
The Potential of Tissue-Resident Memory T Cells for Adoptive Immunotherapy against Cancer

 , ,
, , 
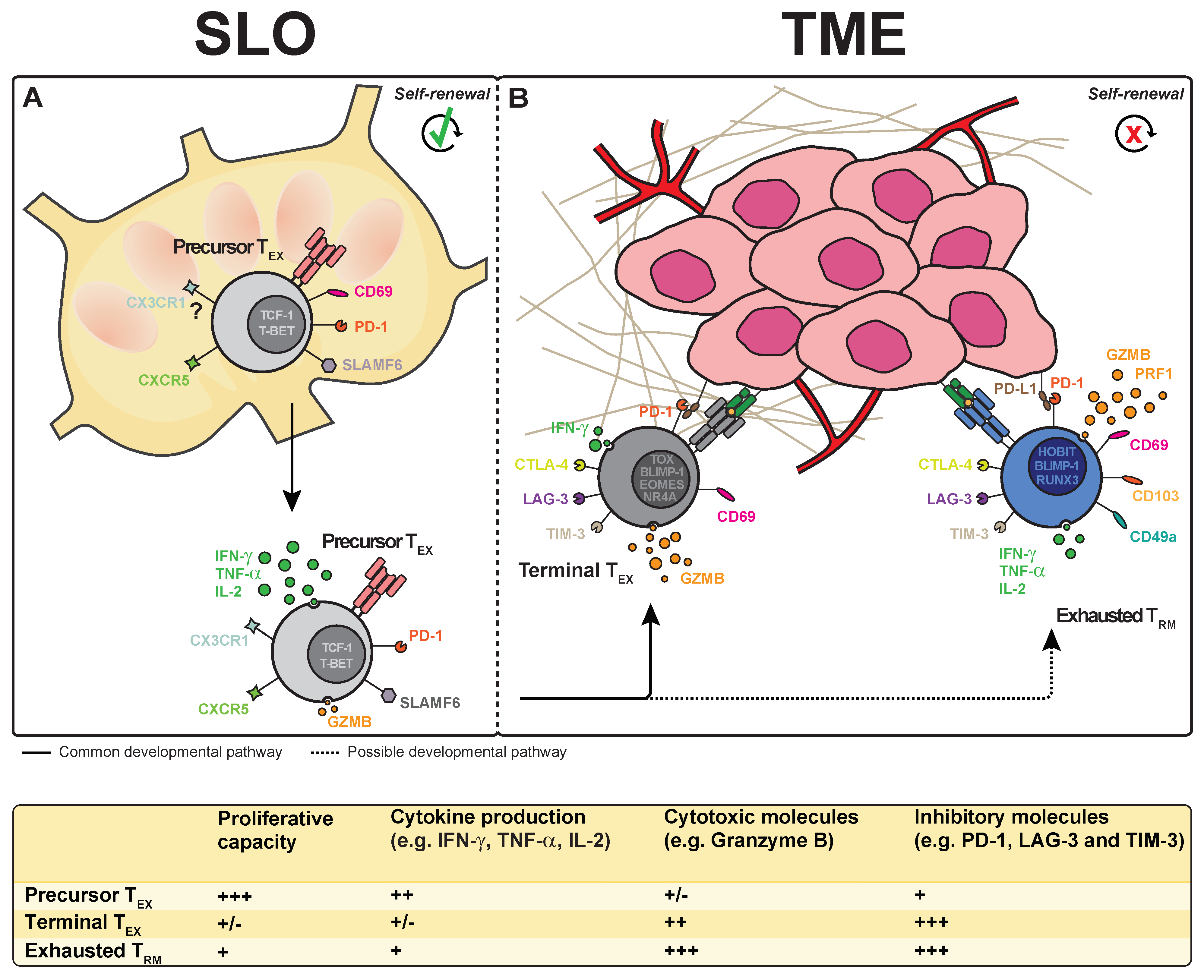{kind=link}
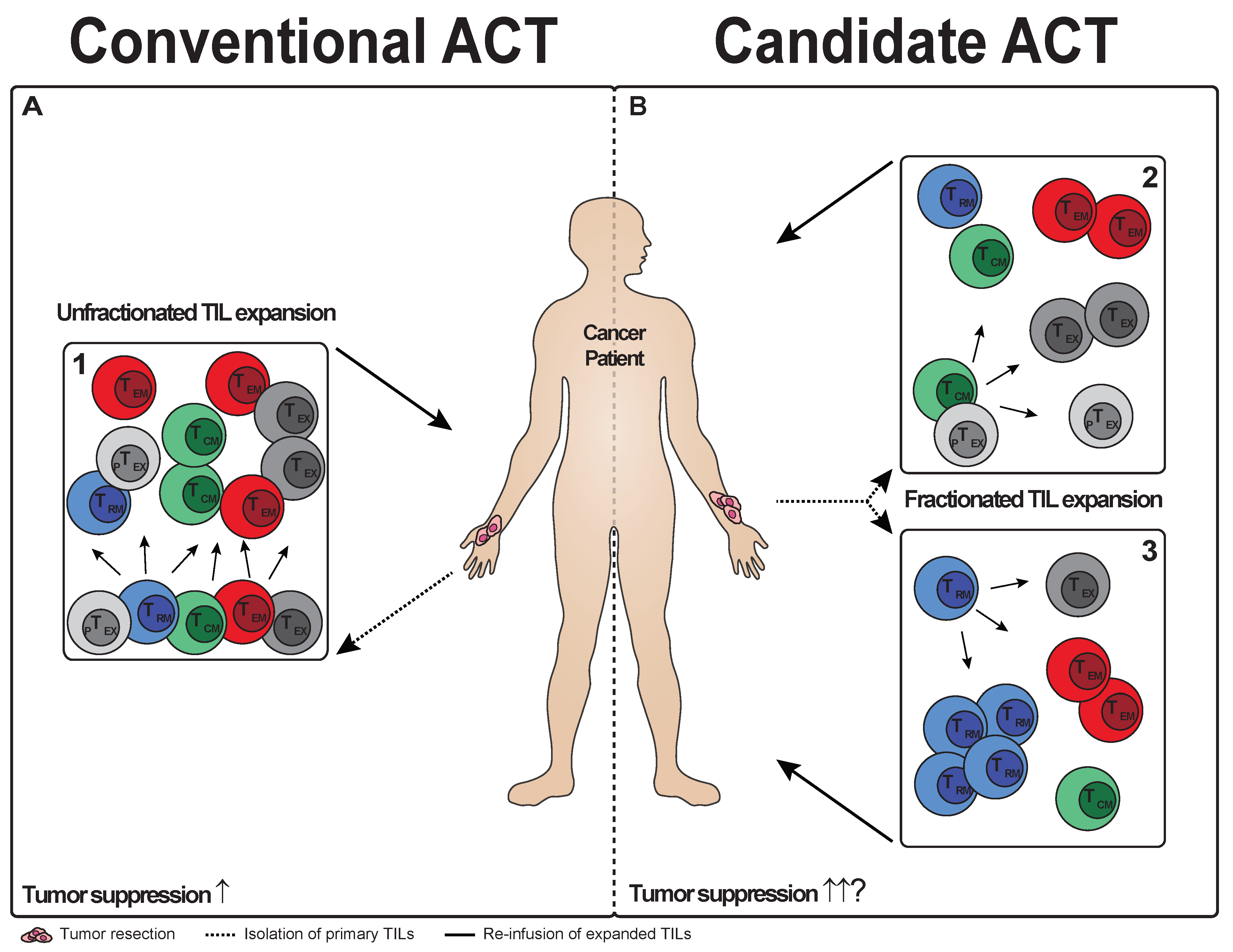{kind=link}
Abstract
:1. Introduction
2. TIL Therapy Is an Important Cancer Immunotherapy
3. Development of T Cell Exhaustion in the Tumor Microenvironment
4. Exhausted T Cell Subsets in Tumor Tissue
5. T Cell Subsets in TIL Therapy
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive cellular therapies: The current landscape. Virchows 2018, 474, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2014, 21, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennock, N.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Restifo, N.P.; Esquivel, F.; Kawakami, Y.; Yewdell, J.W.; Mulé, J.J.; Rosenberg, A.S.; Bennink, J.R. Identification of human cancers deficient in antigen processing. J. Exp. Med. 1993, 177, 265–272. [Google Scholar] [CrossRef]
- Johnsen, A.K.; Templeton, D.J.; Sy, M.; Harding, C. V Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J. Immunol. 1999, 163, 4224–4231. [Google Scholar]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Stary, C.M.; Gao, Q.; Wang, Q.; Zeng, Z.; Jian, Z.; Gu, L.; Xiong, X. Genetically Modified T-Cell-Based Adoptive Immunotherapy in Hematological Malignancies. J. Immunol. Res. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Mayor, M.; Yang, N.; Sterman, D.; Jones, D.R.; Adusumilli, P.S. Immunotherapy for non-small cell lung cancer: Current concepts and clinical trials. Eur. J. Cardio-Thoracic Surg. 2015, 49, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.; Burke, K.P.; Van Allen, E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8+T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore antitumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Edwards, J.; Wilmott, J.; Madore, J.; Gide, T.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103+ Tumor-Resident CD8+ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti–PD-1 Treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef] [Green Version]
- Bösmüller, H.-C.; Wagner, P.; Peper, J.K.; Schuster, H.; Pham, D.L.; Greif, K.; Beschorner, C.; Rammensee, H.-G.; Stevanović, S.; Fend, F.; et al. Combined Immunoscore of CD103 and CD3 Identifies Long-Term Survivors in High-Grade Serous Ovarian Cancer. Int. J. Gynecol. Cancer 2016, 26, 671–679. [Google Scholar] [CrossRef]
- Savas, P.; Kathleen Cuningham Foundation Consortium for research into Familial Breast cancer (kConFab); Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef]
- Workel, H.H.; Komdeur, F.L.; Wouters, M.C.; Plat, A.; Klip, H.G.; Eggink, F.; Wisman, G.B.A.; Arts, H.J.; Oonk, M.H.; Mourits, M.J.; et al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur. J. Cancer 2016, 60, 1–11. [Google Scholar] [CrossRef]
- Okła, K.; Farber, D.L.; Zou, W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 2021, 218, e20201605. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive cellular therapy: A race to the finish line. Sci. Transl. Med. 2015, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Carreno, B.M. Tumor-Infiltrating Lymphocytes in the Checkpoint Inhibitor Era. Curr. Hematol. Malign- Rep. 2019, 14, 286–291. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint Blockade in Cancer Immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer Regression and Autoimmunity in Patients after Clonal Repopulation with Antitumor Lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive Cell Transfer Therapy Following Non-Myeloablative but Lymphodepleting Chemotherapy for the Treatment of Patients with Refractory Metastatic Melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical Responses in a Phase II Study Using Adoptive Transfer of Short-term Cultured Tumor Infiltration Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Riddell, S.R.; Watanabe, K.; Goodrich, J.; Li, C.; Agha, M.; Greenberg, P. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 257, 238–241. [Google Scholar] [CrossRef]
- Gattinoni, L.; Powell, D.J.; Rosenberg, S.A.; Restifo, N.P. Adoptive immunotherapy for cancer: Building on success. Nat. Rev. Immunol. 2006, 6, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Blattman, J.N.; Grayson, J.M.; Wherry, E.J.; Kaech, S.M.; Smith, K.A.; Ahmed, R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 2003, 9, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma: Intent-to-Treat Analysis and Efficacy after Failure to Prior Immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific Lymphocyte Subsets Predict Response to Adoptive Cell Therapy Using Expanded Autologous Tumor-Infiltrating Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilon-Thomas, S.; Kuhn, L.; Ellwanger, S.; Janssen, W.; Royster, E.; Marzban, S.; Kudchadkar, R.; Zager, J.; Gibney, G.; Sondak, V.K.; et al. Efficacy of Adoptive Cell Transfer of Tumor-infiltrating Lymphocytes After Lymphopenia Induction for Metastatic Melanoma. J. Immunother. 2012, 35, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevanovic, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete Regression of Metastatic Cervical Cancer After Treatment With Human Papillomavirus–Targeted Tumor-Infiltrating T Cells. J. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Kim, Y.-A.; Sim, C.K.; Heo, S.-H.; Song, I.H.; Park, H.S.; Park, S.Y.; Bang, W.S.; Park, I.A.; Lee, M.; et al. Expansion of tumor-infiltrating lymphocytes and their potential for application as adoptive cell transfer therapy in human breast cancer. Oncotarget 2017, 8, 113345–113359. [Google Scholar] [CrossRef] [Green Version]
- Thommen, D.S.; Koelzer, V.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- De Groot, R.; Van Loenen, M.M.; Guislain, A.; Nicolet, B.P.; Heeren, J.J.F.-V.; Verhagen, O.J.; Heuvel, M.M.V.D.; De Jong, J.; Burger, P.; Van Der Schoot, C.; et al. Polyfunctional tumor-reactive T cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged T cell profile. OncoImmunology 2019, 8, e1648170. [Google Scholar] [CrossRef]
- Klebanoff, C.; Khong, H.T.; Antony, P.A.; Palmer, D.C.; Restifo, N.P. Sinks, suppressors and antigen presenters: How lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.; Chan, C.-C.; Klebanoff, C.; Overwijk, W.W.; et al. CD8+T Cell Immunity Against a Tumor/Self-Antigen Is Augmented by CD4+T Helper Cells and Hindered by Naturally Occurring T Regulatory Cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Van Der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.; Braber, M.V.D.; Rozeman, E.A.; Haanen, J.B.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2018, 176, 775–789.e18. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Hakeem, M.A.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.-Z.; Liu, Z.-W.; Zhang, J.-Y.; Yang, Y.-P.; Tien, P.; Wang, F.-S. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2010, 128, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, J.; Wada, Y.; Matsumoto, K.; Azuma, M.; Kikuchi, K.; Ueda, S. Overexpression of B7-H1 (PD-L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunol. Immunother. 2006, 56, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, S.; Gong, D.; Qin, Y.; Shen, Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell. Mol. Immunol. 2010, 7, 389–395. [Google Scholar] [CrossRef]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, I.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Schietinger, A. Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Curr. Opin. Immunol. 2019, 58, 98–103. [Google Scholar] [CrossRef]
- Guo, Q.; Huang, F.; Goncalves, C.; Del Rincón, S.V.; Miller Jr, W.H. Translation of cancer immunotherapy from the bench to the bedside. Adv Cancer Res 2019, 143, 1–62. [Google Scholar] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Gallimore, A.; Glithero, A.; Godkin, A.; Tissot, A.C.; Plückthun, A.; Elliott, T.; Hengartner, H.; Zinkernagel, R. Induction and Exhaustion of Lymphocytic Choriomeningitis Virus–specific Cytotoxic T Lymphocytes Visualized Using Soluble Tetrameric Major Histocompatibility Complex Class I–Peptide Complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral Immune Evasion Due to Persistence of Activated T Cells Without Effector Function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Shankar, P.; Russo, M.; Harnisch, B.; Patterson, M.; Skolnik, P.; Lieberman, J. Impaired function of circulating HIV-specific CD8(+) T cells in chronic human immunodeficiency virus infection. Blood 2000, 96, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Kostense, S.; Ogg, G.S.; Manting, E.H.; Gillespie, G.; Joling, J.; Vandenberghe, K.; Veenhof, E.Z.; Van Baarle, D.; Jurriaans, S.; Klein, M.R.; et al. High viral burden in the presence of major HIV-specific CD8+ T cell expansions: Evidence for impaired CTL effector function. Eur. J. Immunol. 2001, 31, 677–686. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Gruener, N.H.; Lechner, F.; Jung, M.-C.; Diepolder, H.; Gerlach, T.; Lauer, G.; Walker, B.; Sullivan, J.; Phillips, R.; Pape, G.R.; et al. Sustained Dysfunction of Antiviral CD8 + T Lymphocytes after Infection with Hepatitis C Virus. J. Virol. 2001, 75, 5550–5558. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef] [Green Version]
- Kallies, A.; Zehn, D.; Utzschneider, D. Precursor exhausted T cells: Key to successful immunotherapy? Nat. Rev. Immunol. 2019, 20, 128–136. [Google Scholar] [CrossRef]
- Fuller, M.J.; Zajac, A.J. Ablation of CD8 and CD4 T Cell Responses by High Viral Loads. J. Immunol. 2003, 170, 477–486. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speiser, D.; Utzschneider, D.; Oberle, S.G.; Münz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thommen, D.S.; Schumacher, T. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, A.A.D.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2008, 10, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Duraiswamy, J.; Ibegbu, C.C.; Masopust, D.; Miller, J.D.; Araki, K.; Doho, G.H.; Tata, P.; Gupta, S.; Zilliox, M.J.; Nakaya, H.; et al. Phenotype, Function, and Gene Expression Profiles of Programmed Death-1hi CD8 T Cells in Healthy Human Adults. J. Immunol. 2011, 186, 4200–4212. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2017, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8 + T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Marraco, S.A.F.; Calderon-Copete, S.; Ferreira, D.P.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef] [Green Version]
