
Glucocorticoid-Induced Leucine Zipper (GILZ) in Cardiovascular Health and Disease




Abstract
:1. Introduction
2. Glucocorticoid Signaling
3. GILZ in the Immune System
4. GCs in Cardiovascular Physiology and Diseases
5. GILZ in the Cardiovascular System
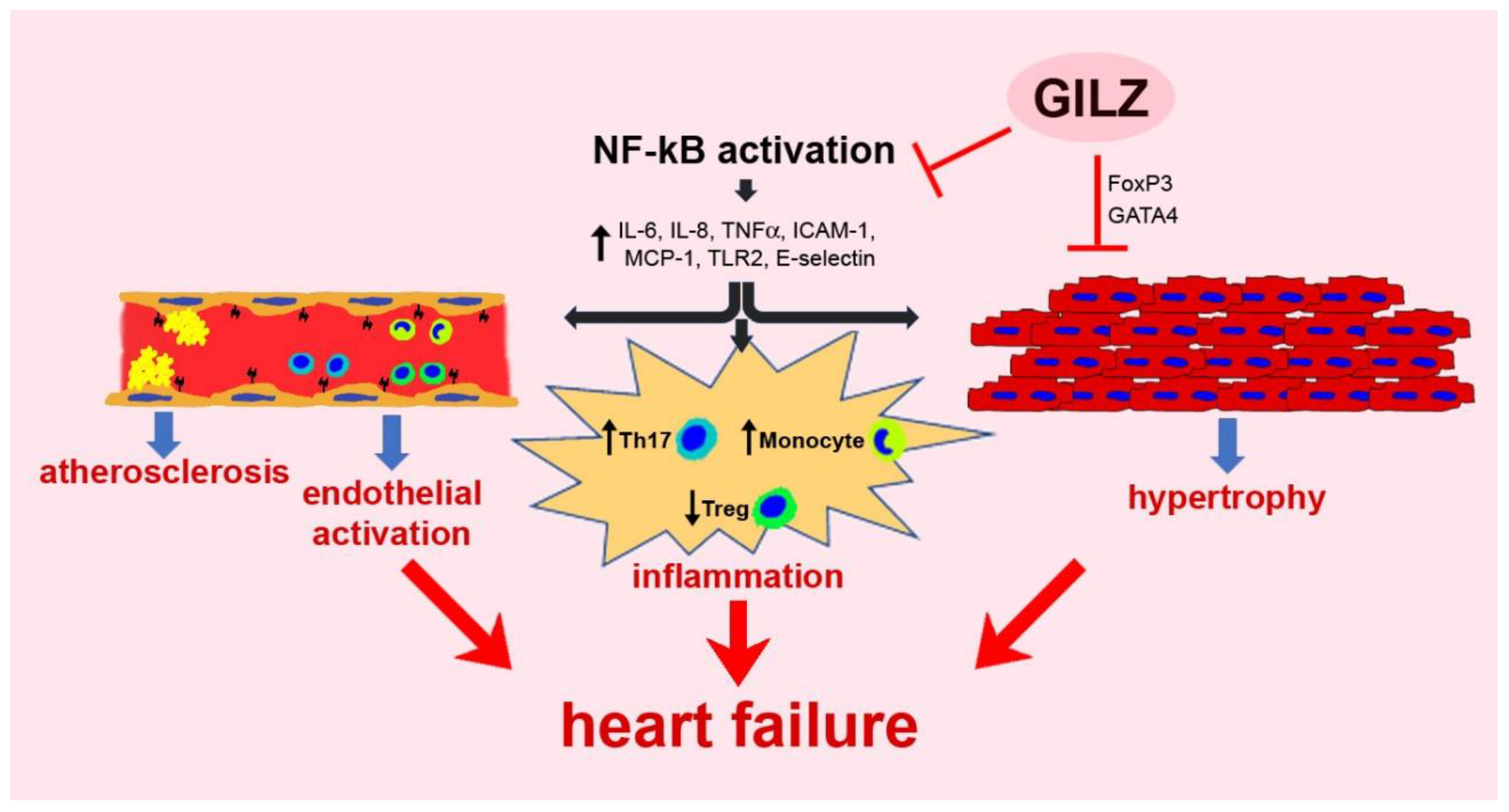
5.1. Chronic Inflammation
5.2. Vascular Dysfunction
5.3. Myocardial Damage and Remodeling
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Steptoe, A.; Kivimäki, M. Stress and cardiovascular disease. Nat. Rev. Cardiol. 2021, 9, 360–370. [Google Scholar] [CrossRef]
- Selye, H. A syndrome produced by diverse nocuous agents. Nature 1936, 138, 32. [Google Scholar] [CrossRef]
- Selye, H.; Fortier, C. Adaptive reactions to stress. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1949, 29, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Mercanoglu, G.; Macit, C.; Mercanoglu, F. Stress as a risk factor for cardiovascular events. Cardiol. Pharmacol. 2015, 4, 2. [Google Scholar] [CrossRef]
- van den Beld, A.W.; Kaufman, J.M.; Zillikens, M.C.; Lamberts, S.W.J.; Egan, J.M.; van der Lely, A.J. The physiology of endocrine systems with ageing. Lancet Diabetes Endocrinol. 2018, 6, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, S.; Souffriau, J.; Libert, C. A General introduction to glucocorticoid biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.R. Glucocorticoids and cardiovascular disease. Eur. J. Endocrinol. 2007, 157, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Z.; Wang, Y.X.; Jiang, C.L. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Rog-Zielinska, E.A.; Richardson, R.V.; Denvir, M.A.; Chapman, K.E. Glucocorticoids and foetal heart maturation; implications for prematurity and foetal programming. J. Mol. Endocrinol. 2014, 52, R125–R135. [Google Scholar] [CrossRef] [Green Version]
- Walejko, J.M.; Antolic, A.; Koelmel, J.P.; Garrett, T.J.; Edison, A.S.; Keller-Wood, M. Chronic maternal cortisol excess during late gestation leads to metabolic alterations in the newborn heart. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E546–E556. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, T.N.; Knight, J.K.; Goodwin, J.E. The Glucocorticoid receptor in cardiovascular health and disease. Cells 2019, 8, 1227. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of action in health and disease. Rheum. Dis. Clin. North Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef]
- Bereshchenko, O.; Bruscoli, S.; Riccardi, C. Glucocorticoids, Sex Hormones, and Immunity. Front. Immunol. 2018, 9, 1332. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Hardy, R.S.; Raza, K.; Cooper, M.S. Therapeutic glucocorticoids: Mechanisms of actions in rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Croxtall, J.D.; Choudhury, Q.; Flower, R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000, 130, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Kfir-Erenfeld, S.; Sionov, R.V.; Spokoini, R.; Cohen, O.; Yefenof, E. Protein kinase networks regulating glucocorticoid-induced apoptosis of hematopoietic cancer cells: Fundamental aspects and practical considerations. Leuk. Lymphoma 2010, 51, 1968–2005. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Kim, H.K.; Youm, J.B.; Dizon, L.A.; Song, I.S.; Jeong, S.H.; Seo, D.Y.; Ko, K.S.; Rhee, B.D.; Kim, N.; et al. Non-genomic effect of glucocorticoids on cardiovascular system. Pflügers Arch. Eur. J. Physiol. 2012, 464, 549–559. [Google Scholar] [CrossRef]
