interferon-gamma in hematopoietic stem cell development, homeostasis, and disease. Curr. Stem Cell Rep. 2018, 4, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Hirche, C.; Frenz, T.; Haas, S.F.; Döring, M.; Borst, K.; Tegtmeyer, P.-K.; Brizić, I.; Jordan, S.; Keyser, K.; Chhatbar, C.; et al. Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Rep. 2017, 19, 2345–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matatall, K.A.; Jeong, M.; Chen, S.; Sun, D.; Chen, F.; Mo, Q.; Kimmel, M.; King, K.Y. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. 2016, 17, 2584–2595. [Google Scholar] [CrossRef] [Green Version]
- Binder, D.; Fehr, J.; Hengartner, H.; Zinkernagel, R.M. Virus-induced transient bone marrow aplasia: Major role of interferon-α/Β during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J. Exp. Med. 1997, 185, 517–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacNamara, K.C.; Oduro, K.; Martin, O.; Jones, D.D.; McLaughlin, M.; Choi, K.; Borjesson, D.L.; Winslow, G.M. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-γ signaling. J. Immunol. 2011, 186, 1032–1043. [Google Scholar] [CrossRef]
- Ghimire, S.; Weber, D.; Mavin, E.; Wang, X.N.; Dickinson, A.M.; Holler, E. Pathophysiology of GvHD and other HSCT-related major complications. Front. Immunol. 2017, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malard, F.; Huang, X.-J.; Sim, J.P.Y. Treatment and unmet needs in steroid-refractory acute graft-versus-host disease. Leukemia 2020, 34, 1229–1240. [Google Scholar] [CrossRef]
- Murphy, K.; Charles, A.; Janeway, J.; Travers, P.; Walport, M. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2012; ISBN 978-0-8153-4243-4. [Google Scholar]
- Erdem Kuruca, S.; Çetin, M.B.; Akgün Dar, K.; Özerkan, D. Protective effects of cytokine combinations against the apoptotic activity of glucocorticoids on CD34+ hematopoietic stem/progenitor cells. Cytotechnology 2019, 71, 67–77. [Google Scholar] [CrossRef]
- Koromila, T.; Baniwal, S.K.; Song, Y.S.; Martin, A.; Xiong, J.; Frenkel, B. Glucocorticoids antagonize RUNX2 during osteoblast differentiation in cultures of ST2 pluripotent mesenchymal cells. J. Cell. Biochem. 2013, 115, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, J.L.M. Cytokine inhibitors and graft-versus-host disease. Ann. N. Y. Acad. Sci. 1995, 770, 227–236. [Google Scholar] [CrossRef]
- Chen, J.; Astle, C.M.; Harrison, D.E. Hematopoietic stem cell functional failure in interleukin-2-deficient mice. J. Hematother. Stem Cell Res. 2002, 11, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Giampaolo, S.; Wójcik, G.; Serfling, E.; Patra, A.K. Interleukin-2-regulatory T cell axis critically regulates maintenance of hematopoietic stem cells. Oncotarget 2017, 8, 29625–29642. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.E. Implications of TNF-α in the pathogenesis and management of GVHD. Int. J. Hematol. 2011, 93, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busca, A.; Locatelli, F.; Marmont, F.; Ceretto, C.; Falda, M. Recombinant human soluble tumor necrosis factor receptor fusion protein as treatment for steroid refractory graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Am. J. Hematol. 2007, 82, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, K.; Nishida, A.; Ota, H.; Ikebe, T.; Tsuji, M.; Yamamoto, H.; Asano-Mori, Y.; Uchida, N.; Izutsu, K.; Taniguchi, S. Infliximab treatment for steroid-refractory acute graft-versus-host disease after reduced-intensity cord blood transplantation in adults. Blood 2011, 118, 4553. [Google Scholar] [CrossRef]
- Dybedal, I.; Bryder, D.; Fossum, A.; Rusten, L.S.; Jacobsen, S.E.W. Tumor necrosis factor (TNF)–mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood 2001, 98, 1782–1791. [Google Scholar] [CrossRef]
- Selleri, C.; Sato, T.; Anderson, S.M.; Young, N.S.; Maciejewski, J.P. Interferon-γ and tumor necrosis factor-α suppress both early and late stages of hematopoiesis and induce programmed cell death. J. Cell. Physiol. 1995, 165, 538–546. [Google Scholar] [CrossRef]
