1. Introduction
Recombinant proteins are foreign proteins produced in various expression systems. They are mainly used as pharmaceuticals for diagnostics, for the vaccination of humans or animals, and as drugs or monoclonal antibodies [
1]. The ever-growing demand for recombinant proteins stimulates the development of various expression systems for the production of these proteins that meet the existing stringent standards. Today, recombinant proteins are mainly produced in traditional prokaryotic and eukaryotic systems such as
Escherichia coli, several yeast species, and mammalian cell cultures. More than 50% of all pharmaceutical recombinant proteins are synthesized in mammalian cells [
2,
3]. Mammalian cells are necessary for the production of complex proteins because bacteria cannot form disulfide bonds efficiently and neither bacteria nor yeast can add human-like post-translational modifications (PTMs) in the secretory pathway [
4]. Protein production in mammalian cells is expensive due to the high cost of the culture media components. Moreover, some recombinant proteins, such as immunotoxins or regulatory proteins, are toxic to mammalian cells. Production of biopharmaceuticals in plant expression systems is becoming a promising alternative to existing platforms based on mammalian and bacterial cells [
2,
5,
6]. The cultivation of plant cells under controlled conditions of bioreactors ensures the production of high-quality protein according to GMP standards. The advantages of plant expression systems are rapid cell growth, low cost of nutrient media components, the low risk of contamination with animal pathogens, as well as the possibility of obtaining recombinant proteins with a modified human-type glycosylation profile.
Plant cell cultures as a system for the production of recombinant proteins, however, have several drawbacks, the main one of which is associated with a low yield of recombinant protein, which rarely exceeds 100 μg/kg of fresh weight, despite significant efforts to optimize protein expression and stability [
6]. In whole plants, the problem of low yield of the target protein can be overcome by transient expression. Transient expression systems have surpassed the challenge of rapid and high-yield expression of recombinant proteins in plants, allowing for gram-sized quantities to be attained in as little as 5 days [
7,
8]. However, so far only a few examples of the use of a transient expression in suspension cell cultures are known in the literature [
9,
10]. One of the reasons for the low yield of recombinant protein is the insertion of the transgene into a random genome region, which leads to different levels of expression due to the positional effects and different numbers of transgene copies. The transgene can integrate into the chromatin region with varying degrees of compaction: into the region of active euchromatin or inactive heterochromatin, as well as in close proximity to native regulatory elements. What is more, epigenetic modifications such as methylation can also reduce transgene expression.
The problems of random integration can be overcome with the use of site-specific endonucleases. Such endonucleases, the most well-known of which is the Cas9 endonuclease, induce double-stranded breaks (DSB) in the target region of the genome, which are repaired by one of the two innate DNA repair systems: the non-homologous end-joining (NHEJ) pathway and the homology-directed repair (HDR) pathway [
11,
12,
13]. If, during homologous repair, a donor template is provided that carries a sequence homologous to the site of a DSB or carries a whole additional gene, then the result of such repair can be a targeted gene change or a targeted insertion of an additional gene into a given region of the genome, the so-called knock-in [
14,
15,
16]. Insertion of the target gene in the transcriptionally active region of the genome may provide it with a high level of expression. The possibility of creating “acceptor lines” in which reporter genes are inserted into regions well characterized for expression and then replaced with target genes using zinc finger nucleases is discussed in the paper of Schiermeyer et al. [
17].
Using a donor format can influence the rate of HDR and editing outcomes. Donor templates can be delivered as dsDNA, such as PCR products, linearized or non-linearized plasmids, or ssDNA. The literature provides examples of the use of all types of templates for knock-in; however, ssDNA templates showed their low efficiency in plants, and linear DNA was usually delivered together with circular DNA, so it is difficult to assess the effect of template composition [
18,
19]. Circular DNA is most often used as a template for knock-in, and flanking the repair template with target sites and the release of linear repair template has been shown to provide a 2- to-5-fold improvement of targeted gene insertion in human culture cells in comparison with circular plasmid DNA [
20,
21]. In our study, we decided to test three types of templates for knock-in-target genes surrounded by homology arms, target genes surrounded by homology arms and flanked by the sequences recognized by Cas9, and target genes flanked only by the sequences recognized by Cas9 without homology arms.