- Boddupalli, C.S.; Bar, N.; Kadaveru, K.; Krauthammer, M.; Pornputtapong, N.; Mai, Z.; Ariyan, S.; Narayan, D.; Kluger, H.; Deng, Y.; et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight 2016, 1, e88955. [Google Scholar] [CrossRef] [PubMed]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpréville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ Tumor–Infiltrating Lymphocytes Are Tumor-Specific Tissue-Resident Memory T Cells and a Prognostic Factor for Survival in Lung Cancer Patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.-P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.; Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017, 18, 940–950. [Google Scholar] [CrossRef]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Oja, A.E.; Piet, B.; Van Der Zwan, D.; Blaauwgeers, H.; Mensink, M.; de Kivit, S.; Borst, J.; Nolte, M.A.; Van Lier, R.A.W.; Stark, R.; et al. Functional Heterogeneity of CD4+ Tumor-Infiltrating Lymphocytes With a Resident Memory Phenotype in NSCLC. Front. Immunol. 2018, 9, 2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wu, S.; Zeng, H.; Liu, Z.; Dong, W.; He, W.; Chen, X.; Dong, X.; Zheng, L.; Lin, T.; et al. CD103 + Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J. Urol. 2015, 194, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Hartana, C.A.; Bergman, E.A.; Broomé, A.; Berglund, S.; Johansson, M.; Alamdari, F.; Jakubczyk, T.; Huge, Y.; Aljabery, F.; Palmqvist, K.; et al. Tissue-resident memory T cells are epigenetically cytotoxic with signs of exhaustion in human urinary bladder cancer. Clin. Exp. Immunol. 2018, 194, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; Milne, K.; Watson, P.; DeLeeuw, R.J.; Nelson, B. Tumor-Infiltrating Lymphocytes Expressing the Tissue Resident Memory Marker CD103 Are Associated with Increased Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2013, 20, 434–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, J.R.; Wick, D.A.; Nielsen, J.S.; Tran, E.; Milne, K.; McMurtrie, E.; Nelson, B. Profound elevation of CD8+ T cells expressing the intraepithelial lymphocyte marker CD103 (αE/β7 Integrin) in high-grade serous ovarian cancer. Gynecol. Oncol. 2010, 118, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Komdeur, F.L.; Prins, T.M.; Van De Wall, S.; Plat, A.; Wisman, G.B.A.; Hollema, H.; Daemen, T.; Church, D.; De Bruyn, M.; Nijman, H.W. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. OncoImmunology 2017, 6, e1338230. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Milne, K.; DeRocher, H.; Webb, J.R.; Nelson, B.; Watson, P.H. CD103 and Intratumoral Immune Response in Breast Cancer. Clin. Cancer Res. 2016, 22, 6290–6297. [Google Scholar] [CrossRef] [Green Version]
- Quinn, E.; Hawkins, N.; Yip, Y.L.; Suter, C.; Ward, R. CD103+ intraepithelial lymphocytes--a unique population in microsatellite unstable sporadic colorectal cancer. Eur. J. Cancer 2003, 39, 469–475. [Google Scholar] [CrossRef]
