
Transcriptomic Analysis of HCN-2 Cells Suggests Connection among Oxidative Stress, Senescence, and Neuron Death after SARS-CoV-2 Infection

 , ,
, ,  ,
,  , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus
2.2. In Vitro HCN-2 SARS-CoV-2 Infection Assay
2.3. RNA-Seq Analysis
2.4. Culture Medium Western Blot
2.5. Statistical Analysis
3. Results
3.1. Virus Replication
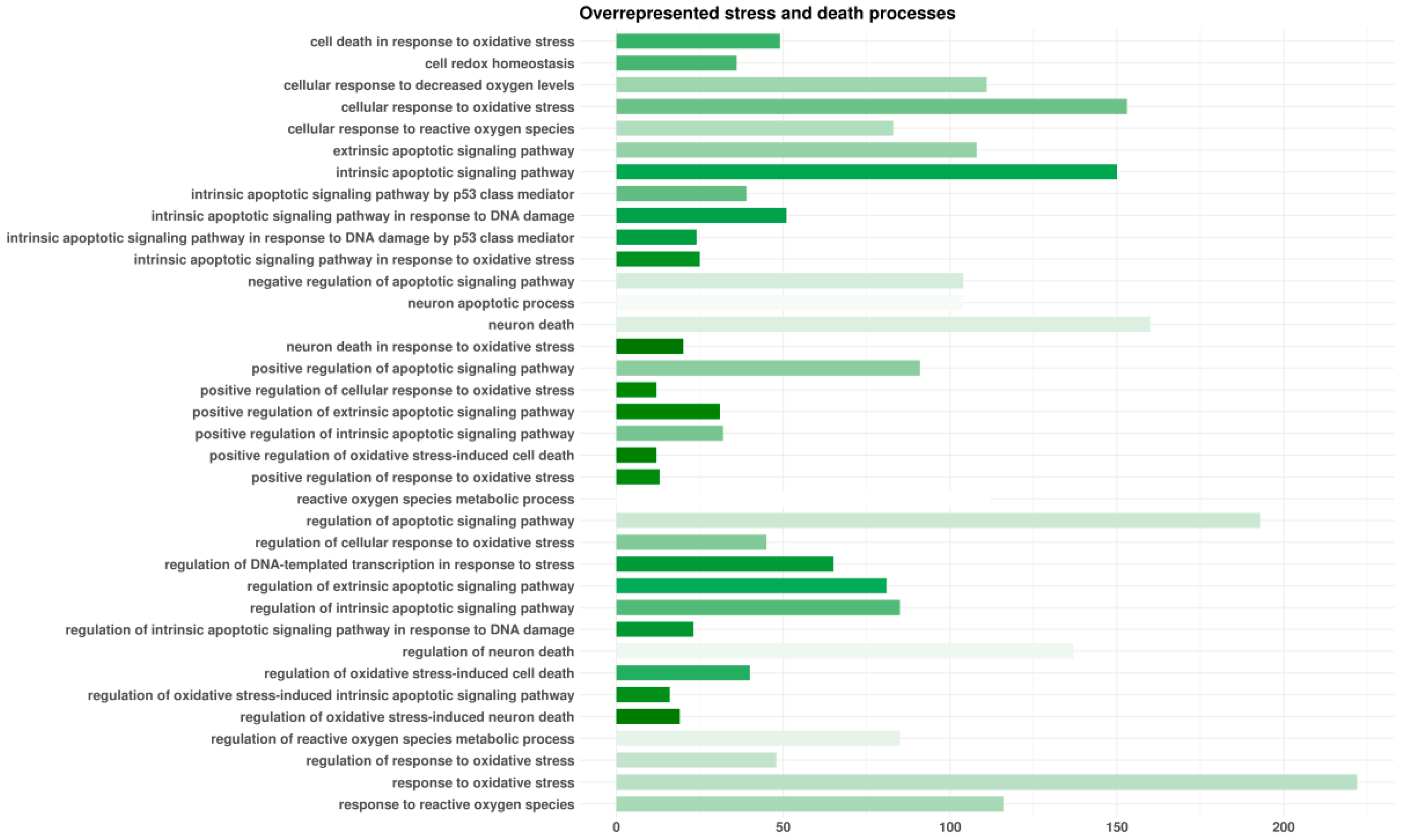
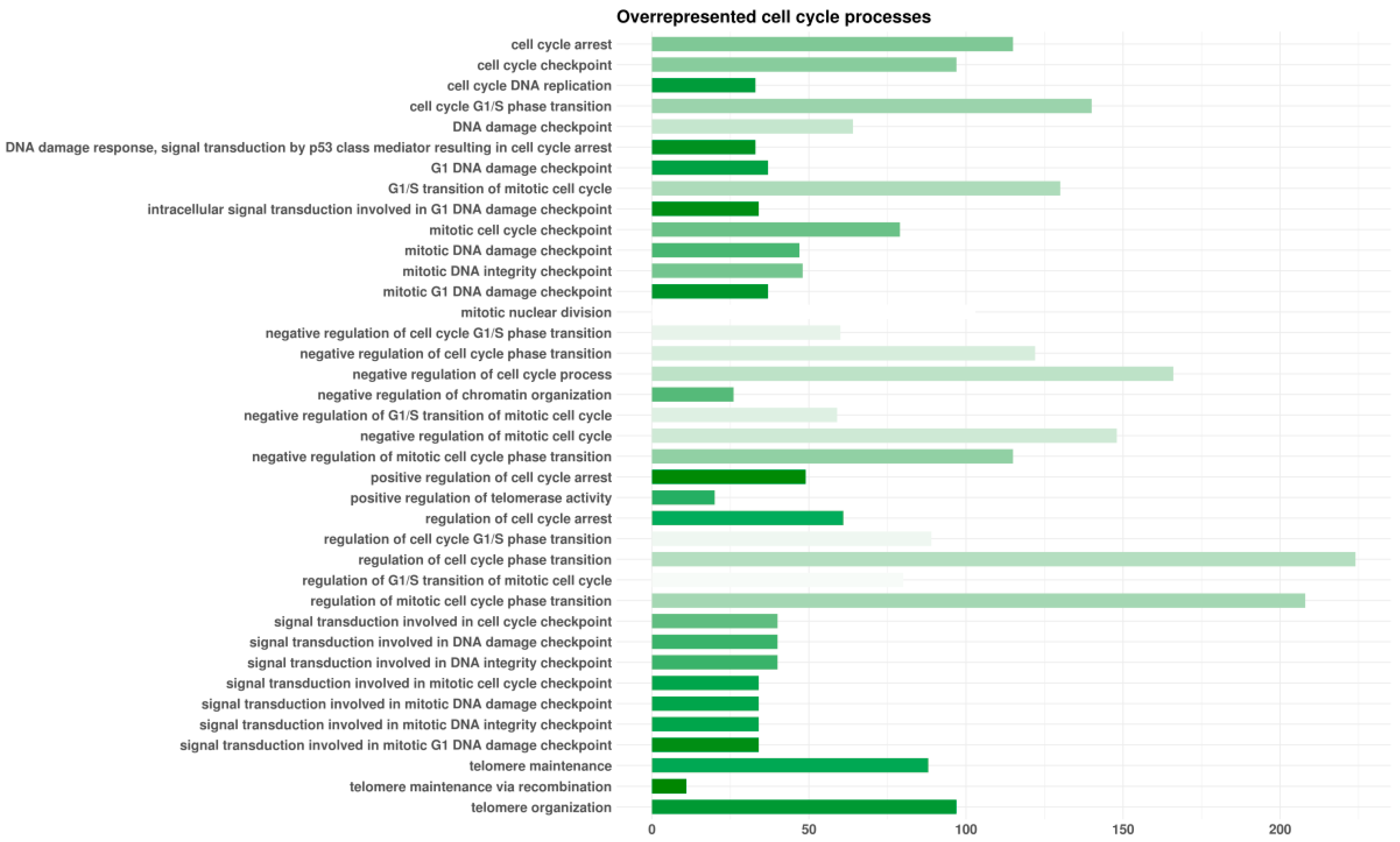
3.2. Enrichment Analyses
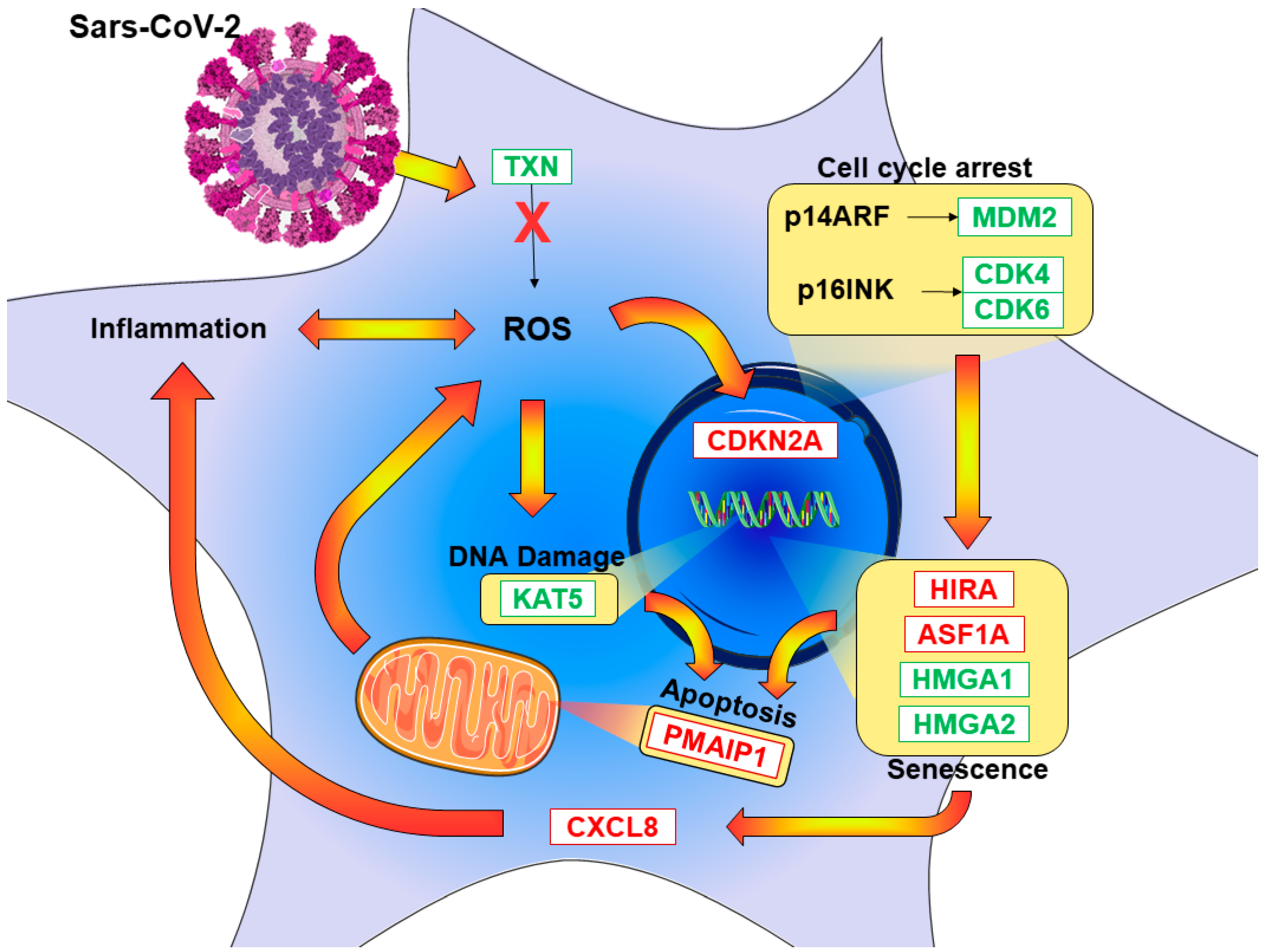
