
Microglia-Derived Small Extracellular Vesicles Reduce Glioma Growth by Modifying Tumor Cell Metabolism and Enhancing Glutamate Clearance through miR-124

 , , ,
, , , 
 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Primary Microglial Cultures
2.3. Microglia Stimulation (Polarization)
2.4. Extraction of sEVs
2.5. Nanoparticle Tracking Analysis (NTA)
2.6. NO Assay
2.7. Glutamate Release
2.8. Lactate Release
2.9. Cell Transfection
2.10. In Vivo Experiments
2.11. Tumor Size Analysis
2.12. Tissue Sampling for Transmission Electron Microscopy
2.13. Transmission Electron Microscopy (TEM)
2.14. Immunofluorescence
2.15. BrdU Proliferation Assay
2.16. qRT-PCR
2.17. NanoString nCounter Analysis
2.18. Statistical Analysis
3. Results
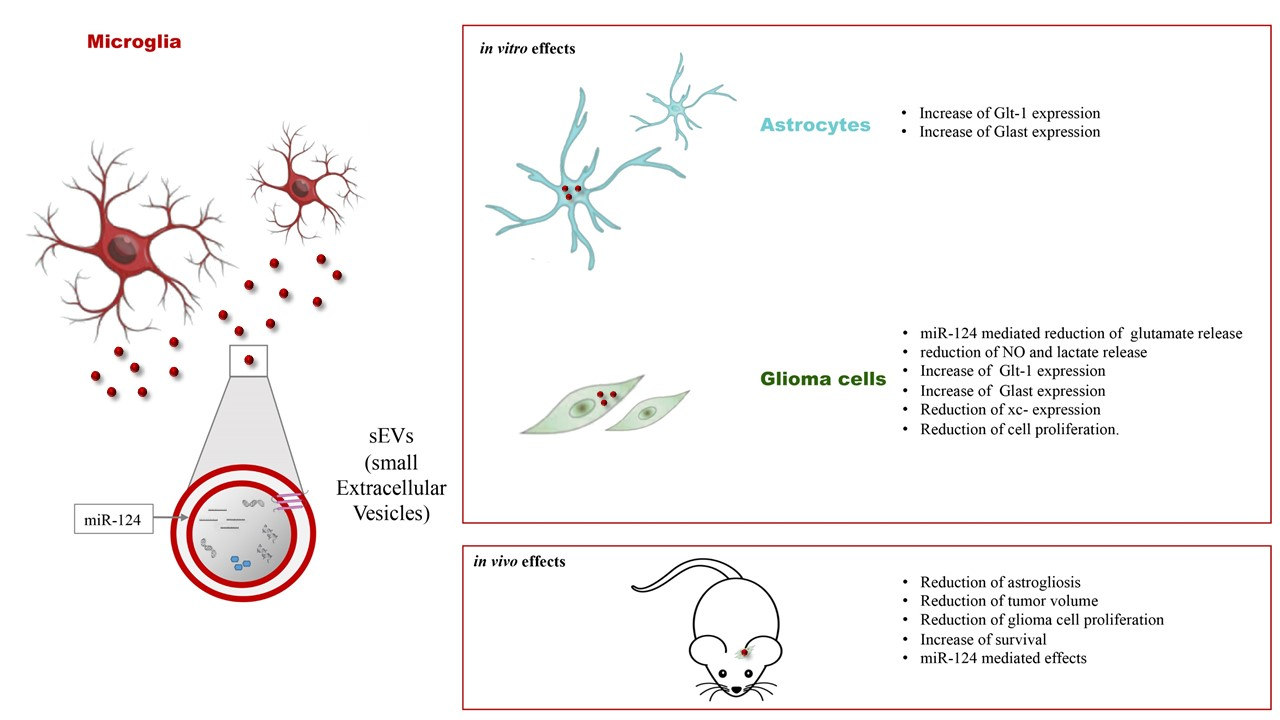
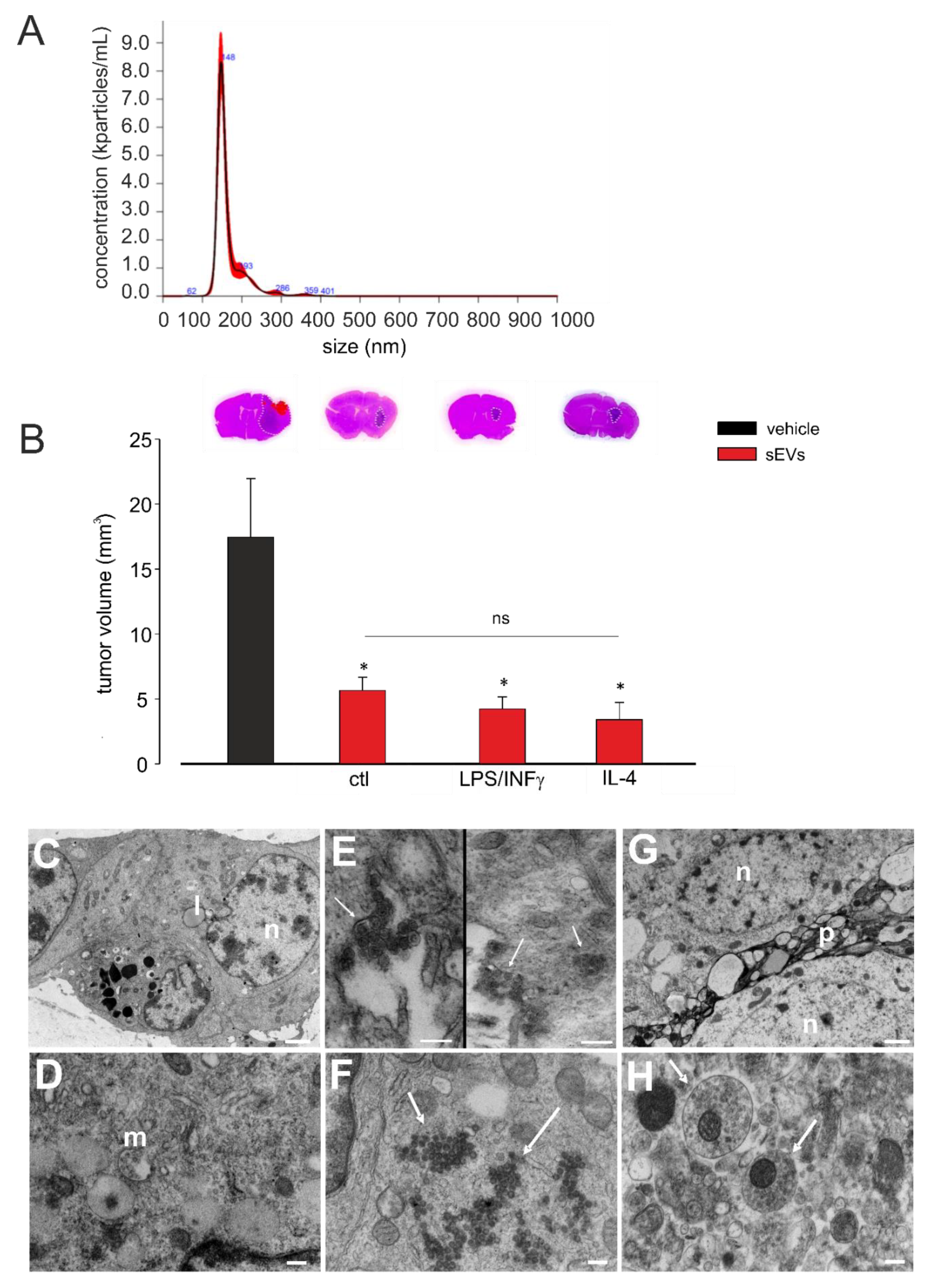
3.1. Microglia-Derived sEVs Exert Anti-Tumoral Activity in Glioma-Bearing Mice Independent of the Inflammatory State of Donor Cells
3.2. Microglia-Derived sEVs Reduce Astrocytosis in Glioma-Bearing Mice
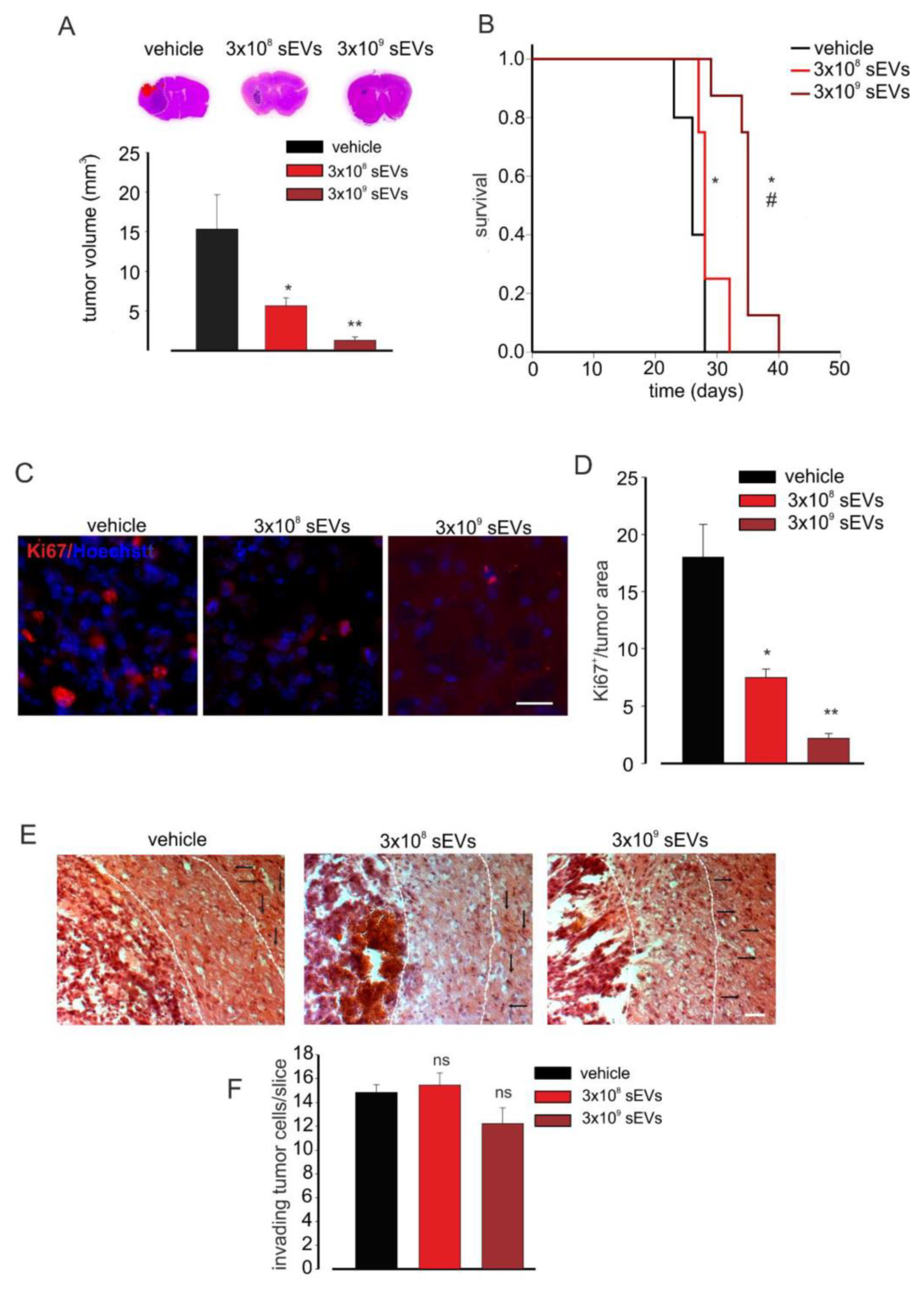
3.3. Microglia-Derived sEVs Exert an Anti-Tumor Effect and Reduce Glioma Cell Proliferation in Mice Depending on the Number
3.4. Microglia-Derived sEVs Reduce the Toxicity of Glioma Cells
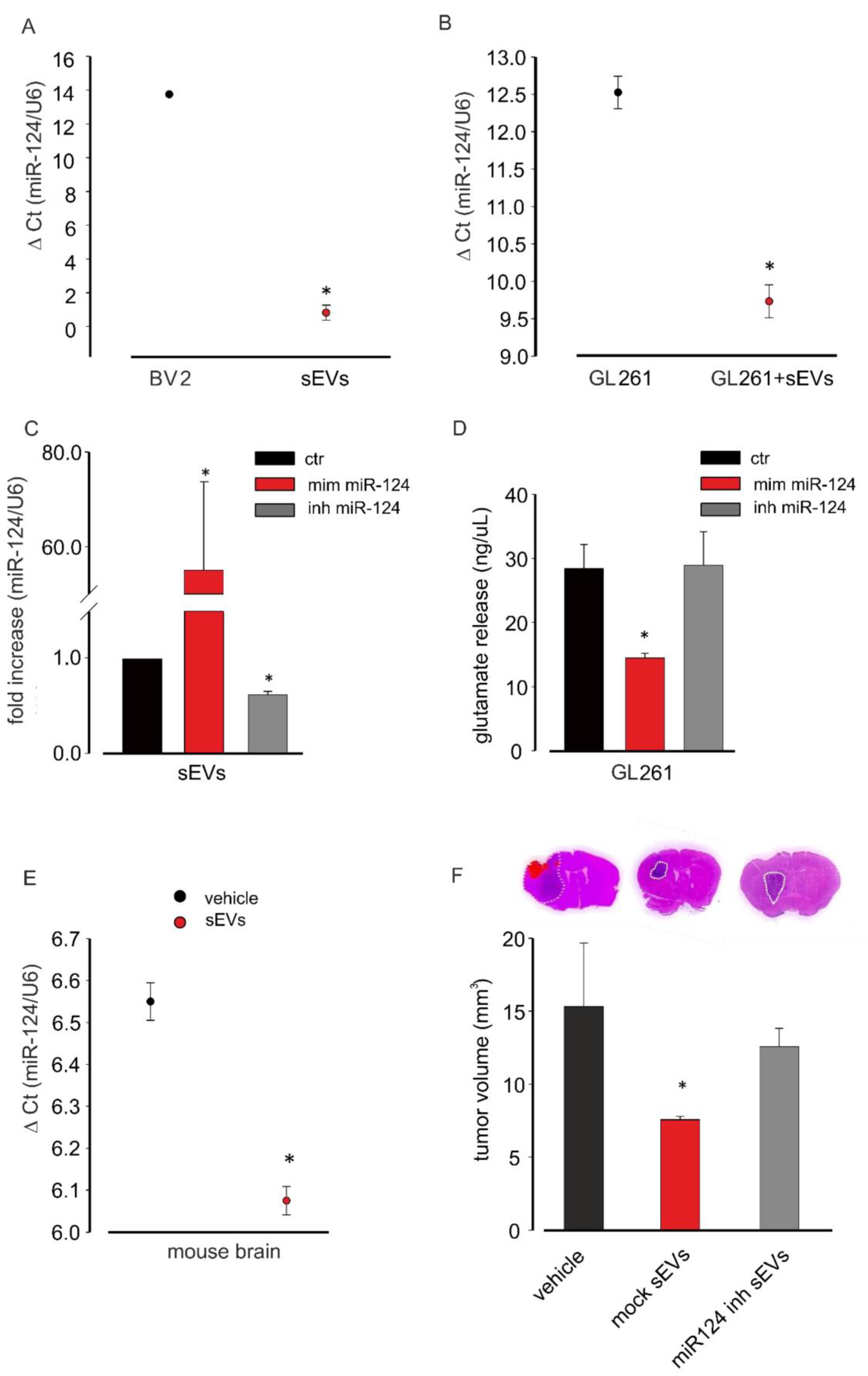
3.5. miR-124 Transferred from Microglia to Glioma Cells via sEVs Is a Key Regulator of the Anti-Tumoral Effect of Microglia-Derived sEVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Théry, C.; Zitvogel, L.; Amigorena, S. MVs and Exo are currently indistinguishable for cargo molecules, both carrying proteins, lipids, mRNA, microRNA and DNA. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, L.A.; Pilk, R.C.; Carter, D.F.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Wang, Y.; Wang, H.; Zhu, Z.; Xiao, Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J. Cell. Biochem. 2010, 11, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Subra, C.; Laulagnier, K.; Perret, B.; Record, M. Exosome lipidomics unravels lipid sorting at the level of multivesicular bodies. Biochimie 2007, 89, 205–212. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle 2009, 8, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Ciafrè, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; Farace, M.G. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem. Biophys. Res. Commun. 2005, 334, 1351–1358. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Zhao, H.; Wang, Y.; Chen, Z. Exosomal shuttled miR-424-5p from ischemic preconditioned microglia mediates cerebral endothelial cell injury through negatively regulation of FGF2/STAT3 pathway. Exp. Neurol. 2020, 333, 113411. [Google Scholar] [CrossRef]