- Behr, F.M.; Chuwonpad, A.; Stark, R.; Van Gisbergen, K.P.J.M. Armed and Ready: Transcriptional Regulation of Tissue-Resident Memory CD8 T Cells. Front. Immunol. 2018, 9, 1770. [Google Scholar] [CrossRef] [Green Version]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, F.M.; Parga-Vidal, L.; Kragten, N.A.M.; Van Dam, T.J.P.; Wesselink, T.H.; Sheridan, B.S.; Arens, R.; Van Lier, R.A.W.; Stark, R.; Van Gisbergen, K.P.J.M. Tissue-resident memory CD8+ T cells shape local and systemic secondary T cell responses. Nat. Immunol. 2020, 21, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.K.; Wijeyesinghe, S.; Thompson, E.A.; Macchietto, M.G.; Rosato, P.C.; Pierson, M.J.; Schenkel, J.; Mitchell, J.S.; Vezys, V.; Fife, B.; et al. T Cells in Nonlymphoid Tissues Give Rise to Lymph-Node-Resident Memory T Cells. Immunity 2018, 48, 327–338.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.J.; Toma, C.; Yu, B.; Zhang, K.; Omilusik, K.; Phan, A.T.; Wang, D.; Getzler, A.; Nguyen, T.; Crotty, S.; et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 2017, 552, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.M.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, L.K.; Braun, A.; Macleod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting Edge: CD69 Interference with Sphingosine-1-Phosphate Receptor Function Regulates Peripheral T Cell Retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef] [Green Version]
- Skon, C.N.; Lee, J.-Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.; Jameson, S. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [Green Version]
- Cepek, K.L.; Shaw, S.K.; Parker, C.M.; Russell, G.J.; Morrow, J.S.; Rimm, D.L.; Brenner, M.B. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature 1994, 372, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Mackay, L. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2015, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, S.H.; Schlums, H.; Sérézal, I.G.; Martini, E.; Chiang, S.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8 + T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef]
- Beltra, J.-C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e8. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Panwar, B.; Madrigal, A.; Singh, D.; Gujar, R.; Wood, O.; Chee, S.J.; Eschweiler, S.; King, E.V.; Awad, A.S.; et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J. Exp. Med. 2019, 216, 2128–2149. [Google Scholar] [CrossRef]
- Le Floc’H, A.; Jalil, A.; Vergnon, I.; Chansac, B.L.M.; Lazar, V.; Bismuth, G.; Chouaib, S.; Mami-Chouaib, F. αEβ7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J. Exp. Med. 2007, 204, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, B.S.; Pham, Q.-M.; Lee, Y.-T.; Cauley, L.S.; Puddington, L.; Lefrançois, L. Oral Infection Drives a Distinct Population of Intestinal Resident Memory CD8+ T Cells with Enhanced Protective Function. Immunity 2014, 40, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, L.; Rahimpour, A.; Ma, J.; Collins, N.C.; Stock, A.T.; Hafon, M.-L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.; Stefanovic, T.; et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Wakim, L.M.; Woodward-Davis, A.; Bevan, M.J. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc. Natl. Acad. Sci. USA 2010, 107, 17872–17879. [Google Scholar] [CrossRef] [Green Version]
- Reilly, E.C.; Emo, K.L.; Buckley, P.M.; Reilly, N.S.; Smith, I.; Chaves, F.A.; Yang, H.; Oakes, P.W.; Topham, D.J. TRMintegrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection. Proc. Natl. Acad. Sci. USA 2020, 117, 12306–12314. [Google Scholar] [CrossRef]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-Independent Differentiation and Maintenance of Effector-like Resident Memory T Cells in Tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef] [Green Version]