3.3. Pathway Inspection
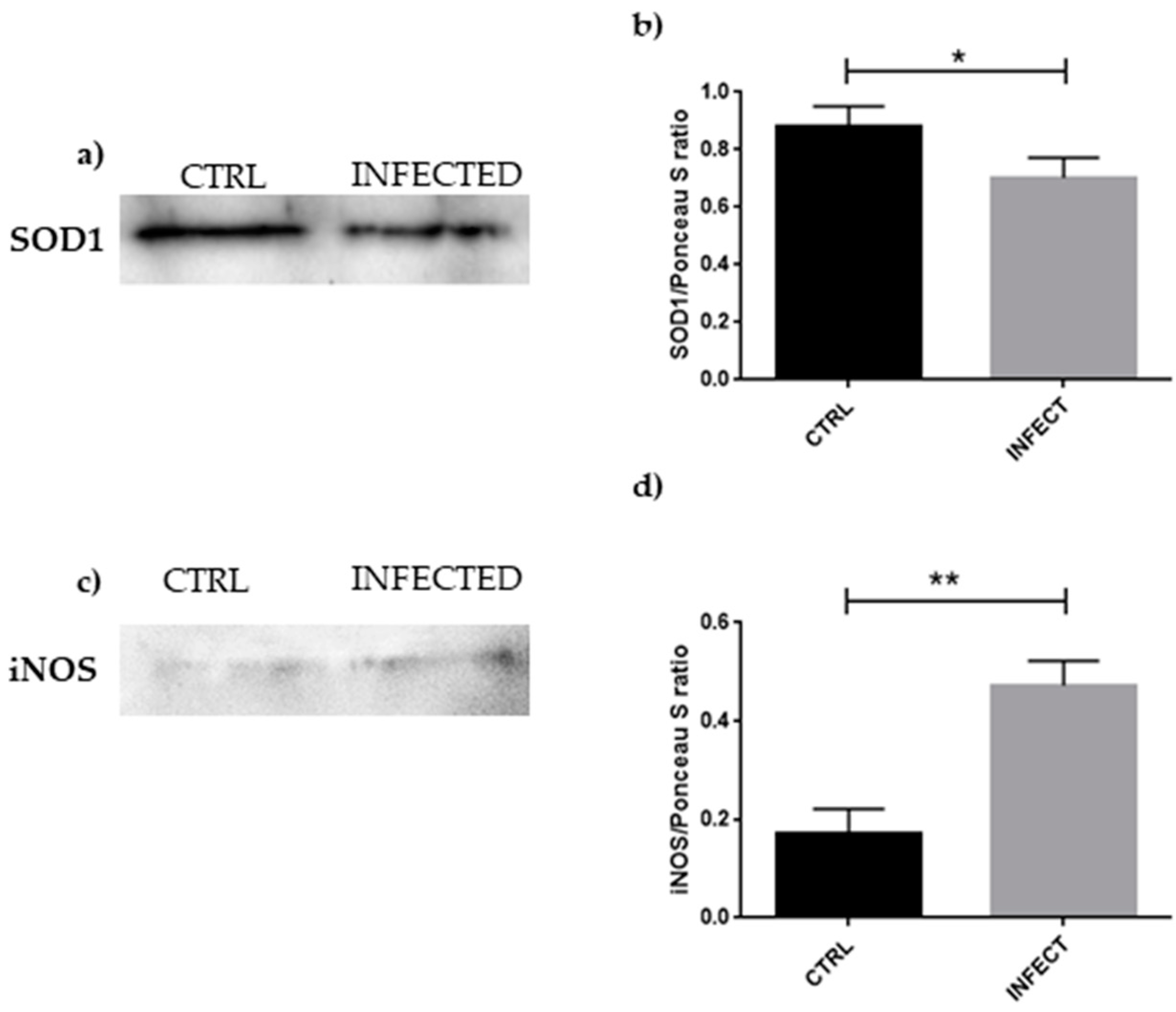
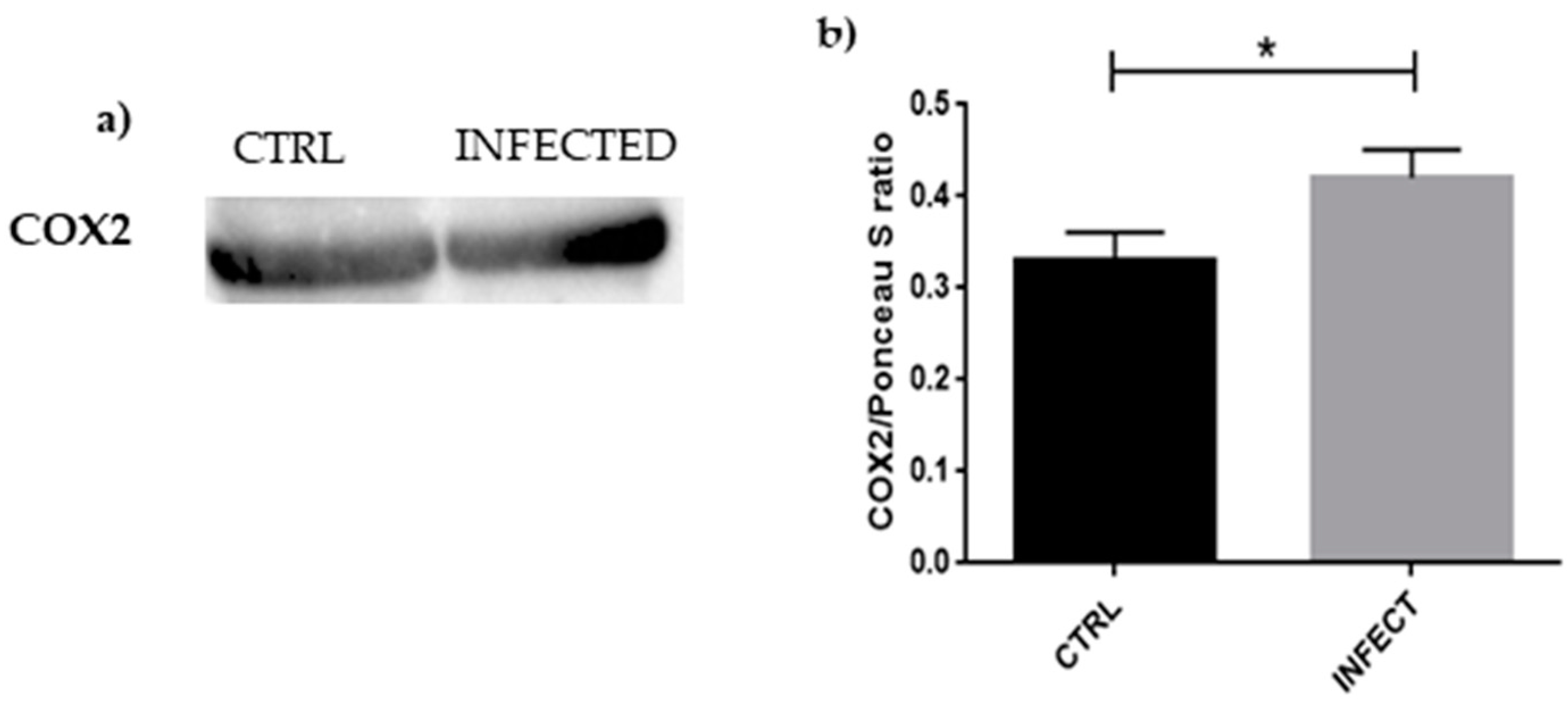
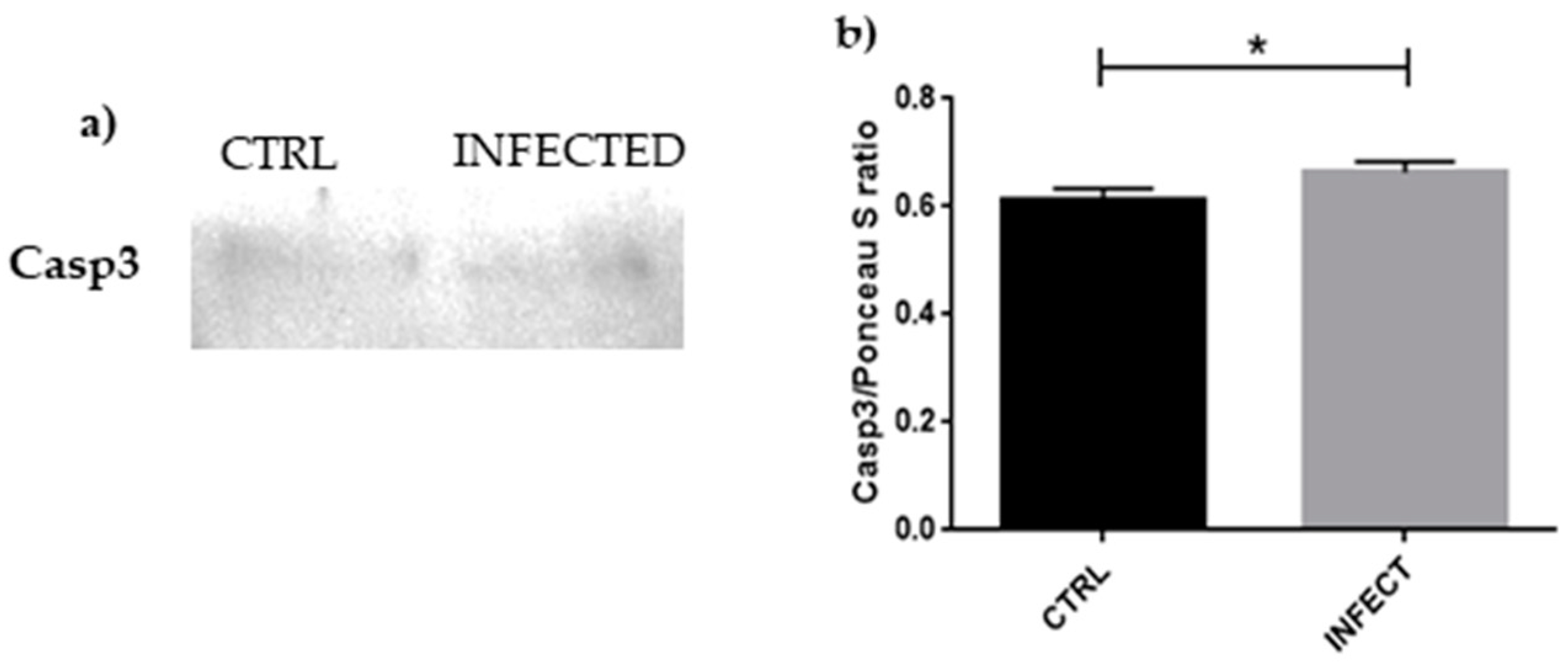
3.4. Western Blot Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cherry, J.; Demmler-Harrison, G.J.; Kaplan, S.L.; Steinbach, W.J.; Hotez, P.J. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2013; Volume 2. [Google Scholar]
- Virology: Coronaviruses. Nature 1968, 220, 650. [CrossRef] [Green Version]
- Clinical Characteristics of COVID-19. Available online: https://www.ecdc.europa.eu/en/covid-19/latest-evidence/clinical (accessed on 4 June 2021).
- Long-Term Effects of COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-ncov/long-term-effects.html (accessed on 12 July 2021).