Our study was aimed to test the possibility of integrating target genes into evolutionarily adapted regions of genes, the expression of which provides vital functions of the plant cell (regions of housekeeping genes). It should be emphasized that site-specific insertion of target genes into the genome of a plant cell is a very difficult task [
15]. Moreover, when choosing such regions, the researcher can fail completely, since the introduction of foreign DNA into the regions of housekeeping genes can disrupt the coordinated expression of its own genes. In this regard, it is of interest to not only assess the possibility of stable expression of the target gene delivered to such a region but also demonstrate the features of expression of other genes in this region, as well as to evaluate the yield of the recombinant target protein.
Deltaferon (dIFN) encoded by the
dIFN gene was chosen as the target recombinant protein. Deltaferon is a recombinant analog of human gamma-interferon, in which Arg-129 is replaced by Gly and Lys-130 is replaced by Ser and 10 C-terminal amino acid residues are deleted. Deltaferon is biologically active and more resistant to proteolysis than natural interferon gamma and has the same phagocytosis-stimulating properties as human IFN-gamma but reduced antiviral activity. According to its biological properties, it can be considered a component of medicines intended for the treatment of severe viral, immune, and oncological diseases [
22,
23]. For the targeted insertion of the dIFN gene, we selected the region of the histone H3.3 gene
HTR5 (At4g40040). Histone genes are the most important housekeeping genes since they provide the compaction of chromatin necessary for all eukaryotes, take part in the epigenetic regulation of gene expression, and are located in areas of actively transcribed chromatin. In plants, the most well-studied are the genes of histone H3 [
24]. The haploid genome of
A. thaliana contains 15 histone H3 genes, including five H3.1 genes, three H3.3 genes, and five H3.3-like genes. Histone H3.1 genes are expressed only in the S phase of the cell cycle and histone H3.3 genes are constitutively expressed during the entire interphase. Two of the three H3.3 genes
HTR5 and
HTR8 (At4g40040 and At5g10980) exhibited replication-independent expression in suspension cells [
25]; among them the
HTR5 gene region looked more convenient for cloning.
Here, we describe the creation of three types of genetic constructs for the delivery of the target gene (dIFN) to the region of the HTR5 gene, aimed at optimizing the frequency of knock-ins, receiving of transgenic cell suspension lines with the help of biolistic transformation, and a comparative analysis of the accumulation of the target recombinant protein (dIFN) in monoclonal cell lines with knock-in and cell lines with random insertions of the target gene. In addition, we evaluate the transcriptional activity of the HTR5 gene and the adjacent gene encoding the 12 KDa subunit of microsomal signal peptidase in the A. thaliana monoclonal cell lines created. In this work, for the first time, the creation of plant cell suspension culture lines with a site-specific integration of the target gene obtained using CRISPR/Cas9 is described.
2. Materials and Methods
The fast-growing cell line of
A. thaliana (L.) Heynh (Columbia ecotype, Col-0 inbred line) was used as the initial plant material. This cell line deposited in the All-Russian collection of higher plant cell cultures (ARCCCHP,
http://www.ippras.ru/cfc/alccmp/, accessed on 16 August 2021) under No. 85 and designated as NFC-0 [
26] was kindly provided by Ph.D. Nosov A.V. (Timiryazev Institute of Plant Physiology RAS, Moscow, Russia). The cell culture was maintained in vitro on SH medium [
27] with the addition of phytohormones (1 mg/L 2,4-D and 0.1 mg/L kinetin).
2.1. Plasmids Carrying Cas9 and Guide RNA
The plasmids pBlu/gRNA (#59188) for an intermediate cloning step and Cas9 MDC123 (#59184) with the Cas9 endonuclease gene under the control of the 2 × 35S CaMV promoter optimized for expression in Glycine max cells were donated by R. Stupar [
28] from the Addgene repository. The pBlu/gRNA plasmid carrying an sgRNA cassette under the control of the
A. thaliana U6 promoter was used as an intermediate vector for inserting selected spacer regions into the sgRNA sequence.
2.2. The Sources of Elements of Genetic Constructs for Knock-In
Nucleotide sequences for creating genetic constructs were obtained by PCR using appropriate oligonucleotides and templates. The genomic DNA of
A. thaliana was used as a template for the amplification of sequences flanking the integration site of the target gene. The signal peptide sequence directing the synthesized protein to the apoplast was amplified on a
Daucus carota genomic DNA template. The plasmid pGEX4T-1 was the source of the
GST (glutathione S-transferase) gene encoding the tag for affinity purification of the target protein. For the amplification of the
nptII gene sequences (neomycin phosphotransferase II) and the CaMV35S promoter of the cauliflower mosaic virus, the pBi121 plasmid was used as a template. The
dIFN (deltaferon) gene, a recombinant analog of human interferon gamma for the plant cells expression, was amplified from the pIFN-γ-trp2-Δ plasmid [
29].
