
Regulation of Paneth Cell Function by RNA-Binding Proteins and Noncoding RNAs




Abstract
:
1. Introduction
2. Paneth Cells in the Intestinal Epithelium Homeostasis
2.1. Paneth Cells Enhance Epithelial Defense by Secreting Antimicrobial Peptides/Proteins
2.2. Paneth Cells Regulate Intestinal Mucosal Growth by Interacting with Intestinal Stem Cells
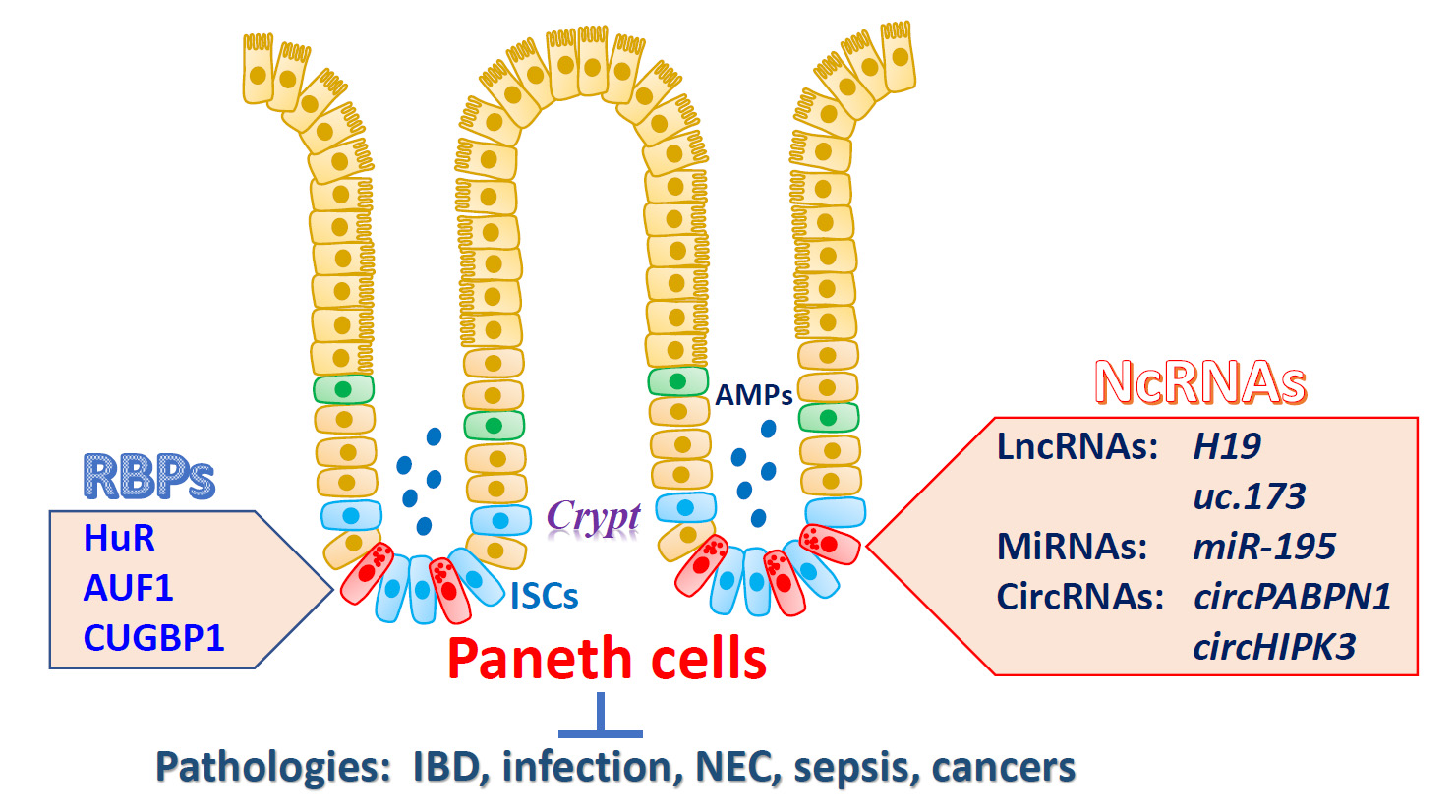
3. Regulation of Paneth Cell Function by RBPs
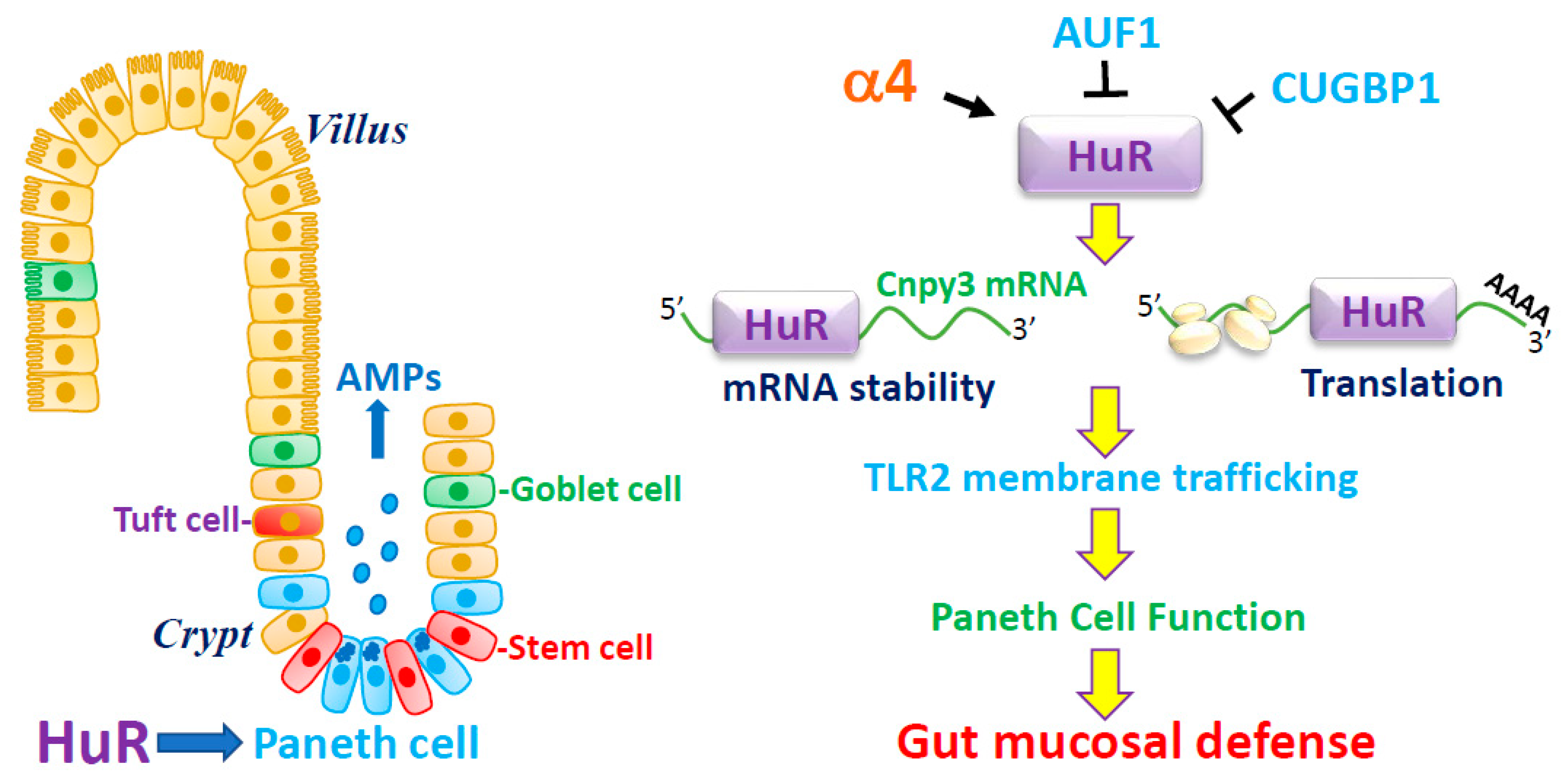
3.1. HuR Is Essential for Paneth Cell Function
3.2. HuR Regulates Paneth Cells by Altering TLR2 Membrane Distribution
3.3. HuR Promotes TLR2 Subcellular Trafficking and Autophagy by Increasing CNPY3
3.4. Regulation of HuR Stability by α4
3.5. Other RBPs in the Regulation of Paneth Cells
4. Regulation of Paneth Cells by NcRNAs
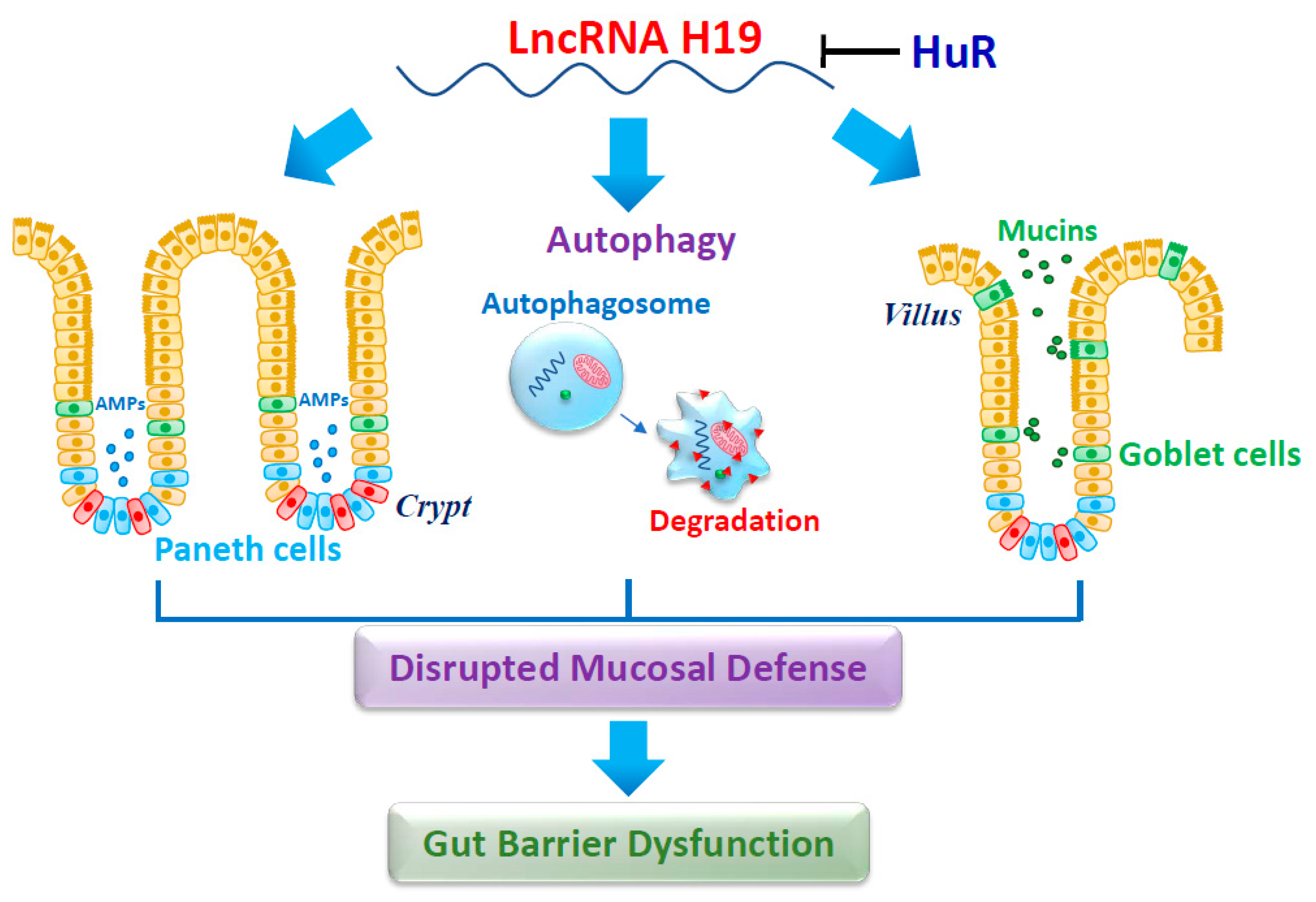
4.1. LncRNA H19 Impairs Paneth Cells and Compromises the Intestinal Barrier Function
4.2. MiR-195 Regulates Paneth and Tuft Cells in the Intestinal Epithelium
4.3. CircRNAs Are Novel Regulators of the Intestinal Epithelium Homeostasis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [Green Version]
- Torow, N.; Marsland, B.J.; Hornef, M.W.; Gollwitzer, E.S. Neonatal mucosal immunology. Mucosal Immunol. 2017, 10, 5–17. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Riba, A.; Olier, M.; Lacroix-Lamande, S.; Lencina, C.; Bacquié, V.; Harkat, C.; Van Langendonck, N.; Gillet, M.; Cartier, C.; Baron, M.; et al. Paneth cell defects induce microbiota dysbiosis in mice and promote visceral hypersensitivity. Gastroenterology 2017, 153, 1594–1606. [Google Scholar] [CrossRef] [Green Version]
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell. Biol. 2021, 22, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Rao, J.N.; Wang, J.Y. RNA-binding proteins and long noncoding RNAs in intestinal epithelial autophagy and barrier function. Tissue Barriers 2021, 9, 1895648. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell. Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian autophagy: How does it work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Gukovsky, I.; Algul, H.; Habtezion, A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology 2017, 153, 1212–1226. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Lueschow, S.R.; McElroy, S.J. The Paneth cell: The curator and defender of the immature small intestine. Front Immunol. 