- Huang, S.; Ge, X.; Yu, J.; Han, Z.; Yin, Z.; Li, Y.; Chen, F.; Wang, H.; Zhang, J.; Lei, P. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. 2018, 32, 512–528. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, A.; Serpe, C.; Chece, G.; Nigro, V.; Sarra, A.; Ruzicka, B.; Relucenti, M.; Familiari, G.; Ruocco, G.; Pascucci, G.R.; et al. Microglia-Derived Microvesicles Affect Microglia Phenotype in Glioma. Front. Cell. Neurosci. 2019, 22, 41. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.; D’Alessandro, G.; Trettel, F.; Limatola, C. Role of Infiltrating Microglia/Macrophages in Glioma. Adv. Exp. Med. Biol. 2020, 1202, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, I.; Thanos, S.; Paulus, W. Microglia promote glioma migration. Acta Neuropathol. 2002, 103, 351–355. [Google Scholar] [CrossRef]
- Lepore, F.; D’Alessandro, G.; Antonangeli, F.; Santoro, A.; Esposito, V.; Limatola, C.; Trettel, F. CXCL16/CXCR6 Axis Drives Microglia/Macrophages Phenotype in Physiological Conditions and Plays a Crucial Role in Glioma. Front. Immunol. 2018, 9, 2750. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Carvalho da Fonseca, A.C.; Wang, H.; Fan, H.; Chen, X.; Zhang, I.; Zhang, L.; Lima, F.R.; Badie, B. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 2014, 274, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Saederup, N.; Cardona, A.E.; Croft, K.; Mizutani, M.; Cotleur, A.C.; Tsou, C.L.; Ransohoff, R.M.; Charo, I.F. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 2017, 12, e0176931. [Google Scholar] [CrossRef]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-beta as an important regulator of glioblastoma Extensive modulation of a set of microRNAs in primary glioblastoma invasion—An inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelzinger, D.B.; Demuth, T.; Berens, M.E. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer Inst. 2007, 99, 1583–1593. [Google Scholar] [CrossRef]
- de Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.C.; Rothstein, J.D.; Sontheimer, H. Compromised glutamate transport in human glioma cells: Reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, S.C.; Robel, S. Glutamate and tumor-associated epilepsy: Glial cell dysfunction in the peritumoral environment. Neurochem. Int. 2013, 63, 696–701. [Google Scholar] [CrossRef] [Green Version]