2.3. Selection of Guide RNA
The region of the histone H3.3 gene
HTR5 was chosen as the region for the integration of the target gene. The CRISPOR software (
http://crispor.tefor.net/crispor.py, accessed on 16 August 2021) was used to select the guide RNA for knock-in into the region of the
HTR5 gene. The sequence for knock-in was located between the locus of histone H3.3 gene
HTR5 (At4g40040) and the adjacent gene encoding the 12 kDa subunit of microsomal signal peptidase (At4g40042), preceding the coding region of the
HTR5 gene.
2.4. Creation of a Cas9 (H3.3) Genetic Construct for Introducing Double-Strand Breaks into Target Regions
The selected sequences that determine the specificity of the targeting sgRNA were transferred into the Cas9 MDC123 plasmid by means of the pBlu/gRNA intermediate plasmid using the corresponding oligonucleotides (
Supplementary Materials Table S1): for the region of the
HTR5 gene–1_gRNA_H Up and 1_gRNA_H Lo. The first stage of the construct assembly consisted of hybridization of the selected phosphorylated oligonucleotides and their subsequent integration into the pBlu/gRNA plasmid treated with the BstV2I restriction enzyme (Sibenzyme, Novosibirsk, Russia). To confirm the insertion of the target sequence, the DNA of the resulting clones was sequenced using a standard T3 primer (
Supplementary Materials, Table S1). The second stage of the construct assembly consisted in the transfer of the obtained sgRNA carrying the targeting sequence into the plasmid Cas9 MDC123. The insertion was performed at the
EcoRI endonuclease restriction site.
2.5. Creation of the Genetic Constructs pIFN (H3.3).1, pIFN (H3.3).2 and pIFN (H3.3).3, Carrying a Template for Homologous Recombination
To deliver the desired gene
dIFN, encoding a human γ-interferon, and the selective
nptII gene, conferring kanamycin resistance to transformed cells, into the location of histone H3.3 gene, three types of genetic constructs, pIFN (H3.3).1, pIFN (H3.3).2 and pIFN (H3.3).3, were designed. Schemes of their structure are presented in
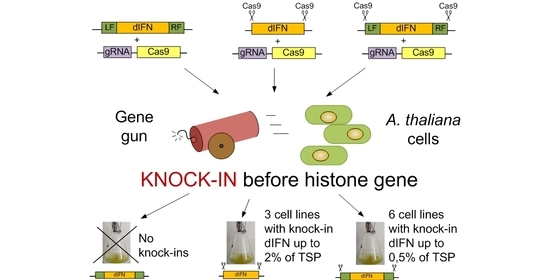
Figure 1. To optimize the integration frequency of the latter two constructs into the target region, they were flanked by the sequences recognized by sgRNA, which ensured their excision from a plasmid in the cell as a linear structure.
To obtain the target deltaferon gene, with a signal peptide and an attached
GST gene, primers Up_sig.BglII(62), Lo_dIFN(60), ifn + Up_GST(60), and Lo_GST(GEX) stopA65I(66) were used (all sequences of the primers used are given in
Supplementary Materials Table S1). The first two primers were used to generate the
dIFN gene with sequence coding of the signal peptide. In order for the chimeric
dIFN-GST gene to be translated entirely, the stop codon was removed from the deltaferon gene in the resulting DNA fragment. Primers ifn + Up_GST and Lo_GST(GEX)stopA65I(66) were used to obtain the
GST gene sequence. Then primers Up_sig.BglII(62), Lo_GST(GEX) stopA65I(66) and both DNA fragments obtained were used in PCR to obtain the target DNA fragment signal--his6-dIFN-GST.
To obtain the three final constructs, three intermediate plasmids were created. All intermediate plasmids were obtained on the basis of the pUC19 vector.