2020, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Hodin, C.M.; Lenaerts, K.; Grootjans, J.; de Haan, J.J.; Hadfoune, M.; Verheyen, F.K.; Kiyama, H.; Heineman, E.; Buurman, W.A. Starvation compromises Paneth cells. Am. J. Pathol. 2011, 179, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell. Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Wang, J.Y. RNA-binding proteins and microRNAs in gastrointestinal epithelial homeostasis and diseases. Curr. Opin. Pharmacol. 2014, 19, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Gorospe, M.; Tominaga, K.; Wu, X.; Fahling, M.; Ivan, M. Post-transcriptional control of the hypoxic response by RNA-binding proteins and microRNAs. Front. Mol. Neurosci. 2011, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA binding proteins and the messages they carry. Nat. Rev. Mol. Cell. Biol. 2002, 3, 195–205. [Google Scholar] [CrossRef]
- Yang, H.; Rao, J.N.; Wang, J.Y. Posttranscriptional regulation of intestinal epithelial tight junction barrier by RNA-binding proteins and microRNAs. Tissue Barriers 2014, 2, e28320. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.; Xiao, L.; Wang, J.Y. Posttranscriptional regulation of intestinal epithelial integrity by noncoding RNAs. Wiley Interdiscip. Rev. RNA 2017, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.; Shen, J. The roles and functions of Paneth cells in Crohn’s disease: A critical review. Cell Prolif. 2021, 54, e12958. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, L.; Wang, B.; Sun, Q.; Zhao, P.; Li, W. The role of autophagy in maintaining intestinal mucosal barrier. J. Cell Physiol. 2019, 234, 19406–19419. [Google Scholar] [CrossRef] [PubMed]
- Cray, P.; Sheahan, B.J.; Dekany, C.M. Secretory Sorcery: Paneth cell control of intestinal repair and homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Wehkamp, J.; Stange, E.F. An Update review on the Paneth cell as key to ileal Crohn’s disease. Front Immunol. 2020, 11, 646. [Google Scholar] [CrossRef] [PubMed]