- Song, I.H.; Buttgereit, F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol. Cell. Endocrinol. 2006, 246, 142–146. [Google Scholar] [CrossRef]
- Clarck, A.R.; Lasa, M. Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr. Opin. Pharmacol. 2003, 3, 404–411. [Google Scholar] [CrossRef]
- D’Adamio, F.; Zollo, O.; Moraca, R.; Ayroldi, E.; Bruscoli, S.; Bartoli, A.; Cannarile, L.; Migliorati, G.; Riccardi, C. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity 1997, 7, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Cannarile, L.; Zollo, O.; D’Adamio, F.; Ayroldi, E.; Marchetti, C.; Tabilio, A.; Bruscoli, S.; Riccardi, C. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ. 2001, 8, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Bereshchenko, O.; Migliorati, G.; Bruscoli, S.; Riccardi, C. Glucocorticoid-induced leucine zipper: A novel anti-inflammatory molecule. Front. Pharmacol. 2019, 10, 308. [Google Scholar] [CrossRef] [Green Version]
- Mittelstadt, P.R.; Ashwell, J.D. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J. Biol. Chem. 2001, 276, 29603–29610. [Google Scholar] [CrossRef] [Green Version]
- Ayroldi, E.; Migliorati, G.; Bruscoli, S.; Marchetti, C.; Zollo, O.; Cannarile, L.; D’Adamio, F.; Riccardi, C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood 2001, 98, 743–753. [Google Scholar] [CrossRef]
- Ayroldi, E.; Zollo, O.; Macchiarulo, A.; Di Marco, B.; Marchetti, C.; Riccardi, C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol. Cell. Biol. 2002, 22, 7929–7941. [Google Scholar] [CrossRef] [Green Version]
- Ayroldi, E.; Zollo, O.; Bastianelli, A.; Marchetti, C.; Agostini, M.; Di Virgilio, R.; Riccardi, C. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J. Clin. Investig. 2007, 117, 1605–1615. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, B.; Massetti, M.; Bruscoli, S.; Macchiarulo, A.; Di Virgilio, R.; Velardi, E.; Donato, V.; Migliorati, G.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: Role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res. 2007, 35, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Ronchetti, S.; Pericolini, E.; Gabrielli, E.; Cari, L.; Gentili, M.; Roselletti, E.; Migliorati, G.; Vecchiarelli, A.; Riccardi, C. Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migration via control of annexin A1 expression. FASEB J. 2017, 31, 3054–3065. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.H.; Aeberli, D.; Dacumos, A.; Xue, J.R.; Morand, E.F. Annexin-1 regulates macrophage IL-6 and TNF via glucocorticoid-induced leucine zipper. J. Immunol. 2009, 183, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Delfino, D.V.; Spinicelli, S.; Pozzesi, N.; Pierangeli, S.; Velardi, E.; Bruscoli, S.; Martelli, M.P.; Pettirossi, V.; Falchi, L.; Kang, T.-B.; et al. Glucocorticoid-induced activation of caspase-8 protects the glucocorticoid-induced protein Gilz from proteasomal degradation and induces its binding to SUMO-1 in murine thymocytes. Cell Death Differ. 