- Takano, T.; Lin, J.H.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Puram, S.V.; Yeung, C.M.; Jahani-Asl, A.; Lin, C.; de la Iglesia, N.; Konopka, G.; Jackson-Grusby, L.; Bonni, A. STAT3-iNOS Signaling Mediates EGFRvIII-Induced Glial Proliferation and Transformation. J. Neurosci. 2012, 32, 7806–7818. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuraca, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, V.; Nowicki, M.O.; Idriss, M.; Jimenez, M.A.; Lugli, G.; Hayes, J.L.; Mahmoud, A.B.; Zane, R.E.; Passaro, C.; Ligon, K.L.; et al. The functional synergism of microRNA clustering provides therapeutically relevant epigenetic interference in glioblastoma. Nat. Commun. 2019, 25, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silber, J.; Lim, D.A.; Petritsch, C.; Persson, A.I.; Maunakea, A.K.; Yu, M.; Vandenberg, S.R.; Ginzinger, D.G.; James, C.D.; Costello, J.F.; et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008, 24, 14. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cheung, W.K.C.; Ng, S.S.; Jiang, X.; Jiang, S.; Sze, J.; Leung, G.K.K.; Lu, G.; Chan, D.T.M.; Bian, X.W.; et al. Loss of brain-enriched miR-124 microRNA enhances stem-like traits and invasiveness of glioma cells. J. Biol. Chem. 2012, 287, 9962–9971. [Google Scholar] [CrossRef] [Green Version]
- Marisetty, A.L.; Singh, S.K.; Nguyen, T.N.; Coarfa, C.; Liu, B.; Majumder, S. REST represses miR-124 and miR-203 to regulate distinct oncogenic properties of glioblastoma stem cells. Neuro Oncol. 2017, 19, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Mucaj, V.; Lee, S.S.; Skuli, N.; Giannoukos, D.N.; Qiu, B.; Eisinger-Mathason, T.S.; Nakazawa, M.S.; Shay, J.E.; Gopal, P.P.; Venneti, S.; et al. MicroRNA-124 expression counteracts pro-survival stress responses in glioblastoma. Oncogene 2015, 34, 2204–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, D.; Wang, J.; Wang, X. miR-124 ameliorates depressive-like behavior by targeting STAT3 to regulate microglial activation. Mol. Cell. Probes 2019, 48, 101470. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, M.; Chen, C.; Gong, W.; Yin, Z.; Li, H.; Fan, J.; Zhang, X.A.; Wang, D.W.; Zuo, H. MiR-124 aggravates failing hearts by suppressing CD151-facilitated angiogenesis in heart. Oncotarget 2018, 9, 14382–14396. [Google Scholar] [CrossRef]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhao, X.; Zhou, Y.; Hu, Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 2012, 28, 1346–1352. [Google Scholar] [CrossRef] [Green Version]
- Li, K.K.; Pang, J.C.; Ching, A.K.; Wong, C.K.; Kong, X.; Wang, Y.; Zhou, L.; Chen, Z.; Ng, H.K. miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum. Pathol. 2009, 40, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhao, Y.; Song, G.Q.; Ma, Y.H.; Jin, X.H.; Jin, S.L.; Fang, Y.H.; Chen, Y.C. Interfering cellular lactate homeostasis overcomes Taxol resistance of breast cancer cells through the microRNA-124-mediated lactate transporter (MCT1) inhibition. Cancer Cell Int. 2019, 19, 193. [Google Scholar] [CrossRef] [Green Version]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Grimaldi, A.; Chece, G.; Porzia, A.; Esposito, V.; Santoro, A.; Salvati, M.; Mainiero, F.; Ragozzino, D.; Di Angelantonio, S.; et al. KCa3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 2016, 7, 30781–30796. [Google Scholar] [CrossRef] [PubMed]