- Caux, C.; Saeland, S.; Favre, C.; Duvert, V.; Mannoni, P.; Banchereau, J. Tumor necrosis factor-alpha strongly potentiates interleukin-3 and granulocyte-macrophage colony-stimulating factor-induced proliferation of human CD34+ hematopoietic progenitor cells. Blood 1990, 75, 2292–2298. [Google Scholar] [CrossRef]
- Pearl-Yafe, M.; Mizrahi, K.; Stein, J.; Yolcu, E.S.; Kaplan, O.; Shirwan, H.; Yaniv, I.; Askenasy, N.; Mizrahi, K.; Kaplan, O. Tnf receptors support murine hematopoietic progenitor function in the early stages of engraftment. Stem Cells 2010, 28, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Pronk, C.J.H.; Veiby, O.P.; Bryder, D.; Jacobsen, S.E.W. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: Involvement of two distinct receptors. J. Exp. Med. 2011, 208, 1563–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Passegué, E. TNF-α coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell 2019, 25, 357–372.e7. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Y.-G. The complex and central role of interferon-γ in graft-versus-host disease and graft-versus-tumor activity. Immunol. Rev. 2014, 258, 30–44. [Google Scholar] [CrossRef]
- Zeiser, R.; Von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Delisle, J.-S.; Gaboury, L.; Bélanger, M.-P.; Tassé, É.; Yagita, H.; Perreault, C. Graft-versus-host disease causes failure of donor hematopoiesis and lymphopoiesis in interferon-γ receptor-deficient hosts. Blood 2008, 112, 2111–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Dybedal, I.; Bryder, D.; Nilsson, L.; Sitnicka, E.; Sasaki, Y.; Jacobsen, S.E.W. IFN-γ negatively modulates self-renewal of repopulating human hemopoietic stem cells. J. Immunol. 2005, 174, 752–757. [Google Scholar] [CrossRef]
- De Bruin, A.M.; Demirel, Ö.; Hooibrink, B.; Brandts, C.H.; Nolte, M.A. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood 2013, 121, 3578–3585. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, F.; Jordan, M.B.; Allen, C.; Cesaro, S.; Rizzari, C.; Rao, A.; Degar, B.; Garrington, T.P.; Sevilla, J.; Putti, M.-C.; et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N. Engl. J. Med. 2020, 382, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Erkelens, M.N.; Nolte, M.A. Impact of viral infections on hematopoiesis: From beneficial to detrimental effects on bone marrow output. Front. Immunol. 2016, 7, 364. [Google Scholar] [CrossRef] [Green Version]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef]
- Srinivasan, A.; Wang, C.; Srivastava, D.K.; Burnette, K.; Shenep, J.L.; Leung, W.; Hayden, R.T. Timeline, epidemiology, and risk factors for bacterial, fungal, and viral infections in children and adolescents after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2013, 19, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Isomura, H.; Yoshida, M.; Oda, M.; Seino, Y.; Ohuchi, R.; Uno, F.; Yamada, M.; Namba, H.; Fujiwara, N. Suppressive effects of human herpesvirus-6 on thrombopoietin-inducible megakaryocytic colony formation in vitro. J. Gen. Virol. 2000, 81, 663–673. [Google Scholar] [CrossRef]
- Maciejewski, J.; Bruening, E.; Donahue, R.; Mocarski, E.; Young, N.; Jeor, S.S. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 1992, 80, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Hadjibabaie, M.; Valadkhani, B.; Kargar, M.; Ashouri, A.; Gholami, K.; Ghavamzadeh, A. The risk factors for cytomegalovirus reactivation following stem cell transplantation. J. Res. Pharm. Pract. 2016, 5, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Pichereau, C.; Desseaux, K.; Janin, A.; Scieux, C.; Peffault de Latour, R.; Xhaard, A.; Robin, M.; Ribaud, P.; Agbalika, F.; Chevret, S.; et al. The complex relationship between human herpesvirus 6 and acute graft-versus-host disease. Biol. Blood Marrow Transplant. 2012, 18, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, P.; Kaushansky, K.; Torok-Storb, B. Mechanisms of cytomegalovirus-mediated myelosuppression: Perturbation of stromal cell function versus direct infection of myeloid cells. Proc. Natl. Acad. Sci. USA 1990, 87, 1386–1390. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Song, J.; Jiang, Z.; Li, Y.; Gao, Y.; Xu, W.; Lu, Z.; Wang, Y.; Xiao, H. Risk-factor analysis of poor graft function after allogeneic hematopoietic stem cell transplantation. Int. J. Med. Sci. 2014, 11, 652–657. [Google Scholar] [CrossRef] [Green Version]