2011, 18, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Mouly, E.; Hamdi, H.; Maillot, M.C.; Pallardy, M.; Godot, V.; Capel, F.; Balian, A.; Naveau, S.; Galanaud, P.; et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood 2006, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Bruscoli, S.; Biagioli, M.; Sorcini, D.; Frammartino, T.; Cimino, M.; Sportoletti, P.; Mazzon, E.; Bereshchenko, O.; Riccardi, C. Lack of glucocorticoid-induced leucine zipper (GILZ) deregulates B-cell survival and results in B-cell lymphocytosis in mice. Blood 2015, 126, 1790–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruscoli, S.; Sorcini, D.; Flamini, S.; Gagliardi, A.; Adamo, F.; Ronchetti, S.; Migliorati, G.; Bereshchenko, O.; Riccardi, C. Glucocorticoid-induced leucine zipper inhibits interferon-gamma production in B cells and suppresses colitis in mice. Front. Immunol. 2018, 9, 1720. [Google Scholar] [CrossRef] [Green Version]
- Vago, J.P.; Tavares, L.P.; Garcia, C.C.; Lima, K.M.; Perucci, L.O.; Vieira, É.L.; Nogueira, C.R.C.; Soriani, F.M.; Martins, J.O.; Silva, P.M.R.; et al. The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J. Immunol. 2015, 194, 4940–4950. [Google Scholar] [CrossRef] [Green Version]
- Cannarile, L.; Fallarino, F.; Agostini, M.; Cuzzocrea, S.; Mazzon, E.; Vacca, C.; Genovese, T.; Migliorati, G.; Ayroldi, E.; Riccardi, C. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood 2006, 107, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Cannarile, L.; Cuzzocrea, S.; Santucci, L.; Agostini, M.; Mazzon, E.; Esposito, E.; Muià, C.; Coppo, M.; di Paola, R.; Riccardi, C. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology 2009, 136, 530–541. [Google Scholar] [CrossRef]
- Bereshchenko, O.; Coppo, M.; Bruscoli, S.; Biagioli, M.; Cimino, M.; Frammartino, T.; Sorcini, D.; Venanzi, A.; Di Sante, M.; Riccardi, C. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-β signaling. Cell Rep. 2014, 7, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Esposito, E.; Bruscoli, S.; Mazzon, E.; Paterniti, I.; Coppo, M.; Velardi, E.; Cuzzocrea, S.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ) over-expression in T lymphocytes inhibits inflammation and tissue damage in spinal cord injury. Neurotherapeutics 2012, 9, 210–225. [Google Scholar] [CrossRef] [Green Version]
- Luz-Crawford, P.; Tejedor, G.; Mausset-Bonnefont, A.L.; Beaulieu, E.; Morand, E.F.; Jorgensen, C.; Noël, D.; Djouad, F. Glucocorticoid-induced leucine zipper governs the therapeutic potential of mesenchymal stem cells by inducing a switch from pathogenic to regulatory Th17 cells in a mouse model of collagen-induced arthritis. Arthritis Rheumatol. 2015, 67, 1514–1524. [Google Scholar] [CrossRef]