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature 2021, 594, 259–264. [Google Scholar] [CrossRef]
- Piekarski, F.; Steinbicker, A.U.; Armann, J.P. The multisystem inflammatory syndrome in children and its association to SARS-CoV-2. Curr. Opin. Anaesthesiol. 2021, 34, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood-Brain Barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood–brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Welcome, M.O.; Mastorakis, N.E. Neuropathophysiology of coronavirus disease 2019: Neuroinflammation and blood brain barrier disruption are critical pathophysiological processes that contribute to the clinical symptoms of SARS-CoV-2 infection. Inflammopharmacology 2021, 29, 939–963. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- Gao, Y.-M.; Xu, G.; Wang, B.; Liu, B.-C. Cytokine storm syndrome in coronavirus disease 2019: A narrative review. J. Intern. Med. 2021, 289, 147–161. [Google Scholar] [CrossRef]
- Pero, R.W.; Roush, G.C.; Markowitz, M.M.; Miller, D.G. Oxidative stress, DNA repair, and cancer susceptibility. Cancer Detect. Prev. 1990, 14, 555–561. [Google Scholar]
- Ahamed, M.; Fareed, M.; Kumar, A.; Siddiqui, W.A.; Siddiqui, M.K. Oxidative stress and neurological disorders in relation to blood lead levels in children. Redox Rep. Commun. Free Radic. Res. 2008, 13, 117–122. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef]
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M.K. Mechanisms of Cell Death in Oxidative Stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonney, S.; Seitz, S.; Ryan, C.A.; Jones, K.L.; Clarke, P.; Tyler, K.L.; Siegenthaler, J.A.; Estes, M.K.; Nath, A.; Fujinami, R. Gamma Interferon Alters Junctional Integrity via Rho Kinase, Resulting in Blood-Brain Barrier Leakage in Experimental Viral Encephalitis. mBio 2019, 10, e01675-19. [Google Scholar] [CrossRef] [Green Version]
- Ronnett, G.V.; Hester, L.D.; Nye, J.S.; Snyder, S.H. Human cerebral cortical cell lines from patients with unilateral megalencephaly and Rasmussen’s encephalitis. Neuroscience 1994, 63, 1081–1099. [Google Scholar] [CrossRef]
- Peyrl, A.; Krapfenbauer, K.; Slavc, I.; Strobel, T.; Lubec, G. Proteomic characterization of the human cortical neuronal cell line HCN-2. J. Chem. Neuroanat. 2003, 26, 171–178. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Cheung, M.-C.; Perera, R.A.P.M.; Ng, K.-C.; Bui, C.H.T.; Ho, J.C.W.; Ng, M.M.T.; Kuok, D.I.T.; Shih, K.C.; Tsao, S.-W.; et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: An analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. 2020, 8, 687–695. [Google Scholar] [CrossRef]
- Clerici, M.; Seminari, E.; Suter, F.; Castelli, F.; Pan, A.; Biasin, M.; Colombo, F.; Trabattoni, D.; Maggiolo, F.; Carosi, G.; et al. Different immunologic profiles characterize HIV infection in highly active antiretroviral therapy-treated and antiretroviral-naïve patients with undetectable viraemia. The Master Group. AIDS 2000, 14, 109–116. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Panda, P.K.; Sharawat, I.K.; Panda, P.; Natarajan, V.; Bhakat, R.; Dawman, L. Neurological Complications of SARS-CoV-2 Infection in Children: A Systematic Review and Meta-Analysis. J. Trop. Pediatr. 2020, 67, fmaa070. [Google Scholar] [CrossRef]
- Schwarz, K.B. Oxidative stress during viral infection: A review. Free. Radic. Biol. Med. 1996, 21, 641–649. [Google Scholar] [CrossRef]
- Collet, J.F.; Messens, J. Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 2010, 13, 1205–1216. [Google Scholar] [CrossRef]
- Singh, J.; Dhindsa, R.S.; Misra, V.; Singh, B. SARS-CoV2 infectivity is potentially modulated by host redox status. Comput. Struct. Biotechnol. J. 2020, 18, 3705–3711. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1 1This review is based on the licentiate thesis. Free. Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, J.; Peng, S.; Li, X.; Fang, J. Securinine disturbs redox homeostasis and elicits oxidative stress-mediated apoptosis via targeting thioredoxin reductase. BBA Mol. Basis Dis. 2017, 1863, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, G.W.; Reodica, M.; Davies, M.J.; Heeren, G.; Jarolim, S.; Pillay, B.; Breitenbach, M.; Higgins, V.J.; Dawes, I.W. Superoxide radicals have a protective role during H2O2 stress. Mol. Biol. Cell 2013, 24, 2876–2884. [Google Scholar] [CrossRef]