- Ayabe, T.; Satchell, D.P.; Wilson, C.L.; Parks, W.C.; Selsted, M.E.; Ouellette, A.J. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 2000, 1, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 357, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Li, X.X.; Chung, H.K.; Kalakonda, S.; Cai, J.Z.; Cao, S.; Chen, N.; Liu, Y.; Rao, J.N.; Wang, H.Y.; et al. RNA-binding protein HuR regulates Paneth cell function by altering membrane localization of TLR2 via post-transcriptional control of CNPY3. Gastroenterology 2019, 157, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjöberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–83. [Google Scholar] [CrossRef]
- Porter, E.M.; Liu, L.; Oren, A.; Anton, P.A.; Ganz, T. Localization of human intestinal defensin 5 in Paneth cell granules. Infect Immun. 1997, 65, 2389–2395. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Li, X.X.; Xiao, L.; Chung, H.K.; Ma, X.X.; Liu, X.; Song, J.L.; Jin, C.Z.; Rao, J.N.; Gorospe, M.; Wang, J.Y. Interaction between HuR and circPABPN1 modulates autophagy in the intestinal epithelium by altering ATG16L1 translation. Mol. Cell. Biol. 2020, 40, e00492-19. [Google Scholar] [CrossRef]
- Gunther, C.; Neumann, H.; Neurath, M.F.; Becker, C. Apoptosis, necrosis and necroptosis: Cell death regulation in the intestinal epithelium. Gut 2013, 62, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Giammanco, A.; Blanc, V.; Montenegro, G.; Klos, C.; Xie, Y.; Kennedy, S.; Luo, J.; Chang, S.H.; Hla, T.; Nalbantoglu, I.; et al. Intestinal epithelial HuR modulates distinct pathways of proliferation and apoptosis and attenuates small intestinal and colonic tumor development. Cancer Res. 2014, 74, 5322–5335. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.; Bu, H.F.; Liu, F.; Wu, L.; Pfeifer, K.; Chou, P.M.; Wang, X.; Sun, J.; Lu, L.; Pandey, A.; et al. In Inflamed intestinal tissues and epithelial cells, interleukin 22 signaling increases expression of H19 long noncoding RNA, which promotes mucosal regeneration. Gastroenterology 2018, 155, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Ma, X.X.; Luo, J.; Chung, H.K.; Kwon, M.S.; Yu, T.X.; Rao, J.N.; Kozar, R.; Gorospe, M.; Wang, J.Y. Circular RNA circHIPK3 promotes homeostasis of the intestinal epithelium by reducing miR-29b function. Gastroenterology 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.; Clevers, H. How the gut feels, smells, and talks. Cell 2017, 170, 10–11. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.X.; Chung, H.K.; Xiao, L.; Piao, J.J.; Lan, S.; Jaladanki, S.K.; Turner, D.J.; Raufman, J.P.; Gorospe, M.; Wang, J.Y. Long noncoding RNA H19 impairs the intestinal barrier by suppressing autophagy and lowering Paneth and Goblet cell function. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 611–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterji, P.; Rustgi, A.K. RNA binding proteins in intestinal epithelial biology and colorectal cancer. Trends. Mol. Med. 2018, 24, 490–506. [Google Scholar] [CrossRef]
- Nighot, P.; Ma, T. Role of autophagy in the regulation of epithelial cell junctions. Tissue Barriers 2016, 4, e1171284. [Google Scholar] [CrossRef]
- Shukla, R.; Medeiros-Silva, J.; Parmar, A.; Vermeulen, B.J.A.; Das, S.; Paioni, A.L.; Jekhmane, S.; Lorent, J.; Bonvin, A.M.J.J.; Baldus, M.; et al. Mode of action of teixobactins in cellular membranes. Nat. Commun. 2020, 11, 2848. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Christodoulou-Vafeiadou, E.; Rao, J.N.; Zou, T.; Xiao, L.; Chung, H.K.; Yang, H.; Gorospe, M.; Kontoyiannis, D.; Wang, J.Y. RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway. Mol. Biol. Cell. 2014, 25, 3308–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Balasubramanian, I.; Zhang, L.; Gao, N. Metaplastic Paneth cells in extra-intestinal mucosal niche indicate a link to microbiome and inflammation. Front Physiol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Mei, X.; Gu, M.; Li, M. Plasticity of Paneth cells and their ability to regulate intestinal stem cells. Stem. Cell. Res. Ther. 2020, 11, 349. [Google Scholar] [CrossRef]
- Kwon, M.S.; Chung, H.K.; Xiao, L.; Yu, T.X.; Wang, S.R.; Piao, J.J.; Rao, J.N.; Gorospe, M.; Wang, J.Y. MicroRNA-195 regulates Tuft cell function in the intestinal epithelium by altering translation of DCLK1. Am. J. Physiol. Cell Physiol. 2021, 320, C1042–C1054. [Google Scholar] [CrossRef]
- Lukong, K.E.; Chang, K.W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends. Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef]
- Hinman, M.N.; Lou, H. Diverse molecular functions of Hu proteins. Cell. Mol. Life. Sci. 2008, 65, 3168–3181. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.A.; Deretic, V. Toll-like receptors in control of immunological autophagy. Cell. Death. Differ. 