- Crisafulli, C.; Bruscoli, S.; Esposito, E.; Mazzon, E.; Di Paola, R.; Genovese, T.; Bramanti, P.; Migliorati, G.; Cuzzocrea, S. PPAR-alpha contributes to the anti- inflammatory activity of 17beta-estradiol. J. Pharmacol. Exp. Ther. 2009, 331, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, B.; Pieri, L.; Donniacuo, M.; Cappetta, D.; Capuano, A.; Domenici, L.; Carnuccio, R.; Romagnoli, P.; Filippelli, A.; Rossi, F. Rosiglitazone reduces the inflammatory response in a model of vascular injury in rats. Shock 2009, 32, 638–644. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Bruscoli, S.; Mazzon, E.; Crisafulli, C.; Donato, V.; Di Paola, R.; Velardi, E.; Esposito, E.; Nocentini, G.; Riccardi, C. Peroxisome proliferator-activated receptor-alpha contributes to the anti-inflammatory activity of glucocorticoids. Mol. Pharmacol. 2008, 73, 323–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert-Nicoud, M.; Flahaut, M.; Elalouf, J.M.; Nicod, M.; Salinas, M.; Bens, M.; Doucet, A.; Wincker, P.; Artiguenave, F.; Horisberger, J.-D.; et al. Transcriptome of a mouse kidney cortical collecting duct cell line: Effects of aldosterone and vasopressin. Proc. Natl. Acad. Sci. USA 2001, 98, 2712–2716. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Fujiki, K.; Shirahige, K.; Gomez-Sanchez, C.E.; Fujita, T.; Nangaku, M.; Nagase, M. Genome-wide analysis of murine renal distal convoluted tubular cells for the target genes of mineralocorticoid receptor. Biochem. Biophys. Res. Commun. 2014, 445, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, V.; Soundararajan, R.; Pao, A.C.; Li, H.; Pearce, D. Disinhibitory pathways for control of sodium transport: Regulation of ENaC by SGK1 and GILZ. Am. J. Physiol. Renal. Physiol. 2006, 291, F714–F721. [Google Scholar] [CrossRef] [Green Version]
- Cari, L.; Ricci, E.; Gentili, M.; Petrillo, M.G.; Ayroldi, E.; Ronchetti, S.; Nocentini, G.; Riccardi, C. A focused Real Time PCR strategy to determine GILZ expression in mouse tissues. Results Immunol. 2015, 5, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruscoli, S.; Donato, V.; Velardi, E.; Di Sante, M.; Migliorati, G.; Donato, R.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ) and long GILZ inhibit myogenic differentiation and mediate anti-myogenic effects of glucocorticoids. J. Biol. Chem. 2010, 285, 10385–10396. [Google Scholar] [CrossRef] [Green Version]
- Ayyar, V.S.; Almon, R.R.; Jusko, W.J.; DuBois, D.C. Quantitative tissue-specific dynamics of in vivo GILZ mRNA expression and regulation by endogenous and exogenous glucocorticoids. Physiol. Rep. 2015, 3, e12382. [Google Scholar] [CrossRef]
- Venanzi, A.; Di Sante, M.; Bruscoli, S.; Biagioli, M.; Sorcini, D.; Cimino, M.; Frammartino, T.; Bereshchenko, O.; Franconi, F.; Riccardi, C. Recombinant long-glucocorticoid-induced leucine zipper (L-GILZ) protein restores the control of proliferation in gilz KO spermatogonia. Eur. J. Pharm. Sci. 2014, 63, 22–28. [Google Scholar] [CrossRef]
- Fowden, A.L.; Li, J.; Forhead, A.J. Glucocorticoids and the preparation for life after birth: Are there long-term consequences of the life insurance? Proc. Nutr. Soc. 1998, 57, 113–122. [Google Scholar] [CrossRef]
- Roberts, D.; Brown, J.; Medley, N.; Dalziel, S.R. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst. Rev. 2017, 3, CD004454. [Google Scholar] [CrossRef]
- Mulder, E.J.; de Heus, R.; Visser, G.H. Antenatal corticosteroid therapy: Short-term effects on fetal behaviour and haemodynamics. Semin. Fetal Neonatal Med. 2009, 14, 151–156. [Google Scholar] [CrossRef]