- Lauro, C.; Cipriani, R.; Catalano, M.; Trettel, F.; Chece, G.; Brusadin, V.; Antonilli, L.; van Rooijen, N.; Eusebi, F.; Fredholm, B.B.; et al. Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against Glu-induced death. Neuropsychopharmacology 2010, 35, 1550–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, A.; D’Alessandro, G.; Golia, M.T.; Grössinger, E.M.; Di Angelantonio, S.; Ragozzino, D.; Santoro, A.; Esposito, V.; Wulff, H.; Catalano, M.; et al. KCa3.1 inhibition switches the phenotype of glioma-infiltrating microglia/macrophages. Cell Death Dis. 2016, 7, e2174. [Google Scholar] [CrossRef] [Green Version]
- Wahnschaffe, U. An improved method for preserving the cellular membrane ultrastructure of in vitro cultivated cells using tannic acid. Exp. Pathol. 1988, 34, 79–84. [Google Scholar] [CrossRef]
- Rider, M.A.; Hurwitz, S.N.; Meckes, D.G., Jr. ExtraPEG: A Polyethylene Glycol-Based Method for Enrichment of Extracellular Vesicles. Sci. Rep. 2016, 6, 23978. [Google Scholar] [CrossRef]
- Garcia-Segura, L.M.; Lafarga, M.; Berciano, M.T.; Hernandez, P.; Andres, M.A. Distribution of nuclear pores and chromatin organization in neurons and glial cells of the rat cerebellar cortex. J. Comp. Neurol. 1989, 290, 440–450. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Dietz, S.; Madhu, B.; Griffiths, J.; Price, S.J.; Collins, V.P.; Watts, C. Fluorescence-guided surgical sampling of glioblastoma identifies phenotypically distinct tumour-initiating cell populations in the tumour mass and margin. Br. J. Cancer 2012, 107, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, D.P.; Palmieri, D.; Hua, E.; Hargrave, E.; Herring, J.M.; Qian, Y.; Vega-Valle, E.; Weil, R.J.; Stark, A.M.; Vortmeyer, A.O.; et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin. Exp. Metastasis 2008, 25, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jin, X.; Sohn, Y.W.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; Yin, J.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef]
- Lemée, J.M.; Clavreul, A.; Menei, P. Intratumoral heterogeneity in glioblastoma: Don’t forget the peritumoral brain zone. Neuro Oncol. 2015, 17, 1322–1332. [Google Scholar] [CrossRef]