- Steinert, E.M.; Schenkel, J.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyártó, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.-Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Fraser, K.A.; Casey, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. IL-15–Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. J. Immunol. 2016, 196, 3920–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Kobayashi, T.; Sugihara, E.; Yamada, T.; Ikuta, K.; Pittaluga, S.; Saya, H.; Amagai, M.; Nagao, K. Hair follicle–derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med. 2015, 21, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Beura, L.K.; Quarnstrom, C.F.; Ghoneim, H.E.; Fan, Y.; Zebley, C.C.; Scott, M.C.; Fares-Frederickson, N.J.; Wijeyesinghe, S.; Thompson, E.A.; et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol. 2020, 21, 412–421. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.-B.; Wang, W.; et al. Fatty acid oxidation controls CD8+ tissue-resident memory T cell survival in gastric adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Rosario, A.; Wang, R.; Solomon, S.; Srinivasan, G.; Nelson, M.S.; Huang, Y.; Lim, M.H.; et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight 2019, 4, e130000. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; Milne, K.; Nelson, B. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Huang, P.; Luo, Y.; Wang, B.; Luo, X.; Zheng, Z.; Yuan, K.; Huang, Z.; Peng, S.; Yu, H.; et al. CD103 expression in normal epithelium is associated with poor prognosis of colorectal cancer patients within defined subgroups. Int. J. Clin. Exp. Pathol. 2017, 10, 6624–6634. [Google Scholar]
- Amsen, D.; van Gisbergen, K.; Hombrink, P.; Van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef]
- Lohneis, P.; Sinn, M.; Bischoff, S.; Jühling, A.; Pelzer, U.; Wislocka, L.; Bahra, M.; Sinn, B.V.; Denkert, C.; Oettle, H.; et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur. J. Cancer 2017, 83, 290–301. [Google Scholar] [CrossRef]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workel, H.H.; Van Rooij, N.; Plat, A.; Spierings, D.C.; Fehrmann, R.S.N.; Nijman, H.W.; De Bruyn, M. Transcriptional Activity and Stability of CD39+CD103+CD8+ T Cells in Human High-Grade Endometrial Cancer. Int. J. Mol. Sci. 2020, 21, 3770. [Google Scholar] [CrossRef] [PubMed]
- Komdeur, F.L.; Wouters, M.; Workel, H.H.; Tijans, A.M.; Terwindt, A.L.; Brunekreeft, K.L.; Plat, A.; Klip, H.G.; Eggink, F.; Leffers, N.; et al. CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRαβ+ CD8αβ+ T cells that can be targeted for cancer immunotherapy. Oncotarget 2016, 7, 75130–75144. [Google Scholar] [CrossRef] [Green Version]
- Gamradt, P.; Laoubi, L.; Nosbaum, A.; Mutez, V.; Lenief, V.; Grande, S.; Redoulès, D.; Schmitt, A.-M.; Nicolas, J.F.; Vocanson, M. Inhibitory checkpoint receptors control CD8+ resident memory T cells to prevent skin allergy. J. Allergy Clin. Immunol. 2019, 143, 2147–2157.e9. [Google Scholar] [CrossRef]
- Shwetank, A.; Abdelsamed, H.; Frost, E.L.; Schmitz, H.M.; Mockus, T.E.; Youngblood, B.A.; Lukacher, A.E. Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol. Cell Biol. 2017, 95, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martínez-Cano, S.; Mejías-Pérez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 2017, 8, 16073. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.C.; Wijeyesinghe, S.; Stolley, J.M.; Nelson, C.E.; Davis, R.L.; Manlove, L.S.; Pennell, C.A.; Blazar, B.R.; Chen, C.C.; Geller, M.A.; et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Farber, D.; Yudanin, N.; Restifo, N.P. Human memory T cells: Generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 2013, 14, 24–35. [Google Scholar] [CrossRef]