- Lin, X.; Wang, R.; Zou, W.; Sun, X.; Liu, X.; Zhao, L.; Wang, S.; Jin, M. The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression. Viruses 2016, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Honjo, T.; Otsui, K.; Shiraki, R.; Kawashima, S.; Sawamura, T.; Yokoyama, M.; Inoue, N. Essential role of NOXA1 in generation of reactive oxygen species induced by oxidized low-density lipoprotein in human vascular endothelial cells. Endothel. J. Endothel. Cell Res. 2008, 15, 137–141. [Google Scholar] [CrossRef]
- Mattera, L.; Courilleau, C.; Legube, G.; Ueda, T.; Fukunaga, R.; Chevillard-Briet, M.; Canitrot, Y.; Escaffit, F.; Trouche, D. The E1A-associated p400 protein modulates cell fate decisions by the regulation of ROS homeostasis. PLoS Genet. 2010, 6, e1000983. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Kim, H.S.; Lee, J.; Chung, J.; Lee, J.S.; Choi, J.S.; Yoon, T.R.; Kim, H.K.; Chung, H.Y. Down-regulation of oxidative stress and COX-2 and iNOS expressions by dimethyl lithospermate in aged rat kidney. Arch. Pharm. Res. 2014, 37, 1032–1038. [Google Scholar] [CrossRef]
- Koppula, S.; Kumar, H.; Kim, I.S.; Choi, D.-K. Reactive Oxygen Species and Inhibitors of Inflammatory Enzymes, NADPH Oxidase, and iNOS in Experimental Models of Parkinson’s Disease. Mediat. Inflamm. 2012, 2012, 823902. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 Histone Acetylase Complex in DNA Repair and Apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Yasuda, H. Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eymin, B.; Leduc, C.; Coll, J.-L.; Brambilla, E.; Gazzeri, S. p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene 2003, 22, 1822–1835. [Google Scholar] [CrossRef] [Green Version]
- Voncken, J.W.; Niessen, H.; Neufeld, B.; Rennefahrt, U.; Dahlmans, V.; Kubben, N.; Holzer, B.; Ludwig, S.; Rapp, U.R. MAPKAP Kinase 3pK Phosphorylates and Regulates Chromatin Association of the Polycomb Group Protein Bmi1. J. Biol. Chem. 2005, 280, 5178–5187. [Google Scholar] [CrossRef] [Green Version]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 2002, 1602, 73–87. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Gopinathan, L.; Tan, S.L.W.; Padmakumar, V.C.; Coppola, V.; Tessarollo, L.; Kaldis, P. Loss of Cdk2 and cyclin A2 impairs cell proliferation and tumorigenesis. Cancer Res. 2014, 74, 3870–3879. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Yu, Q.; Sicinska, E.; Das, M.; Schneider, J.E.; Bhattacharya, S.; Rideout, W.M.; Bronson, R.T.; Gardner, H.; Sicinski, P. Cyclin E ablation in the mouse. Cell 2003, 114, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-Containing Senescence-Associated Heterochromatin Foci and Senescence Driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nuñez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A Novel Role for High-Mobility Group A Proteins in Cellular Senescence and Heterochromatin Formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.-L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Gao, M.; Fan, H.; Wang, Y.; Xu, X.; Chen, C.; Liu, J.; Kim, J.; Aliyari, R.; et al. Interleukin-8 as a Biomarker for Disease Prognosis of Coronavirus Disease-2019 Patients. Front. Immunol. 2021, 11, 602395. [Google Scholar] [CrossRef]
- Simon, L.S. Role and regulation of cyclooxygenase-2 during inflammation. Am. J. Med. 1999, 106, 37S–42S. [Google Scholar] [CrossRef]
- Zdanov, S.; Bernard, D.; Debacq-Chainiaux, F.; Martien, S.; Gosselin, K.; Vercamer, C.; Chelli, F.; Toussaint, O.; Abbadie, C. Normal or stress-induced fibroblast senescence involves COX-2 activity. Exp. Cell Res. 2007, 313, 3046–3056. [Google Scholar] [CrossRef]
- Chen, J.S.; Alfajaro, M.M.; Chow, R.D.; Wei, J.; Filler, R.B.; Eisenbarth, S.C.; Wilen, C.B.; Gallagher, T. Nonsteroidal Anti-inflammatory Drugs Dampen the Cytokine and Antibody Response to SARS-CoV-2 Infection. J. Virol. 2021, 95, e00014–e00021. [Google Scholar] [CrossRef]
- Casolini, P.; Catalani, A.; Zuena, A.R.; Angelucci, L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J. Neurosci. Res. 2002, 68, 337–343. [Google Scholar] [CrossRef]
- Liu, Y.; Min, W. Thioredoxin Promotes ASK1 Ubiquitination and Degradation to Inhibit ASK1-Mediated Apoptosis in a Redox Activity-Independent Manner. Circ. Res. 2002, 90, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Im, J.-Y.; Lee, K.-W.; Woo, J.-M.; Junn, E.; Mouradian, M.M. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estus, S.; Zaks, W.J.; Freeman, R.S.; Gruda, M.; Bravo, R.; Johnson, E.M., Jr. Altered gene expression in neurons during programmed cell death: Identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 1994, 127, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, D.; McQuade, J.S.; Behbehani, M.; Tsien, J.Z.; Xu, M. c-fos regulates neuronal excitability and survival. Nat. Genet. 2002, 30, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Bullitt, E. Expression of C-fos-like protein as a marker for neuronal activity following noxious stimulation in the rat. J. Comp. Neurol. 1990, 296, 517–530. [Google Scholar] [CrossRef]