2009, 16, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, R.; Rao, J.N.; Zou, T.; Liu, L.; Xiao, L.; Cao, S.; Hansraj, N.Z.; Gorospe, M.; Wang, J.Y. miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res. 2013, 41, 7905–7919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, M.; Ditsworth, D.; Lindsten, T.; Thompson, C.B. α4 is an essential regulator of PP2A phosphatase activity. Mol. Cell 2009, 36, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.K.; Wang, S.R.; Xiao, L.; Rathor, N.; Turner, D.J.; Yang, P.; Gorospe, M.; Rao, J.N.; Wang, J.Y. α4 coordinates small intestinal epithelium homeostasis by regulating stability of HuR. Mol. Cell. Biol. 2018, 38, e00631-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.X.; Rao, J.N.; Zou, T.; Liu, L.; Xiao, L.; Ouyang, M.; Cao, S.; Gorospe, M.; Wang, J.Y. Competitive binding of CUGBP1 and HuR to occludin mRNA controls its translation and modulates epithelial barrier function. Mol. Biol. Cell. 2013, 24, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.X.; Gu, B.L.; Yan, J.K.; Zhu, J.; Yan, W.H.; Chen, J.; Qian, L.X.; Cai, W. CUGBP1 and HuR regulate E-cadherin translation by altering recruitment of E-cadherin mRNA to processing bodies and modulate epithelial barrier function. Am. J. Physiol. Cell. Physiol. 2016, 310, C54–C65. [Google Scholar] [CrossRef] [Green Version]
- Zou, T.; Rao, J.N.; Liu, L.; Xiao, L.; Yu, T.X.; Jiang, P.; Gorospe, M.; Wang, J.Y. Polyamines regulate the stability of JunD mRNA by modulating the competitive binding of its 3′ untranslated region to HuR and AUF1. Mol. Cell. Biol. 2010, 30, 5021–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, T.; Jaladanki, S.K.; Liu, L.; Xiao, L.; Chung, H.K.; Wang, J.Y.; Xu, Y.; Gorospe, M.; Wang, J.Y. H19 Long noncoding RNA regulates intestinal epithelial barrier function via microRNA 675 by interacting with RNA-binding protein HuR. Mol. Cell. Biol. 2016, 36, 1332–1341. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Wu, J.; Wang, J.Y.; Chung, H.K.; Kalakonda, S.; Rao, J.N.; Gorospe, M.; Wang, J.Y. Long noncoding RNA uc.173 promotes renewal of the intestinal mucosa by inducing degradation of microRNA 195. Gastroenterology 2018, 154, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L.; et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by circPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.; Li, W.; Zhuge, Y.; Xu, S.; Wang, Y.; Chen, Y.; Shen, G.; Wang, F. Inhibition of circHIPK3 prevents angiotensin II-induced cardiac fibrosis by sponging miR-29b-3p. Int. J. Cardiol. 