- Tanaka, R.; Tainaka, M.; Ota, T.; Mizuguchi, N.; Kato, H.; Urabe, S.; Chen, Y.; Fustin, J.M.; Yamaguchi, Y.; Doi, M.; et al. Accurate determination of S-phase fraction in proliferative cells by dual fluorescence and peroxidase immunohistochemistry with 5-bromo-2’-deoxyuridine (BrdU) and Ki67 antibodies. J. Histochem. Cytochem. 2011, 59, 791–798. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.C.; Sontheimer, H. Glioma cells release neurotoxic concentrations of glutamate. Cancer Res. 1999, 59, 4383–4391. [Google Scholar]
- Grimaldi, A.; D’Alessandro, G.; Di Castro, M.A.; Lauro, C.; Singh, V.; Pagani, F.; Sforna, L.; Grassi, F.; Di Angelantonio, S.; Catacuzzeno, L.; et al. Kv1.3 activity perturbs the homeostatic properties of astrocytes in glioma. Sci. Rep. 2018, 8, 7654. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef]
- Vanhoutte, N.; Hermans, E. Glutamate-induced glioma cell proliferation is prevented by functional expression of the glutamate transporter GLT-1. FEBS Lett. 2008, 11, 1847–1852. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Hwang, S.Y.; Hwang, J.S.; Lee, J.W.; Oh, E.S.; Han, I.O. C6 glioma cell insoluble matrix components enhance interferon-gamma-stimulated inducible nitric-oxide synthase/nitric oxide production in BV2 microglial cells. J. Biol. Chem. 2008, 283, 2526–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases: From genes to function to therapy. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Bell, H.S.; Shinoda, J.; Holmes, M.C.; Wharton, S.B.; Whittle, I.R. Glioma tumourgenicity is decreased by iNOS knockout: Experimental studies using the C6 striatal implantation glioma model. Br. J. Neurosurg. 2002, 16, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Lam-Himlin, D.; Espey, M.G.; Perry, G.; Smith, M.A.; Castellani, R.J. Malignant glioma progression and nitric oxide. Neurochem. Int. 2006, 49, 764–768. [Google Scholar] [CrossRef]
- Tran, A.N.; Boyd, N.H.; Walker, K.; Hjelmeland, A.B. NOS Expression and NO Function in Glioma and Implications for Patient Therapies. Antioxid Redox Signal. 2017, 26, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Tsugawa, K.; Tarnawski, A.S.; Baatar, D. Dual actions of nitric oxide on angiogenesis: Possible roles of PKC, ERK, and AP-1. Biochem. Biophys. Res. Commun. 2004, 318, 520–528. [Google Scholar] [CrossRef]
- Verderio, C.; Muzio, L.; Turola, E.; Bergami, A.; Novellino, L.; Ruffini, F.; Riganti, L.; Corradini, I.; Francolini, M.; Garzetti, L.; et al. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 2012, 72, 610–624. [Google Scholar] [CrossRef] [Green Version]