The intermediate plasmid pInt_var.1 carries sequences from the A. thaliana genome flanking the insertion region and a kanamycin resistance gene with a promoter and terminator. To obtain pInt_var.1, three DNA fragments were preliminarily obtained. The first two fragments, LF and RF (flank sequences), were synthesized by two sequential PCRs on a template of A. thaliana genomic DNA with primers: Up_L_HIII and Lo_L_Acc_Sfr (LF) and Up_R_Sfr_Acc and Lo_R_Sal (RF). The third fragment (nptII gene with p35S promoter) was obtained by PCR on the pBi121 plasmid template with primers Lo_A-B_pBI(58) and Up_Xho-pBI(60). Using the LF and RF fragments, as well as the primers Up_Xho-pBI(60), Up_L_HIII, and Lo_R_Sal, an LF-RF DNA fragment containing recognition sites for the Acc65I, Sfr274I restrictases between LF and RF was obtained, which was then inserted into pUC19. A DNA fragment carrying the nptII gene was cloned into pUC-LF-RF at the Sfr274I/Acc65I restriction enzyme recognition sites.
The intermediate plasmid pInt_var.2, instead of the flanking DNA of Arabidopsis, carries the Cas9 endonuclease recognition sites identical to the site of the double-stranded break in front of the A. thaliana HTR5 gene as well as the nptII gene with a promoter and terminator. To obtain the nptII fragment, the primers Up_Sal_gRNA-pBI(60) and Lo_A-B_pBI(58) were used instead of the primers Lo_A-B_pBI(58) and Up_Xho-pBI(60). The resulting DNA fragment was cloned into pUC19 at the SalI and Acc65I restriction enzyme recognition sites. The target signal-his6-dIFN-GST fragment was obtained using the Lo_Acc_gRNA_GST(60) primer instead of Lo_GST(GEX)stopA65I(66).
The intermediate plasmid pInt_var.3 carries sequences from the Arabidopsis genome flanking the insertion region, recognition sites for the Cas9 endonuclease that are identical to the double-strand break site upstream of the A. thaliana HTR5 gene, and the nptII gene with a promoter and terminator. Plasmid pInt_var.3 was obtained similarly to plasmid pInt_var.1, but instead of primers Up_L_HIII and Lo_R_Sal, primers Up_L_HIII_gRNA and Lo_R_Sal_gRNA were used for cloning.
The resulting target DNA fragment signal-his6-dIFN-GST was used for cloning within the intermediate plasmids pInt_var.1, pInt_var.2, and pInt_var.3 at the recognition sites of the BglII/Acc65I restriction enzymes to form three target plasmids, pIFN (H3.3).1, pIFN (H3.3).2 and pIFN (H3.3).3.
2.6. Biolistic Transformation of A. thaliana Cells and Obtaining Transgenic Suspension Cultures
Delivery of the target dIFN gene to the region of the HTR5 gene of A. thaliana was carried out using biolistic transformation with the immobilization of plasmids pIFN (H3.3).1, pIFN (H3.3).2 or pIFN (H3.3).3 together with the plasmid Cas9H33 on the gold particles. In total, 10 biolistic transformations were performed for each construct.
For the transformation by the biolistic method, 1 mL of cell suspension was applied to the surface of the SH medium [
27] with the addition of phytohormones (1 mg/L 2,4-D and 0.1 mg/L kinetin), distributing it evenly, and grown in the dark within 3 days for the formation of a callus cell layer. Calluses were transformed using a PDS1000/He system (Bio-Rad, Hercules, CA, USA), using the following biolistic parameters: gold particle size—0.6 µm; membrane rupture pressure—1100 psi; vacuum pressure in the chamber—27 inches Hg; distance to the explant—6 cm. Immobilization onto gold particles of an equimolar mixture of two plasmids, one of which included the target gene, and the second with the Cas9H3.3, was carried out according to the method of the gene gun manufacturer. For each transformation, 6 Petri dishes with prepared calluses were used; each callus was fired on twice.
Three days after the biolistic transformations, the calluses were transferred to a selective SH medium of the same composition supplied with kanamycin at a concentration of 100 mg/L and cultured in a light with an intensity of 20,000 lux at a photoperiod of 18/6 h (day/night). Transfer of the calluses to fresh media of the same composition were carried out weekly. The resulting antibiotic resistant calluses were used to obtain cell suspensions. The suspensions were cultured in the dark, using an orbital shaker at 160× g. Furthermore, monoclones were obtained from these cell cultures. For this, on the 4th day of cultivation, cell suspensions diluted with the culture medium were seeded on Petri dishes with SH medium supplied with the antibiotic kanamycin (100 mg/L), and microcalluses were grown from individual cells/cell aggregates.