- Ye, Q.; Song, D.-G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J. CD137 Accurately Identifies and Enriches for Naturally Occurring Tumor-Reactive T Cells in Tumor. Clin. Cancer Res. 2013, 20, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; De Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2018, 25, 89–94. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Teichgräber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Bouneaud, C.; Garcia, Z.; Kourilsky, P.; Pannetier, C. Lineage relationships, homeostasis, and recall capacities of central– and effector–memory CD8 T cells in vivo. J. Exp. Med. 2005, 201, 579–590. [Google Scholar] [CrossRef]
- Ahlers, J.D.; Belyakov, I.M. Memories that last forever: Strategies for optimizing vaccine T-cell memory. Blood 2010, 115, 1678–1689. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Huster, K.M.; Koffler, M.; Stemberger, C.; Schiemann, M.; Wagner, H.; Busch, D.H. Unidirectional development of CD8+ central memory T cells into protectiveListeria-specific effector memory T cells. Eur. J. Immunol. 2006, 36, 1453–1464. [Google Scholar] [CrossRef]
- Behr, F.M.; Beumer-Chuwonpad, A.; Kragten, N.A.M.; Wesselink, T.H.; Stark, R.; Van Gisbergen, K.P. Circulating memory CD8 + T cells are limited in forming CD103 + tissue-resident memory T cells at mucosal sites after reinfection. Eur. J. Immunol. 2020, 51, 151–166. [Google Scholar] [CrossRef]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Höfer, T.; Riddell, S.R.; Busch, D.H. Serial Transfer of Single-Cell-Derived Immunocompetence Reveals Stemness of CD8+ Central Memory T Cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Yunger, S.; Bar El, A.; Zeltzer, L.-A.; Fridman, E.; Raviv, G.; Laufer, M.; Schachter, J.; Markel, G.; Itzhaki, O.; Besser, M.J. Tumor-infiltrating lymphocytes from human prostate tumors reveal antitumor reactivity and potential for adoptive cell therapy. OncoImmunology 2019, 8, e1672494. [Google Scholar] [CrossRef] [Green Version]
- Beura, L.K.; Mitchell, J.S.; Thompson, E.A.; Schenkel, J.; Mohammed, J.; Wijeyesinghe, S.; Fonseca, R.; Burbach, B.J.; Hickman, H.; Vezys, V.; et al. Intravital mucosal imaging of CD8+ resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 2018, 19, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Waithman, J.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Dendritic Cell-Induced Memory T Cell Activation in Nonlymphoid Tissues. Science 2008, 319, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Zaid, A.; Hor, J.L.; Christo, S.N.; Prier, J.; Davies, B.; Alexandre, Y.O.; Gregory, J.L.; Russell, T.; Gebhardt, T.; et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018, 19, 183–191. [Google Scholar] [CrossRef]
- Kumar, B.V.; Kratchmarov, R.; Miron, M.; Carpenter, D.J.; Senda, T.; Lerner, H.; Friedman, A.; Reiner, S.L.; Farber, D. Functional heterogeneity of human tissue-resident memory T cells based on dye efflux capacities. JCI Insight 2018, 3, e123568. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Osborn, J.F.; Hobbs, S.J.; Mooster, J.L.; Khan, T.N.; Kilgore, A.M.; Harbour, J.; Nolz, J.C. Central memory CD8+ T cells become CD69+ tissue-residents during viral skin infection independent of CD62L-mediated lymph node surveillance. PLOS Pathog. 2019, 15, e1007633. [Google Scholar] [CrossRef]