- Garzetti, L.; Menon, R.; Finardi, A.; Bergami, A.; Sica, A.; Martino, G.; Comi, G.; Verderio, C.; Farina, C.; Furlan, R. Activated macrophages release microvesicles containing polarized M1 or M2 mRNAs. J. Leukoc. Biol. 2014, 95, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Mahajan, S.D. Transmigration of Tetraspanin 2 (Tspan2) siRNA Via Microglia Derived Exosomes across the Blood Brain Barrier Modifies the Production of Immune Mediators by Microglia Cells. J. Neuroimmune Pharmacol. 2020, 15, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Balas, M.; Alli, S.; Spears, J.; Zador, Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci. Rep. 2020, 10, 19542. [Google Scholar] [CrossRef] [PubMed]
- Caponegro, M.D.; Oh, K.; Madeira, M.M.; Radin, D.; Sterge, N.; Tayyab, M.; Moffitt, R.A.; Tsirka, S.E. A distinct microglial subset at the tumor-stroma interface of glioma. Glia 2021, 69, 1767–1781. [Google Scholar] [CrossRef]
- Potolicchio, I.; Carven, G.J.; Xu, X.; Stipp, C.; Riese, R.J.; Stern, L.J.; Santambrogio, L. Proteomic analysis of microglia-derived exosomes: Metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J. Immunol. 2005, 175, 2237–2243. [Google Scholar] [CrossRef] [Green Version]
- Venturini, A.; Passalacqua, M.; Pelassa, S.; Pastorino, F.; Tedesco, M.; Cortese, K.; Gagliani, M.C.; Leo, G.; Maura, G.; Guidolin, D.; et al. Exosomes from Astrocyte Processes: Signaling to Neurons. Front. Pharmacol. 2019, 10, 1452. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.X.; Liu, Z.; Deng, F.; Wang, D.D.; Li, X.W.; Tian, T.; Zhang, J.; Tang, J.H. MiR-145: A potential biomarker of cancer migration and invasion. Am. J. Transl. Res. 2019, 11, 6739–6753. [Google Scholar]
- Lee, J.K.; Park, S.R.; Jung, B.K.; Jeon, Y.K.; Lee, Y.S.; Kim, M.K.; Kim, Y.G.; Jang, Y.J.; Kim, C.W. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS ONE 2013, 8, e84256. [Google Scholar] [CrossRef] [Green Version]
- Grigorian-Shamagian, L.; Fereydooni, S.; Liu, W.; Echavez, A.; Marbán, E. Harnessing the heart’s resistance to malignant tumors: Cardiac-derived extracellular vesicles decrease fibrosarcoma growth and leukemia-related mortality in rodents. Oncotarget 2017, 8, 99624–99636. [Google Scholar] [CrossRef] [Green Version]
- Peak, T.C.; Praharaj, P.P.; Panigrahi, G.K.; Doyle, M.; Su, Y.; Schlaepfer, I.R.; Singh, R.; Vander Griend, D.J.; Alickson, J.; Hemal, A.; et al. Exosomes secreted by placental stem cells selectively inhibit growth of aggressive prostate cancer cells. Biochem. Biophys. Res. Commun. 2018, 499, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; Brand, F., 3rd; Adamczak, S.; Lee, S.W.; Perez-Barcena, J.; Wang, M.Y.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W. Exosome-mediated inflammasome signaling after central nervous system injury. J. Neurochem. 2016, 136 (Suppl. 1), 39–48. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Su, F.; Lin, L.; Liu, Y.; Huang, J.D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.L.N.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef]