2019, 292, 188–196. [Google Scholar] [CrossRef]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Inui, S.; Kuwahara, K.; Mizutani, J.; Maeda, K.; Kawai, T.; Nakayasu, H.; Sakaguchi, N. Molecular cloning of a cDNA clone encoding a phosphoprotein component related to the Ig receptor-mediated signal transduction. J. Immunol. 1995, 154, 2714–2723. [Google Scholar] [PubMed]
- Kong, M.; Fox, C.J.; Mu, J.; Solt, L.; Xu, A.; Cinalli, R.M.; Birnbaum, M.J.; Lindsten, T.; Thompson, C.B. The PP2A-associated protein α4 is an essential inhibitor of apoptosis. Science 2004, 306, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Srikantan, S.; Yang, X.; Lal, A.; Kim, H.H.; Kuwano, Y.; Galban, S.; Becker, K.G.; Kamara, D.; de Cabo, R.; et al. Ubiquitin-mediated proteolysis of HuR by heat shock. EMBO J. 2009, 28, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Chu, P.C.; Chuang, H.C.; Kulp, S.K.; Chen, C.S. The mRNA-stabilizing factor HuR protein is targeted by β-TrCP protein for degradation in response to glycolysis inhibition. J. Biol. Chem. 2012, 287, 43639–43650. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Rao, J.N.; Zou, T.; Lan, L.; Xiao, L.; Bellavance, E.; Gorospe, M.; and Wang, J.Y. JunD represses transcription and translation of the tight junction protein zona occludens-1 modulating intestinal epithelial barrier function. Mol. Biol. Cell. 2008, 19, 3701–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, L.; Chen, L.L. The Biogenesis, functions, and challenges of circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [Green Version]
- Wesselhoeft, R.A.; Kowalski, P.S.; Anderson, D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018, 9, 2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Piwecka, M.; Glazar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabory, A.; Ripoche, M.A.; Le Digarcher, A.; Watrin, F.; Ziyyat, A.; Forne, T.; Jammes, H.; Ainscough, J.F.; Surani, M.A.; Journot, L.; et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 2009, 136, 3413–3421. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Zhou, J.; Gao, Y.; Ghazal, S.; Lu, L.; Bellone, S.; Yang, Y.; Liu, N.; Zhao, X.; Santin, A.D.; et al. Regulation of tumor cell migration and invasion by the H19/let-7 axis is antagonized by metformin-induced DNA methylation. Oncogene 2015, 34, 3076–3084. [Google Scholar] [CrossRef]