- Lau, C.M.; Adams, N.M.; Geary, C.D.; Weizman, O.-E.; Rapp, M.; Pritykin, Y.; Leslie, C.S.; Sun, J.C. Epigenetic control of innate and adaptive immune memory. Nat. Immunol. 2018, 19, 963–972. [Google Scholar] [CrossRef]
- Yu, B.; Zhang, K.; Milner, J.J.; Toma, C.; Chen, R.; Scott-Browne, J.P.; Pereira, R.; Crotty, R.C.S.; Chang, J.; Pipkin, M.; et al. Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol. 2017, 18, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Miyazono, K. RUNX transcription factors as key targets of TGF-β superfamily signaling. Curr. Opin. Genet. Dev. 2003, 13, 43–47. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Usherwood, E.J.; Cauley, L.S.; Olson, S.; Marzo, A.L.; Ward, R.L.; Woodland, D.L.; Lefrançois, L. Activated Primary and Memory CD8 T Cells Migrate to Nonlymphoid Tissues Regardless of Site of Activation or Tissue of Origin. J. Immunol. 2004, 172, 4875–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, M.; Pircher, H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc. Natl. Acad. Sci. USA 2011, 108, 16741–16746. [Google Scholar] [CrossRef] [Green Version]
- Mazzolini, G.; Alfaro, C.; Sangro, B.; Feijoó, E.; Ruiz, J.; Benito, A.; Tirapu, I.; Arina, A.; Sola, J.; Herraiz, M.; et al. Intratumoral Injection of Dendritic Cells Engineered to Secrete Interleukin-12 by Recombinant Adenovirus in Patients with Metastatic Gastrointestinal Carcinomas. J. Clin. Oncol. 2005, 23, 999–1010. [Google Scholar] [CrossRef]
- Yao, W.; Li, Y.; Zeng, L.; Zhang, X.; Zhou, Z.; Zheng, M.; Wan, H. Intratumoral injection of dendritic cells overexpressing interleukin-12 inhibits melanoma growth. Oncol. Rep. 2019, 42, 370–376. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sakabe, T.; Chiba, A.; Nakajima, A.; Okamoto, M.; Shimodaira, S.; Yonemitsu, Y.; Shibamoto, Y.; Suzuki, N.; Nagaya, M.; et al. Therapeutic effect of intratumoral injections of dendritic cells for locally recurrent gastric cancer: A case report. World J. Surg. Oncol. 2014, 12, 390. [Google Scholar] [CrossRef] [Green Version]
- Mackay, L.; Wakim, L.; Van Vliet, C.J.; Jones, C.; Mueller, S.; Bannard, O.; Fearon, D.T.; Heath, W.R.; Carbone, F.R. Maintenance of T Cell Function in the Face of Chronic Antigen Stimulation and Repeated Reactivation for a Latent Virus Infection. J. Immunol. 2012, 188, 2173–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beumer-Chuwonpad, A.; Taggenbrock, R.L.R.E.; Ngo, T.A.; van Gisbergen, K.P.J.M. The Potential of Tissue-Resident Memory T Cells for Adoptive Immunotherapy against Cancer. Cells 2021, 10, 2234. https://doi.org/10.3390/cells10092234
Beumer-Chuwonpad A, Taggenbrock RLRE, Ngo TA, van Gisbergen KPJM. The Potential of Tissue-Resident Memory T Cells for Adoptive Immunotherapy against Cancer. Cells. 2021; 10(9):2234. https://doi.org/10.3390/cells10092234
Chicago/Turabian StyleBeumer-Chuwonpad, Ammarina, Renske L. R. E. Taggenbrock, T. An Ngo, and Klaas P. J. M. van Gisbergen. 2021. "The Potential of Tissue-Resident Memory T Cells for Adoptive Immunotherapy against Cancer" Cells 10, no. 9: 2234. https://doi.org/10.3390/cells10092234
APA StyleBeumer-Chuwonpad, A., Taggenbrock, R. L. R. E., Ngo, T. A., & van Gisbergen, K. P. J. M. (2021). The Potential of Tissue-Resident Memory T Cells for Adoptive Immunotherapy against Cancer. Cells, 10(9), 2234. https://doi.org/10.3390/cells10092234