- Katakowski, M.; Chopp, M. Exosomes as Tools to Suppress Primary Brain Tumor. Cell. Mol. Neurobiol. 2016, 36, 343–352. [Google Scholar] [CrossRef]
- Sinha, D.; Roy, S.; Saha, P.; Chatterjee, N.; Bishayee, A. Trends in Research on Exosomes in Cancer Progression and Anticancer Therapy. Cancers 2021, 13, 326. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Zhu, Y.L.; Zhou, Y.Y.; Liang, G.F.; Wang, Y.Y.; Hu, F.H.; Xiao, Z.D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [Green Version]
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Mörgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Gao, F.B. Context-dependent functions of specific microRNAs in neuronal development. Neural Dev. 2010, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makeyev, E.V.; Zhang, J.; Carrasco, M.A.; Maniatis, T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 2007, 27, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wang, X.Y.; Li, C.; Xu, S.J. Downregulation of microRNA-124 predicts poor prognosis in glioma patients. Neurol. Sci. 2015, 36, 131–135. [Google Scholar] [CrossRef]
- Liu, X.; Kang, J.; Sun, S.; Luo, Y.; Ji, X.; Zeng, X.; Zhao, S. iASPP, a microRNA 124 target, is aberrantly expressed in astrocytoma and regulates malignant glioma cell migration and viability. Mol. Med. Rep. 2018, 17, 1970–1978. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, Q.; Li, C.; Wang, L.; Qian, X.; Jiang, C.; Liu, X.; Wang, X.; Li, H.; Kang, C.; et al. MiR-124 governs glioma growth and angiogenesis and enhances chemosensitivity by targeting R-Ras and N-Ras. Neuro Oncol. 2014, 16, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- An, L.; Liu, Y.; Wu, A.; Guan, Y. microRNA-124 inhibits migration and invasion by down-regulating ROCK1 in glioma. PLoS ONE 2013, 8, e694782013. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef]
- Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Guo, M.L.; Buch, S. Cocaine-mediated downregulation of miR-124 activates microglia by targeting KLF4 and TLR4 signaling. Mol. Neurobiol. 2018, 55, 3196–3210. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, Y.; Kong, C.; Su, X.; Zhang, X.; Bai, W.; He, X. MiR-124 Enriched Exosomes Promoted the M2 Polarization of Microglia and Enhanced Hippocampus Neurogenesis After Traumatic Brain Injury by Inhibiting TLR4 Pathway. Neurochem. Res. 2019, 44, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Ning, R.; Venkat, P.; Chopp, M.; Zacharek, A.; Yan, T.; Cui, X.; Seyfried, D.; Chen, J. D-4F increases microRNA-124a and reduces neuroinflammation in diabetic stroke rats. Oncotarget 2017, 8, 95481–95494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.; Zhang, T.; Duan, H.; Pan, Y.; Zhang, X.; Yang, G.; Wang, J.; Deng, Y.; Yang, Z. MiR-124 contributes to M2 polarization of microglia and confers brain inflammatory protection via the C/EBP-alpha pathway in intracerebral hemorrhage. Immunol. Lett. 2017, 182, 25. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Arg1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| Cd163 | GCTAGACGAAGTCATCTGCACTGGG | TCAGCCTCAGAGACATGAACTCGG |
| Cd206 | CAAGGAAGGTTGGCATTTGT | CCTTTCAGTCCTTTGCAAGT |
| Fizz1 | CCAATCCAGCTAACTATCCCTCC | ACCCAGTAGCAGTCATCCCA |
| Gapdh | TCGTCCCGTAGACAAAATGG | TTGAGGTCAATGAAGGGGTC |
| Il1β | CAACCAACAAGTGATATTCTCCATG | GATCCACACTCTCCAGCTGCA |
| Slc1a2 | CTGGTGCAAGCCTGTTTCC | GCCTGTTCACCCATCTTCC |
| Slc1a3 | AGCAGGGAGTCCGTAAACG | AGCATTCCGAAACAGGTAACTTT |
| Sc7a11 | CCTCTATTCGGACCCATTTAG | CTGGGTTTCTTGTCCCATATA |
| Slc16a1 | GCTTGGTGACCATTGTTGGAAT | CCCAGTCGTGTATTTGTAGTCTCCAT |
| Tnf-α | GTGGAACTGGCAGAAGAG | CCATAGAACTGATGAGAGG |
| Ym1 | CAGGTCTGGCAATTCTTCTGAA | GTCTTGCTCATGTGTGTAAGTGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serpe, C.; Monaco, L.; Relucenti, M.; Iovino, L.; Familiari, P.; Scavizzi, F.; Raspa, M.; Familiari, G.; Civiero, L.; D’Agnano, I.; et al. Microglia-Derived Small Extracellular Vesicles Reduce Glioma Growth by Modifying Tumor Cell Metabolism and Enhancing Glutamate Clearance through miR-124. Cells 2021, 10, 2066. https://doi.org/10.3390/cells10082066
Serpe C, Monaco L, Relucenti M, Iovino L, Familiari P, Scavizzi F, Raspa M, Familiari G, Civiero L, D’Agnano I, et al. Microglia-Derived Small Extracellular Vesicles Reduce Glioma Growth by Modifying Tumor Cell Metabolism and Enhancing Glutamate Clearance through miR-124. Cells. 2021; 10(8):2066. https://doi.org/10.3390/cells10082066
Chicago/Turabian StyleSerpe, Carmela, Lucia Monaco, Michela Relucenti, Ludovica Iovino, Pietro Familiari, Ferdinando Scavizzi, Marcello Raspa, Giuseppe Familiari, Laura Civiero, Igea D’Agnano, and et al. 2021. "Microglia-Derived Small Extracellular Vesicles Reduce Glioma Growth by Modifying Tumor Cell Metabolism and Enhancing Glutamate Clearance through miR-124" Cells 10, no. 8: 2066. https://doi.org/10.3390/cells10082066
APA StyleSerpe, C., Monaco, L., Relucenti, M., Iovino, L., Familiari, P., Scavizzi, F., Raspa, M., Familiari, G., Civiero, L., D’Agnano, I., Limatola, C., & Catalano, M. (2021). Microglia-Derived Small Extracellular Vesicles Reduce Glioma Growth by Modifying Tumor Cell Metabolism and Enhancing Glutamate Clearance through miR-124. Cells, 10(8), 2066. https://doi.org/10.3390/cells10082066