- Luo, M.; Li, Z.; Wang, W.; Zeng, Y.; Liu, Z.; Qiu, J. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett. 2013, 333, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ghazal, S.; McKinnon, B.; Zhou, J.; Mueller, M.; Men, Y.; Yang, L.; Mueller, M.; Flannery, C.; Huang, Y.; Taylor, H.S. H19 lncRNA alters stromal cell growth via IGF signaling in the endometrium of women with endometriosis. EMBO Mol. Med. 2015, 7, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Matouk, I.J.; DeGroot, N.; Mezan, S.; Ayesh, S.; Abu-lail, R.; Hochberg, A.; Galun, E. The H19 non-coding RNA is essential for human tumor growth. PLoS ONE 2007, 2, e845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Li, Y.; Zhuang, M.; Zhu, B.; Zhang, W.; Yan, H.; Zhang, P.; Li, D.; Yang, J.; Sun, Y.; et al. Long noncoding RNA H19 act as a competing endogenous RNA of Let-7g to facilitate IEC-6 cell migration and proliferation via regulating EGF. J. Cell. Physiol. 2021, 236, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, W.; Lin, G.; Zhong, S. MicroRNA-195 inhibits epithelial-mesenchymal transition via downregulating CDK4 in bladder cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 3891–3902. [Google Scholar]
- Yuan, T.; Zhang, L.; Yao, S.; Deng, S.Y.; Liu, J.Q. miR-195 promotes LPS-mediated intestinal epithelial cell apoptosis via targeting SIRT1/eIF2a. Int. J. Mol. Med. 2020, 45, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, Y.; Xiao, L.; Yu, T.X.; Li, J.Z.; Rao, J.N.; Turner, D.J.; Gorospe, M.; Wang, J.Y. Cooperative repression of insulin-like growth factor type 2 receptor translation by microRNA 195 and RNA-binding protein CUGBP1. Mol. Cell. Biol. 2017, 37, e00225-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Function | Mechanisms | References |
|---|---|---|
| Host defense | AMPs: Lysozyme α-defensin C-type lectins Phospholipase A2 Reg-III MMP-7 CRIP Xanthine oxidase Autophagy Apoptosis | Bel et al. [26]; Xiao et al. [27] Ayabe et at. [25]; Salzman et al. [28]; Porter et al. [29] Riba et al. [5] Boya et al. [8] Bel et al. [26] Peterson et al. [30] Yang et al. [21] Lueschow et al. [13] Clevers et al. [11] Bel et al. [26]; Dikic et al. [31]; Li et al. [32] Gunther et al. [33]; Giammanco et al. [34] |