2.7. Identification of Monoclonal Cell Lines with the Target Insertion of Genes (Knock-Ins)
The detection of knock-ins was carried out using PCR and sequencing DNA of kanamycin-resistant calluses. Genomic DNA was isolated from the calluses using CTAB buffer according to the Allen protocol [
30]. PCR analysis and sequencing (at ZAO Evrogen, Moscow, Russia) were performed using the appropriate primers (
Supplementary Materials Table S1). The monoclonal cell lines were tested using primers for the selective
nptII gene, for the target
dIFN gene, and for the event of target insertion—when one primer is located in the target region of plant DNA, and the other is located in the transgenic construct.
2.8. Identification of Monoclonal Cell Lines with Random Insertion of Transgenes
The control group with random insertion of transgenes included cell lines identified by resistance to the antibiotic kanamycin, in which the presence of the marker
nptII gene and the target
dIFN gene was confirmed by PCR analysis, but insertion into the specified region of the genome was not confirmed. The detection of insertions of target genes was carried out using PCR and DNA sequencing of calluses resistant to selective agents. Genomic DNA was isolated using CTAB buffer according to the Allen protocol [
30]. PCR analysis and DNA sequencing (at ZAO Evrogen, Moscow, Russia) were performed using the appropriate primers (
Supplementary Materials, Table S1). The monoclonal cell lines with random insertion of transgenes were tested using primers for the selective
nptII gene, for the target
dIFN gene, and for the event of target insertion—when one primer is located in the target region of plant DNA, and the other is located in the transgenic construct.
2.9. Analysis of A. thaliana Gene Expression Surrounding the Target Site
Total RNA from the cell biomass of each monoclonal line was isolated using the ExtractRNA reagent (Evrogen, Russia), RNA was treated with DNase I (Thermo Scientific, Riga, Latvia), and 4 µg RNA was used to obtain cDNA (Thermo Scientific RevertAid First Strand cDNA Synthesis Kit, Latvia). Expression analysis was performed using real-time PCR on a CFX96 amplifier (Bio-Rad, CA, USA). The reaction was carried out in 20 μL of a reaction mixture of the following composition: 15 ng of the studied cDNA, 50 mM Tris-SO
4, pH 9.0, 30 mM KCl, 10 mM ammonium sulfate, 0.01% Tween 20, 3 mM MgCl
2, 0.2 mM of each deoxyribonucleoside triphosphate, 0.4 μM of each primer, 0.4 μM of each probe, and 0.1 u.a./μL Hot Start DNA polymerase (Biosan, Novosibirsk, Russia). To create the probes, we used pairs of fluorophore-quencher: FAM-BHQ1 and Cy5-BHQ2. Real-time PCR was carried out in the multiplex version. The level of target gene expression was investigated relative to the
HTR5 gene (primers and probe HIS3.3_f, HIS3.3_r, HIS3.3_p), the gene encoding the 12 kDa subunit of microsomal signal peptidase (primers and probe VAP27-1_f VAP27 -1_r VAP27-1_p), as well as
A. thaliana’s own
PARP2 gene (primers and probe PARP2_f, PARP2_f, PARP2_r). The structures of primers and probes are shown in
Supplementary Materials (Table S1). Amplification was carried out according to the following scheme: 3 min at 95 °C, then 40 cycles with fluorescence detection at the annealing stage: 10 s at 95 °C, 20 s at 61 °C. The total RNA of the initial non-transgenic cell line served as a control in this experiment. All measurements were performed in three biological replicates.
Data processing was carried out using Bio-Rad CFX Manager 3.1 software. Normalized expression was calculated using the formula:
where
E is the PCR efficiency and Ct the threshold fluorescence cycle in the test and control samples, respectively.
2.10. ELISA of Target Protein (dIFN) in Monoclonal Cell Lines
For ELISA, we used protein extracts obtained from a series of A. thaliana monoclonal cell lines with target insertions of the dIFN gene in the region of the HTR5 gene and 7 cell lines with insertions of the studied gene into the random regions of the genome. Protein extracts of A. thaliana non-transgenic cell culture served as a negative control. Protein extract of a non-transgenic cell culture of A. thaliana supplied with 1 μg/μL of dIFN, which we had previously synthesized in E. coli, was used as a positive control. dIFN in E. coli cells was synthesized on the same matrix that was used in genetic constructs for the production of knock-ins in plant cells.