- Heneka, M.T.; Feinstein, D.L.; Galea, E.; Gleichmann, M.; Wüllner, U.; Klockgether, T. Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J. Neuroimmunol. 1999, 100, 156–168. [Google Scholar] [CrossRef]
- Uehara, T.; Kikuchi, Y.; Nomura, Y. Caspase Activation Accompanying Cytochrome c Release from Mitochondria Is Possibly Involved in Nitric Oxide-Induced Neuronal Apoptosis in SH-SY5Y Cells. J. Neurochem. 1999, 72, 196–205. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, F.; Liu, Y.; Lei, X.; Li, H.; Ji, G.; Yuan, Z.; Jiao, J. Ezh2 regulates adult hippocampal neurogenesis and memory. J. Neurosci. 2014, 34, 5184–5199. [Google Scholar] [CrossRef]
- Paradis-Isler, N. Regulation and function of Argonaute proteins in dendrites of hippocampal neurons. Post-. Post-Doctoral’s Thesis, Département de Neurosciences Faculté de Médecine, Université de Montréal, Montrèal, Italy, April 2017. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | HCN2-CTR Expression | HCN2-SARS-CoV-2 Expression | Fold Change | q-Value | Biological Process |
|---|---|---|---|---|---|
| CDKN2A | 53.60 | 86.25 | 0.69 | 3.10 × 10−4 | Oxidative stress Induced senescence |
| EZH2 | 96.00 | 53.75 | −0.84 | 1.73 × 10−5 | Oxidative stress Induced senescence |
| FOS | 172.00 | 11.25 | −3.93 | 1.75 × 10−28 | Oxidative stress Induced senescence |
| JUN | 413.61 | 209.99 | −0.98 | 6.81 × 10−26 | Oxidative stress Induced senescence |
| MAP3K5 | 50.40 | 35.00 | −0.53 | 4.48 × 10−2 | Oxidative stress Induced senescence |
| MOV10 | 493.61 | 324.99 | −0.60 | 1.42 × 10−14 | Oxidative stress Induced senescence |
| ASF1A | 46.40 | 67.50 | 0.54 | 1.18 × 10−2 | DNA damage/telomere stress-induced senescence |
| CCNA2 | 92.00 | 53.75 | −0.78 | 8.33 × 10−5 | DNA damage/telomere stress-induced senescence |
| CCNE1 | 82.40 | 53.75 | −0.62 | 2.50 × 10−3 | DNA damage/telomere stress-induced senescence |
| CCNE2 | 40.80 | 5.00 | −3.03 | 6.73 ×10−8 | DNA damage/telomere stress-induced senescence |
| HIRA | 11.20 | 21.25 | 0.92 | 2.54 × 10−2 | DNA damage/telomere stress-induced senescence |
| HMGA1 | 5586.56 | 3476.15 | −0.68 | 1.20 × 10−194 | DNA damage/telomere stress-induced senescence |
| HMGA2 | 1301.64 | 338.74 | −1.94 | 4.68 × 10−183 | DNA damage/telomere stress-induced senescence |
| CXCL8 | 25.60 | 73.75 | 1.53 | 7.78 × 10−11 | Oxidative stress Alteration |
| NOXA1 | 3.20 | 8.75 | 1.45 | 4.49 × 10−2 | Oxidative stress Alteration |
| PMAIP1 | 23.20 | 37.50 | 0.69 | 1.97 × 10−2 | Oxidative stress Alteration |
| PTGS2 | 21.6 | 343.74 | 3.99 | 3.73 × 10−82 | Oxidative stress Alteration |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valeri, A.; Chiricosta, L.; Calcaterra, V.; Biasin, M.; Cappelletti, G.; Carelli, S.; Zuccotti, G.V.; Bramanti, P.; Pelizzo, G.; Mazzon, E.; et al. Transcriptomic Analysis of HCN-2 Cells Suggests Connection among Oxidative Stress, Senescence, and Neuron Death after SARS-CoV-2 Infection. Cells 2021, 10, 2189. https://doi.org/10.3390/cells10092189
Valeri A, Chiricosta L, Calcaterra V, Biasin M, Cappelletti G, Carelli S, Zuccotti GV, Bramanti P, Pelizzo G, Mazzon E, et al. Transcriptomic Analysis of HCN-2 Cells Suggests Connection among Oxidative Stress, Senescence, and Neuron Death after SARS-CoV-2 Infection. Cells. 2021; 10(9):2189. https://doi.org/10.3390/cells10092189
Chicago/Turabian StyleValeri, Andrea, Luigi Chiricosta, Valeria Calcaterra, Mara Biasin, Gioia Cappelletti, Stephana Carelli, Gian Vincenzo Zuccotti, Placido Bramanti, Gloria Pelizzo, Emanuela Mazzon, and et al. 2021. "Transcriptomic Analysis of HCN-2 Cells Suggests Connection among Oxidative Stress, Senescence, and Neuron Death after SARS-CoV-2 Infection" Cells 10, no. 9: 2189. https://doi.org/10.3390/cells10092189
APA StyleValeri, A., Chiricosta, L., Calcaterra, V., Biasin, M., Cappelletti, G., Carelli, S., Zuccotti, G. V., Bramanti, P., Pelizzo, G., Mazzon, E., & Gugliandolo, A. (2021). Transcriptomic Analysis of HCN-2 Cells Suggests Connection among Oxidative Stress, Senescence, and Neuron Death after SARS-CoV-2 Infection. Cells, 10(9), 2189. https://doi.org/10.3390/cells10092189