- Steffens, H.-P.; Podlech, J.; Kurz, S.; Angele, P.; Dreis, D.; Reddehase, M. Cytomegalovirus inhibits the engraftment of donor bone marrow cells by downregulation of hemopoietin gene expression in recipient stroma. J. Virol. 1998, 72, 5006–5015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, G.R.; Ferrara, J.L.M. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: Rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood 2000, 95, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Regoes, R.R.; Boddupalli, C.S.; Bonhoeffer, S.; Manz, M.G. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J. Exp. Med. 2011, 208, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Sioud, M.; Fløisand, Y.; Forfang, L.; Lund-Johansen, F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J. Mol. Biol. 2006, 364, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunology 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esplin, B.L.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Nie, L.; Zhang, Q.; Humphrey, M.B.; Yang, Q.; Borghesi, L.A.; Kincade, P.W. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J. Immunol. 2011, 186, 5367–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danziger-Isakov, L.; Baillie, M.G. Hematologic complications of anti-CMV therapy in solid organ transplant recipients. Clin. Transplant. 2009, 23, 295–304. [Google Scholar] [CrossRef]
- McKeny, P.T.; Nessel, T.A.; Zito, P.M. Antifungal Antibiotics; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Gomez, M.; Maraqa, N.; Alvarez, A.; Rathore, M. Complications of outpatient parenteral antibiotic therapy in childhood. Pediatr. Infect. Dis. J. 2001, 20, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Andersohn, F.; Konzen, C.; Garbe, E. Systematic review: Agranulocytosis induced by nonchemotherapy drugs. Ann. Intern. Med. 2007, 146, 657–665. [Google Scholar] [CrossRef]
- Bjornson, B.H.; McIntyre, A.P.; Harvey, J.M.; Tauber, A.I.; Mclntyre, A.P. Studies of the effects of trimethoprim and sulfamethoxazole on human granulopoiesis. Am. J. Hematol. 1986, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Baldridge, M.T.; King, K.Y. Hematopoiesis and the bacterial microbiome. Blood 2018, 132, 559–564. [Google Scholar] [CrossRef]
- Iwamura, C.; Bouladoux, N.; Belkaid, Y.; Sher, A.; Jankovic, D. Sensing of the microbiota by NOD1 in mesenchymal stromal cells regulates murine hematopoiesis. Blood 2017, 129, 171–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staffas, A.; Burgos da Silva, M.; Slingerland, A.E.; Lazrak, A.; Bare, C.J.; Holman, C.D.; Docampo, M.D.; Shono, Y.; Durham, B.; Pickard, A.; et al. Nutritional support from the intestinal microbiota improves hematopoietic reconstitution after bone marrow transplantation in mice. Cell Host Microbe 2018, 23, 447–457.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenq, R.R.; Taur, Y.; Devlin, S.M.; Ponce, D.M.; Goldberg, J.D.; Ahr, K.F.; Littmann, E.R.; Ling, L.; Gobourne, A.C.; Miller, L.C.; et al. Intestinal blautia is associated with reduced death from graft-versus-host disease. Biol. Blood Marrow Transplant. 2015, 21, 1373–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peled, J.U.; Devlin, S.M.; Staffas, A.; Lumish, M.; Khanin, R.; Littmann, E.R.; Ling, L.; Kosuri, S.; Maloy, M.; Slingerland, J.B.; et al. Intestinal microbiota and relapse after hematopoietic-cell transplantation. J. Clin. Oncol. 2017, 35, 1650–1659. [Google Scholar] [CrossRef] [Green Version]