- Giraud, G.D.; Louey, S.; Jonker, S.; Schultz, J.; Thornburg, K.L. Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology 2006, 147, 3643–3649. [Google Scholar] [CrossRef] [Green Version]
- Rog-Zielinska, E.A.; Thomson, A.; Kenyon, C.J.; Brownstein, D.G.; Moran, C.M.; Szumska, D.; Michailidou, Z.; Richardson, J.; Owen, E.; Watt, A.; et al. Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 2013, 22, 3269–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, R.H.; Ren, R.; Cruz-Topete, D.; Bird, G.S.; Myers, P.H.; Boyle, M.C.; Schneider, M.D.; Willis, M.; Cidlowski, J.A. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc. Natl. Acad. Sci. USA 2013, 110, 17035–17040. [Google Scholar] [CrossRef] [Green Version]
- Rog-Zielinska, E.A.; Craig, M.A.; Manning, J.R.; Richardson, R.V.; Gowans, G.J.; Dunbar, D.R.; Gharbi, K.; Kenyon, C.J.; Holmes, M.C.; Hardie, D.G.; et al. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: A role for PGC-1α. Cell Death Differ. 2015, 22, 1106–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rossum, E.F.; Koper, J.W.; Huizenga, N.A.; Uitterlinden, A.G.; Janssen, J.A.; Brinkmann, A.O.; Grobbee, D.E.; de Jong, F.H.; van Duyn, C.M.; Pols, H.A.; et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes 2002, 51, 3128–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussel, R.; Reis, A.F.; Dubois-Laforgue, D.; Bellanné-Chantelot, C.; Timsit, J.; Velho, G. The N363S polymorphism in the glucocorticoid receptor gene is associated with overweight in subjects with type 2 diabetes mellitus. Clin. Endocrinol. 2003, 59, 237–241. [Google Scholar] [CrossRef]
- Lin, R.C.; Wang, X.L.; Morris, B.J. Association of coronary artery disease with glucocorticoid receptor N363S variant. Hypertension 2003, 41, 404–407. [Google Scholar] [CrossRef] [Green Version]
- Rosmond, R.; Chagnon, Y.C.; Holm, G.; Chagnon, M.; Pérusse, L.; Lindell, K.; Carlsson, B.; Bouchard, C.; Björntorp, P.A. A glucocorticoid receptor gene marker is associated with abdominal obesity, leptin, and dysregulation of the hypothalamic-pituitary-adrenal axis. Obes. Res. 2000, 8, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Güder, G.; Bauersachs, J.; Frantz, S.; Weismann, D.; Allolio, B.; Ertl, G.; Angermann, C.E.; Störk, S. Complementary and incremental mortality risk prediction by cortisol and aldosterone in chronic heart failure. Circulation 2007, 115, 1754–1761. [Google Scholar] [CrossRef] [Green Version]
- Ren, R.; Oakley, R.H.; Cruz-Topete, D.; Cidlowski, J.A. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology 2012, 153, 5346–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severinova, E.; Alikunju, S.; Deng, W.; Dhawan, P.; Sayed, N.; Sayed, D. Glucocorticoid receptor-binding and transcriptome signature in cardiomyocytes. J. Am. Heart Assoc. 2019, 8, e011484. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of vascular aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Ong, S.L.; Whitworth, J.A. Glucocorticoid-induced hypertension and the nitric oxide system. Expert Rev. Endocrinol. Metab. 2012, 7, 273–280. [Google Scholar] [CrossRef]
- Nauck, M.; Karakiulakis, G.; Perruchoud, A.P.; Papakonstantinou, E.; Roth, M. Corticosteroids inhibit the expression of the vascular endothelial growth factor gene in human vascular smooth muscle cells. Eur. J. Pharmacol. 1998, 341, 309–315. [Google Scholar] [CrossRef]