To extract proteins, 40 mL of a 7-day cell suspension culture was centrifuged for 10 min at 12,000× g (Allegra X-30R, Beckman Coulter, Loveland, CO, USA). The cell precipitate was triturated in liquid nitrogen and a 0.7 g sample was transferred to a 2 mL tube with the addition of 1400 μL of PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 with 8 M urea). Then, the extracts were homogenized on Vibra-Cell™ Ultrasonic Liquid Processors VCX130 (USA) according to the program: 5 cycles, amplitude 45%, 10 s on, 15 s off. The extracts were centrifuged for 10 min at 14,000× g (Eppendorf, Hamburg, Germany) and the supernatant was collected in a separate tube.
The protein extract (100 μL) in four replicates was introduced into the wells of a 96-well MICROLON
® plate (Greiner Bio-One, Frickenhausen, Germany). The sorption on the plate was carried out overnight at a temperature of 10 °C. After sorption, the wells were washed with 250 μL of PBST (PBS buffer with the addition of 0.1% Tween 20) and then 250 μL of 0.5% fat-free dry milk in PBS was added to each well and incubated for 30 min at room temperature with shaking, after which the wells were rinsed again with 250 μL PBST. The remaining liquid was removed with a water-jet pump. In total, 100 μL of primary antibodies (anti-interferon gamma (human) antibodies IMS01-145-319, Agrisera, Sweden) diluted in PBS buffer (1:5000) was added to each well and the plates were incubated with shaking for 1 h at room temperature. After washing three times with 300 μL PBST, 100 μL of secondary antibodies (Goat Anti-Chicken IgY (H&L), HRP conjugated, AS09 603, Agrisera, Sweden) diluted in PBS (1:10000) was added to each well and incubated with shaking for 1 h. After washing the wells three times with 300 μL PBST, 100 μL TMB (3,3′,5,5′-tetramethylbenzidine; Abcam, UK) was added to each well and incubated with shaking for 30 min in the dark at room temperature. The reaction was stopped by adding 100 μL of 0.1 M HCl. Within 15 min after stopping the reaction, the optical density was measured on a Victor X3 2030 device (PerkinElmer, Akron, OH, USA) at a wavelength of 450 nm. All measurements were carried out in three biological replicates. The optical density (OD) value of the negative control was subtracted from the OD of all experimental values and the positive control. The accumulation of the target dIFN protein in the analyzed cell lines was estimated as a percentage of the TSP. The concentration of TSP was determined by the Bradford method [
31] using BSA solution in the PBS buffer as standards. The measurements were carried out in three biological replicates. The ELISA results were statistically processed using the Statistica package.
4. Discussion
Despite the attractiveness of using higher plant cell cultures for the commercial production of biopharmaceuticals, the yield of recombinant protein in this expression system is still low compared to animal cell cultures. Typically, the yield of the recombinant protein is about 1% of the TSP [
32,
33]. In whole plants, the problem of low yield can be solved by transient expression or expression of the target gene in chloroplasts [
1,
8]. To increase the biosynthesis of recombinant proteins in cell suspension culture, researchers use various methods, including optimizing the expression of target genes due to regulatory elements in expression cassettes, reducing the degradation of target proteins, optimizing the conditions for cultivating plant cells, etc. [
34,
35]. In general, the problem of increasing the biosynthesis of recombinant proteins is solved through the search (screening) of the most “successful” events of insertion of the target gene into the plant genome and selection of “elite” highly productive cells. Then, based on “elite” cells, monoclonal suspension cell cultures are formed [
36]. After successful delivery of the target gene into the genome, the plant cell culture is a heterogeneous mass of genetically and epigenetically different cells with different expression levels, the number of transgenic copies, and different insertion sites, all of which significantly affect their productivity. Only a small proportion of cells from the original transformants remain capable of producing the recombinant protein with high efficiency.
The observed variability in the ability to biosynthesize and accumulate recombinant proteins in plant cells is largely determined by the organization of the transgene insertion region. The optimally high level of target gene expression, planned by the researchers during the design of the genetic construct, may not be realized when the transgene enters the transcriptionally inactive regions of the genome. Thus, the identification of transcriptionally active regions of the genome and directed integration of target genes into these regions opens up new prospects for researchers to increase the synthetic capabilities of plant cells for the production of recombinant proteins.