| Mucosal renewal | Stem cell niches Niche signaling (Wnt, Notch) | Beumer et al. [6]; Clevers et al. [11] Sato et al. [12] |
| Impact on diseases | IBD Bacterial Infection Sepsis Cancers NEC | Yang et al. [21]; Geng et al. [35] Xiao et al. [36] Riba et al. [5]; Beumer et al. [37]; Salzman et al. [28] Yu et al. [38] Chatterji et al. [39]; Giammanco et al. [34] Torow et al. [2]; Gunther et al. [33] |
| Names | Functions | Targets | References |
|---|---|---|---|
| RBPs HuR | ↑ Paneth cell function | Enhancing TLR2 membrane distribution and activity via CNPY3 Inhibiting miR-195 activity Interacting with α4 via IKKα | Xiao et al. [7,27] Delgado et al. [49] Zhuang et al. [50] Kong et al. [51] Chung et al. [52] |
| CUGBP1 | ↓ Paneth cell function | Competing with HuR to interact with target mRNAs | Yu et al. [53] Yu et al. [54] |
| AUF1 | ↓ Paneth cell function | Inhibiting HuR binding to target mRNAs | Zou et al. [55] |
| NcRNAs H19 | ↓ Paneth cell function | Inhibiting autophagy, reducing lysozyme, and impairing barrier | Yu et al. [38] Zou et al. [56] |
| uc.173 | ↑ Paneth cell function | Increasing miR-195 degradation | Xiao et al. [57] |
| miR-195 | ↓ Paneth and Tuft cells | Inhibiting HuR binding to mRNAs | Kwon et al. [46] |
| circPABPN1 | ↓ Paneth cell function | Preventing HuR binding to mRNAs Inhibiting ATG16L1 translation | Abdelmohsen et al. [58] Li et al. [32] |
| circHIPK3 | ↑ Paneth cell function | Enhancing epithelial homeostasis via interacting with miR-29b | Xiao et al. [36] Ni et al. [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, H.K.; Xiao, L.; Jaladanki, K.C.; Wang, J.-Y. Regulation of Paneth Cell Function by RNA-Binding Proteins and Noncoding RNAs. Cells 2021, 10, 2107. https://doi.org/10.3390/cells10082107
Chung HK, Xiao L, Jaladanki KC, Wang J-Y. Regulation of Paneth Cell Function by RNA-Binding Proteins and Noncoding RNAs. Cells. 2021; 10(8):2107. https://doi.org/10.3390/cells10082107
Chicago/Turabian StyleChung, Hee K., Lan Xiao, Krishna C. Jaladanki, and Jian-Ying Wang. 2021. "Regulation of Paneth Cell Function by RNA-Binding Proteins and Noncoding RNAs" Cells 10, no. 8: 2107. https://doi.org/10.3390/cells10082107
APA StyleChung, H. K., Xiao, L., Jaladanki, K. C., & Wang, J. -Y. (2021). Regulation of Paneth Cell Function by RNA-Binding Proteins and Noncoding RNAs. Cells, 10(8), 2107. https://doi.org/10.3390/cells10082107