- Jenq, R.R.; Ubeda, C.; Taur, Y.; Menezes, C.C.; Khanin, R.; Dudakov, J.A.; Liu, C.; West, M.L.; Singer, N.V.; Equinda, M.J.; et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J. Exp. Med. 2012, 209, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Eriguchi, Y.; Takashima, S.; Oka, H.; Shimoji, S.; Nakamura, K.; Uryu, H.; Shimoda, S.; Iwasaki, H.; Shimono, N.; Ayabe, T.; et al. Graft-versus-host disease disrupts intestinal microbial ecology by inhibiting Paneth cell production of α-defensins. Blood 2012, 120, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holler, E.; Butzhammer, P.; Schmid, K.; Hundsrucker, C.; Koestler, J.; Peter, K.; Zhu, W.; Sporrer, D.; Hehlgans, T.; Kreutz, M.; et al. Metagenomic analysis of the stool microbiome in patients receiving allogeneic stem cell transplantation: Loss of diversity is associated with use of systemic antibiotics and more pronounced in gastrointestinal graft-versus-host disease. Biol. Blood Marrow Transplant. 2014, 20, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Gavriilaki, M.; Sakellari, I.; Anagnostopoulos, A.; Gavriilaki, E. The impact of antibiotic-mediated modification of the intestinal microbiome on outcomes of allogeneic hematopoietic cell transplantation: Systematic review and meta-analysis. Biol. Blood Marrow Transplant. 2020, 26, 1738–1746. [Google Scholar] [CrossRef]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.-M.; Kong, Y.; Song, Y.; Sun, Y.-Q.; Wang, Y.; Zhang, X.-H.; Xu, L.-P.; Liu, K.-Y.; Huang, X.J. Atorvastatin enhances endothelial cell function in posttransplant poor graft function. Blood 2016, 128, 2988–2999. [Google Scholar] [CrossRef] [PubMed]
- Bassani, B.; Tripodo, C.; Portararo, P.; Gulino, A.; Botti, L.; Chiodoni, C.; Jachetti, E.; Bolli, N.; Ciciarello, M.; Joehrens, K.; et al. CD40 activity on mesenchymal cells negatively regulates OX40L to maintain bone marrow immune homeostasis under stress conditions. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.; Simmons, P.J.; Wang, C.-Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Hanoun, M.; Maryanovich, M.; Arnal-Estapé, A.; Frenette, P.S. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 2015, 86, 360–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, A.C.; Yamazaki, S. The hematopoietic stem cell diet. Int. J. Hematol. 2018, 107, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilic, E.; Delimar, V.; Desnica, L.; Pulanic, D.; Bilic, E.; Bakovic, M.; Curtis, L.M.; Seiwerth, R.S.; Stipetic, M.M.; Ceovic, R.; et al. High prevalence of small- and large-fiber neuropathy in a prospective cohort of patients with moderate to severe chronic GvHD. Bone Marrow Transplant. 2016, 51, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meij, B.S.; De Graaf, P.; Wierdsma, N.J.; Langius, J.A.E.; Janssen, J.J.W.M.; Van Leeuwen, P.A.M.; Visser, O.J. Nutritional support in patients with GVHD of the digestive tract: State of the art. Bone Marrow Transplant. 2012, 48, 474–482. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müskens, K.F.; Lindemans, C.A.; Belderbos, M.E. Hematopoietic Dysfunction during Graft-Versus-Host Disease: A Self-Destructive Process? Cells 2021, 10, 2051. https://doi.org/10.3390/cells10082051
Müskens KF, Lindemans CA, Belderbos ME. Hematopoietic Dysfunction during Graft-Versus-Host Disease: A Self-Destructive Process? Cells. 2021; 10(8):2051. https://doi.org/10.3390/cells10082051
Chicago/Turabian StyleMüskens, Konradin F., Caroline A. Lindemans, and Mirjam E. Belderbos. 2021. "Hematopoietic Dysfunction during Graft-Versus-Host Disease: A Self-Destructive Process?" Cells 10, no. 8: 2051. https://doi.org/10.3390/cells10082051
APA StyleMüskens, K. F., Lindemans, C. A., & Belderbos, M. E. (2021). Hematopoietic Dysfunction during Graft-Versus-Host Disease: A Self-Destructive Process? Cells, 10(8), 2051. https://doi.org/10.3390/cells10082051