- Oliver, A.; Ciulla, T.A. Corticosteroids as antiangiogenic agents. Ophthalmol. Clin. North Am. 2006, 19, 345–351. [Google Scholar] [CrossRef]
- Seidel, T.; Fiegle, D.J.; Baur, T.J.; Ritzer, A.; Nay, S.; Heim, C.; Weyand, M.; Milting, H.; Oakley, R.H.; Cidlowski, J.; et al. Glucocorticoids preserve the t-tubular system in ventricular cardiomyocytes by upregulation of autophagic flux. Basic Res. Cardiol. 2019, 114, 47. [Google Scholar] [CrossRef]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tønnessen, T.; Kryshtal, D.O.; et al. Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of inflammation in heart failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Lee, J.; Durand, J.B.; Bies, R.D.; Young, J.B.; Mann, D.L. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 1996, 93, 704–711. [Google Scholar] [CrossRef]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Kessler, S.M.; Bruscoli, S.; Huwer, H.; Riccardi, C.; Kiemer, A.K. Glucocorticoid-induced leucine zipper: A critical factor in macrophage endotoxin tolerance. J. Immunol. 2015, 194, 6057–6067. [Google Scholar] [CrossRef] [Green Version]
- Kaptoge, S.; Seshasai, S.R.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New therapeutic implications of endothelial nitric oxide synthase (eNOS) function/dysfunction in cardiovascular disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [Green Version]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [Green Version]
- McCormick, S.M.; Eskin, S.G.; McIntire, L.V.; Teng, C.L.; Lu, C.M.; Russell, C.G.; Chittur, K.K. DNA microarray reveals changes in gene expression of shear stressed human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. USA 2001, 98, 8955–8960. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Fan, H.; Ngo, D.; Beaulieu, E.; Leung, P.; Lo, C.Y.; Burgess, R.; Van Der Zwan, Y.G.; White, S.J.; Khachigian, L.M.; et al. GILZ overexpression inhibits endothelial cell adhesive function through regulation of NF-κB and MAPK activity. J. Immunol. 2013, 191, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Hahn, R.T.; Hoppstädter, J.; Hirschfelder, K.; Hachenthal, N.; Diesel, B.; Kessler, S.M.; Huwer, H.; Kiemer, A.K. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis 2014, 234, 391–400. [Google Scholar] [CrossRef]
- Gu, R.; Lei, B.; Jiang, C.; Xu, G. Glucocorticoid-induced leucine zipper suppresses ICAM-1 and MCP-1 expression by dephosphorylation of NF-κB p65 in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 631–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Liu, J.Y.; Abdelsayed, R.; Shi, X.; Yu, J.C.; Mozaffari, M.S.; Baban, B. The status of glucocorticoid-induced leucine zipper protein in the salivary glands in Sjögren’s syndrome: Predictive and prognostic potentials. EPMA J. 2016, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, D.C.; Strom, J.; Xu, B.; Kappeler, K.; Chen, Q.M. Expression of glucocorticoid-induced leucine zipper (GILZ) in cardiomyocytes. Cardiovasc. Toxicol. 2013, 13, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, D.; Strom, J.; Chen, Q.M. Glucocorticoid induced leucine zipper inhibits apoptosis of cardiomyocytes by doxorubicin. Toxicol. Appl. Pharmacol. 