The rapid development of genomic editing methods using CRISPR/Cas9 opens up the opportunity for researchers to deliver target genes to constitutively transcribed regions of the genome and to obtain highly productive lines producing recombinant proteins. Plant cell housekeeping genes that are actively expressed during the entire interphase of the cell cycle can be potential target sites for the integration of target genes. We tried to implement this approach in this study and evaluate the possibilities of increasing the accumulation of recombinant protein in the case of integration of the target dIFN gene into the region of one of the housekeeping genes—the histone H3.3 gene HTR5.
Despite a significant increase in the number of works on editing the plant cell genome, the number of experimental works devoted to gene insertion using the knock-in method is still small [
13,
16]. The DNA DSB, which is formed as a result of the action of Cas9, initiates cellular repair mechanisms, thereby making the break site available for insertion of donor DNA. In plant cells, as in most eukaryotic cells, DSB are usually repaired through the NHEJ mechanism, which functions throughout the entire cell cycle, except for mitosis. The HDR pathway, which provides accurate sequencing, is only possible in the presence of a sister chromatid (homologous template) and can only occur at the end of the S and G2 phases of the cell cycle [
14,
15,
37]. Thus, the predominance of the first mechanism over the second in plants poses very difficult tasks for researchers in the case of attempts to carry out genomic editing in the knock-in variant. Indeed, in plants, the frequency of knock-out obtained during the restoration of DSB by the NHEJ mechanism is 30–70%, and in some cases up to 100%, while the knock-in frequency is usually at a level of a part of a percent or a few percent [
15,
16].
As a rule, most of the genetic constructs intended for the delivery of transgenes to the target region using the CRISPR/Cas9 genomic editing technology include flanking DNA regions homologous to the insertion site [
21,
38,
39,
40,
41]. To increase the efficiency of delivery of the target construct to a given region of the genome of
A. thaliana plants [
38] and
Zea mays [
21,
41], a technique was used that allows the release of the target fragment in the cell in the form of a linear template from the circular plasmid DNA with the help of Cas9 nuclease. In experiments of Peterson and colleagues, direct comparison of T-DNA vectors with and without target sites flanking repair templates consistently demonstrated an approximately three-fold increase in targeted gene insertion frequency [
21]. The use of this approach made it possible to deliver target genes to the target region with a site-specific insertion efficiency of 0.14% for
A. thaliana and 4.7% for
Zea mays [
38,
41]. The advancements in Agrobacterium-mediated maize transformation, combined with optimized vector design, enabled an approximately 100-fold improvement in the efficiency of targeted gene insertion [
21]. Adding Cas9 nuclease recognition sites was effective not only in the case of flanking DNA homologous to the insertion site (pIFN(H3.3)3 construct) but also in the absence of such flanks (pIFN(H3.3)2 construct). Thus, in genomic editing of cell cultures in the knock-in variant, the presence of plant DNA homology arms alone is not enough for successful site-specific insertion of the target gene. When the pIFN(H3.3).1 genetic construct was used, as a result of 10 biolistic transformations, not a single event of
dIFN gene delivery to the region of the
HTR5 gene was detected. At the same time, the inclusion of Cas9 nuclease recognition sites in the flanking regions in the pIFN(H3.3).3 construct made it possible to identify six events of transgene delivery to the target region with the same number of biolistic transformations performed. Although in the case of using the pIFN(H3.3)2 construct, only three desired events were identified, this approach can also be successfully used in the work. The absence of flanking DNA simplifies the process of creating a genetic construct associated with the addition of homology regions to the construct or, on the contrary, their replacement when the target site is changed. Thus, as the experiments have shown, the introduction of Cas9 recognition sites into the genetic construct, intended for its excision from the circular plasmid in the cell, seems to be a very effective approach for integrating transgenes into target regions of plant cell cultures. It seems more promising to us to use a template without flanking homology arms and carrying only the Cas9 recognition sites, since such a design, based on insertion by the NHEJ mechanism, excludes additional stages of construction and, with a fairly high frequency, can lead to the successful production of lines with knock-in.