2014, 276, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, B.; Yin, L.; Qin, X.; Liu, J.Y.; Shi, X.; Mozaffari, M.S. The role of GILZ in modulation of adaptive immunity in a murine model of myocardial infarction. Exp. Mol. Pathol. 2017, 102, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Flamini, S.; Cozzolino, A.; Bereshchenko, O.; Ronchetti, S.; Cianflone, E.; Gagliardi, A.; Ricci, E.; Rafaniello, C.; et al. Deficit of glucocorticoid-induced leucine zipper amplifies angiotensin-induced cardiomyocyte hypertrophy and diastolic dysfunction. J. Cell. Mol. Med. 2021, 25, 217–228. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| In Vitro/In Vivo Model | Treatment | Results | Reference |
|---|---|---|---|
| HUVECs(GILZ overexpression) | TNF-α | Reduction of IL-6, IL-8, E-selectin, ICAM-1, MCP-1 | [87] |
| HUVECs(GILZ silencing) | TNF-α | No effect | [87] |
| HUVECs(GILZ silencing) | TNF-α | Enhancing of NF-κB nuclear translocation; elevation of TLR2, ICAM-1, E-selectin | [88] |
| RMECs(GILZ overexpression) | LPS | Inhibition of NF-κB nuclear translocation; reduction of ICAM-1, MCP-1 | [89] |
| RMECs (GILZ silencing) | LPS | Elevation of ICAM-1, MCP-1 | [89] |
| Mouse salivary gland cells | IL-23 | Inhibition of GILZ; elevation of IL-17; reduction of Del-1 | [90] |
| Primary rat cardiomyocytes, H9c2 cells | Dexamethasone | Elevation of GILZ | [91] |
| C57BL/6 mice | Dexamethasone | Elevation of myocardial GILZ | [91] |
| H9c2 cells(GILZ overexpression) | Doxorubicin | Decreased apoptosis | [92] |
| H9c2 cells(GILZ silencing) | Doxorubicin | Increased apoptosis | [92] |
| Balb/C mice(myocardial infarction) | Injection of GILZ-overexpressing MSCs | Elevation of Tregs, IL-10+ cells; reduction of IL-17+ cells, necrotic cells | [93] |
| GILZ-knockout mice | Angiotensin II infusion | Increased cardiomyocyte hypertrophy, diastolic dysfunction | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappetta, D.; Bereshchenko, O.; Cianflone, E.; Rossi, F.; Riccardi, C.; Torella, D.; Berrino, L.; Urbanek, K.; De Angelis, A.; Bruscoli, S. Glucocorticoid-Induced Leucine Zipper (GILZ) in Cardiovascular Health and Disease. Cells 2021, 10, 2155. https://doi.org/10.3390/cells10082155
Cappetta D, Bereshchenko O, Cianflone E, Rossi F, Riccardi C, Torella D, Berrino L, Urbanek K, De Angelis A, Bruscoli S. Glucocorticoid-Induced Leucine Zipper (GILZ) in Cardiovascular Health and Disease. Cells. 2021; 10(8):2155. https://doi.org/10.3390/cells10082155
Chicago/Turabian StyleCappetta, Donato, Oxana Bereshchenko, Eleonora Cianflone, Francesco Rossi, Carlo Riccardi, Daniele Torella, Liberato Berrino, Konrad Urbanek, Antonella De Angelis, and Stefano Bruscoli. 2021. "Glucocorticoid-Induced Leucine Zipper (GILZ) in Cardiovascular Health and Disease" Cells 10, no. 8: 2155. https://doi.org/10.3390/cells10082155
APA StyleCappetta, D., Bereshchenko, O., Cianflone, E., Rossi, F., Riccardi, C., Torella, D., Berrino, L., Urbanek, K., De Angelis, A., & Bruscoli, S. (2021). Glucocorticoid-Induced Leucine Zipper (GILZ) in Cardiovascular Health and Disease. Cells, 10(8), 2155. https://doi.org/10.3390/cells10082155