It is worth emphasizing that an important problem that remains is the low efficiency of the selection of cells carrying knock-in since plant cells in suspension culture are prone to the formation of cell aggregates. Separating knock-in cells from cells in which the same construct is integrated into a random region of the genome is a laborious task. It is desirable to monitor the presence of knock-in regularly even among cells that have successfully passed selection and carry a confirmed knock-in, since it is known that cells that produce foreign proteins at a high level tend to be displaced from a heterogeneous culture [
36,
42]. To reduce the heterogeneity of the cell culture and obtain monoclonal lines with knock-ins, it is necessary to control the cell population for the presence of knock-in, since the heterogeneity of the culture also often leads to loss of cells carrying the transgene [
43,
44,
45] or knock-in. We faced such a problem in our experiment—some of the cell lines with knock-ins were lost during their selection and creation of monoclonal cell lines based on them. Seven kanamycin-resistant cell lines, in which PCR with genomic DNA did not reveal fragments of the corresponding size, indicating the insertion of the transgene into the target region of the
HTR5 gene, formed the basis for the creation of monoclonal cell lines with random insertions.
The created series of monoclonal cell lines served as a convenient model for analyzing the expression features of the target
dIFN gene integrated into the region of the
HTR5 gene, characterized by high transcriptional activity, and assessing the possibility of increasing the accumulation of the recombinant protein. Contrary to expectations, a high level of accumulation of the dIFN protein was found only among monoclones of line 1, which reached more than 2% of the TSP in the case of monoclone 1.4. In general, the accumulation of the target protein varied both between cell lines and between individual monoclones derived from this line. The region of the
HTR5 gene was used as a target for the delivery of the
dIFN gene due to its high transcriptional activity in the cells of the
A. thaliana suspension culture [
15]; however, the results of RT-PCR did not reveal a direct correlation between protein accumulation and the amount of mRNA with which this protein is synthesized. Moreover, for lines 38 and 4, mRNA was not detected at all, but the protein was detected in monoclones.
The observed high variability in the level of accumulation of the dIFN protein in lines with knock-in can be possibly explained by the fact that during insertion, mutations occur in the regulatory region of the target genes that affect their expression. The contribution of somaclonal variability in the diversity between cell lines in target gene expression and protein accumulation is also not excluded. The variability in the expression of several genes integrated into the rice genome using recombinase is shown in the work [
46]. Forsyth et al. [
47] did not observe any variability in the expression of marker genes integrated into the promoter region of the potato
Ubi7 gene using TALEN. However, in this work, the target marker genes were inserted into the genome in such a way that they are placed under the control of the native promoter (the
Ubi7 gene promoter), and it is possible that the absence of expression variability is associated with this fact. The absence of expression variability within a single insertion site was demonstrated in a study on site-specific gene insertion into the tobacco genome using the Cre/Lox system [
48]. However, the authors noted the silencing of transgenes, including mosaic silencing, among the resulting transgenic plants. The authors found that the level of transgene expression differed up to 10 times depending on the insertion site [
48]. The absence of detectable transcription of the target gene in the presence of a detectable target protein for lines 38 and 4 may be due to the instability of the target protein mRNA synthesized in these lines. For these cell lines, the possibility of integration of target gene DNA fragments into random regions of the genome is not excluded, which could act as a trigger for the formation of short interfering RNAs that trigger its silence. The possibility of formation of a mosaic population of cells in plants with insertions of DNA fragments in inverse orientation to a target gene was shown by us earlier for tobacco plants [
49]. Further work will be aimed at identifying factors associated with the variability of expression and accumulation of the target recombinant protein among site-specific events of integration of the target gene into regions of the genome with high transcriptional activity.
It should be emphasized that the integration of foreign genes in some cases is accompanied by a change in the expression of own plant genes located in the insertion region. It was found that insertions of transgenes can cause both large-scale chromosomal rearrangements and point mutations, as well as affect the epigenetic landscape of the surrounding chromatin [
50]. Our analysis of the expression level of both plant genes surrounding the insertion site did not reveal any significant changes in their expression. The results obtained indicate that the site-specific insertion of the transgene did not disrupt the expression of the
HTR5 and 12 kDa subunit of microsomal peptidase genes, a change in the expression of which could reduce the viability of cells, as well as their ability to reproduce.
Even though the accumulation of dIFN protein in cell lines with targeted insertions into the target region of the HTR5 gene does not statistically differ from the level of accumulation of dIFN protein in the group of lines with random integration of the transgene, it is among the knock-ins that lines 1–4 were identified, a monoclone of line 1, the accumulation of protein in which is more than 2% of the TSP. This cell line is of interest as a bioproducer for the accumulation of recombinant interferon gamma and can be used to reveal the potential for increasing the biosynthesis of recombinant protein by optimizing the conditions for cultivating plant cells and optimizing the composition of nutrient media.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}