
Quasar: Easy Machine Learning for Biospectroscopy



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
- Visual programming: Orange does not impose a preset order of operations. Instead, it offers components—widgets—that either process, visualize or model inputs. Users can connect them as they see fit as long as they share connection types. This approach allows the creation of flexible workflows;
- Immediate feedback: Orange follows the general principle that being able to observe the effects of actions, and adapt immediately, improves efficiency. The option to inspect the results at every step of the analysis helps users gain confidence in results and familiarity with the analysis procedures [14];
- Interactive visualizations: Orange allows interaction with visualized elements, which can influence further analysis—for example, selecting a point on a scatter plot sends the associated data to the output. This principle empowers users to further explore interesting elements identified in the visualization;
- Machine learning: Orange includes components for unsupervised and supervised modeling and their evaluation. Mainly, it wraps established machine learning libraries for Python, such as scikit-learn or XGBoost, into user-friendly GUI components. Methods in Orange include various clustering methods, t-SNE, random forests, support vector machines, gradient boosting, and neural networks;
- Extendability and modularity: Orange is mainly written in Python, with computationally intensive parts in C [13]. It builds on Python data science libraries, such as NumPy, SciPy, Pandas, and scikit-learn. If predefined components do not suffice, the Python Script widget allows adding custom Python code into the workflow. Additionally, Orange provides a well-documented programming interface for adding new components and modules. More than a dozen specialized modules currently exist, including text processing, gene expression data analysis [15], image analytics [14], and time series analysis.
- Data input: Quasar supports native data reading from major instrument manufacturers, commonly used exchange data formats and some specialized instruments;
- Spectral preprocessing: Commonly used preprocessing methods for smoothing, derivatives, baseline removal, normalization, EMSC, ME-EMSC, integration, peak fitting, etc.;
- Visualization: Plotting widgets for individual spectra, hyperspectral maps with visible image overlays, and maps of spectral series are available.
3. Results and Discussion
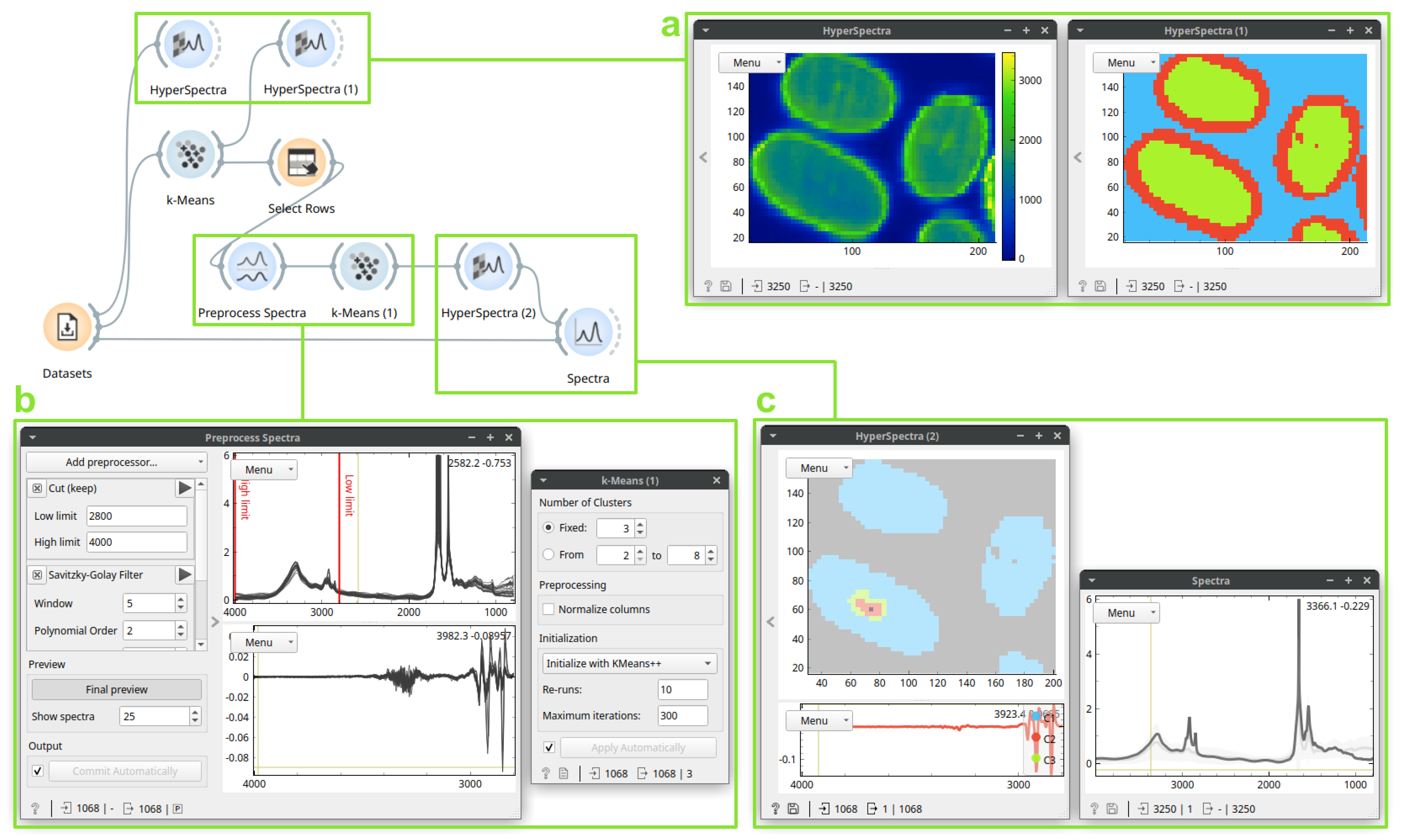
3.1. Case Study 1: Identification and Localization of the Medullas in Human Hair Sections
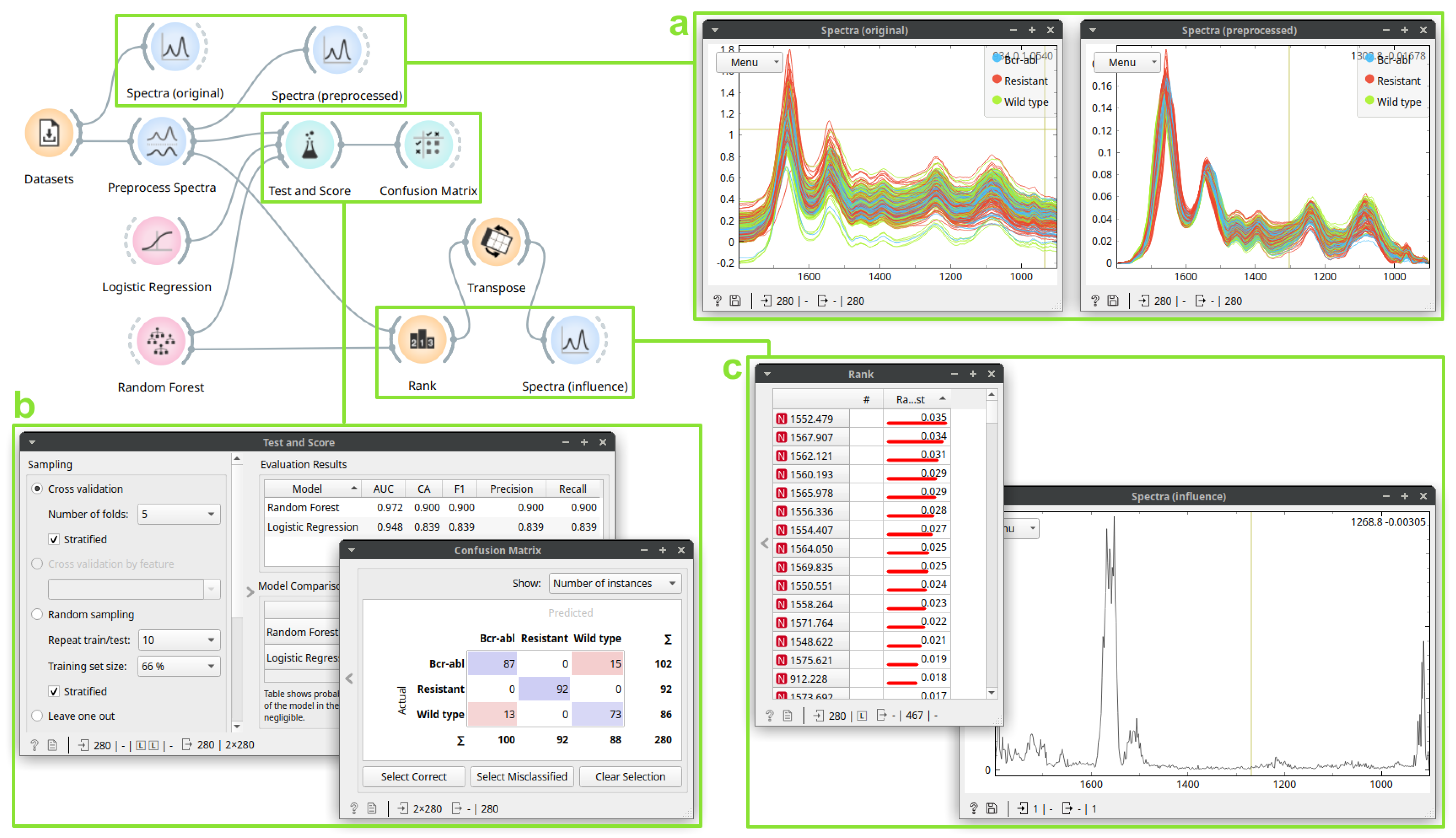
3.2. Case Study 2: Building a Drug Resistance Prediction Model
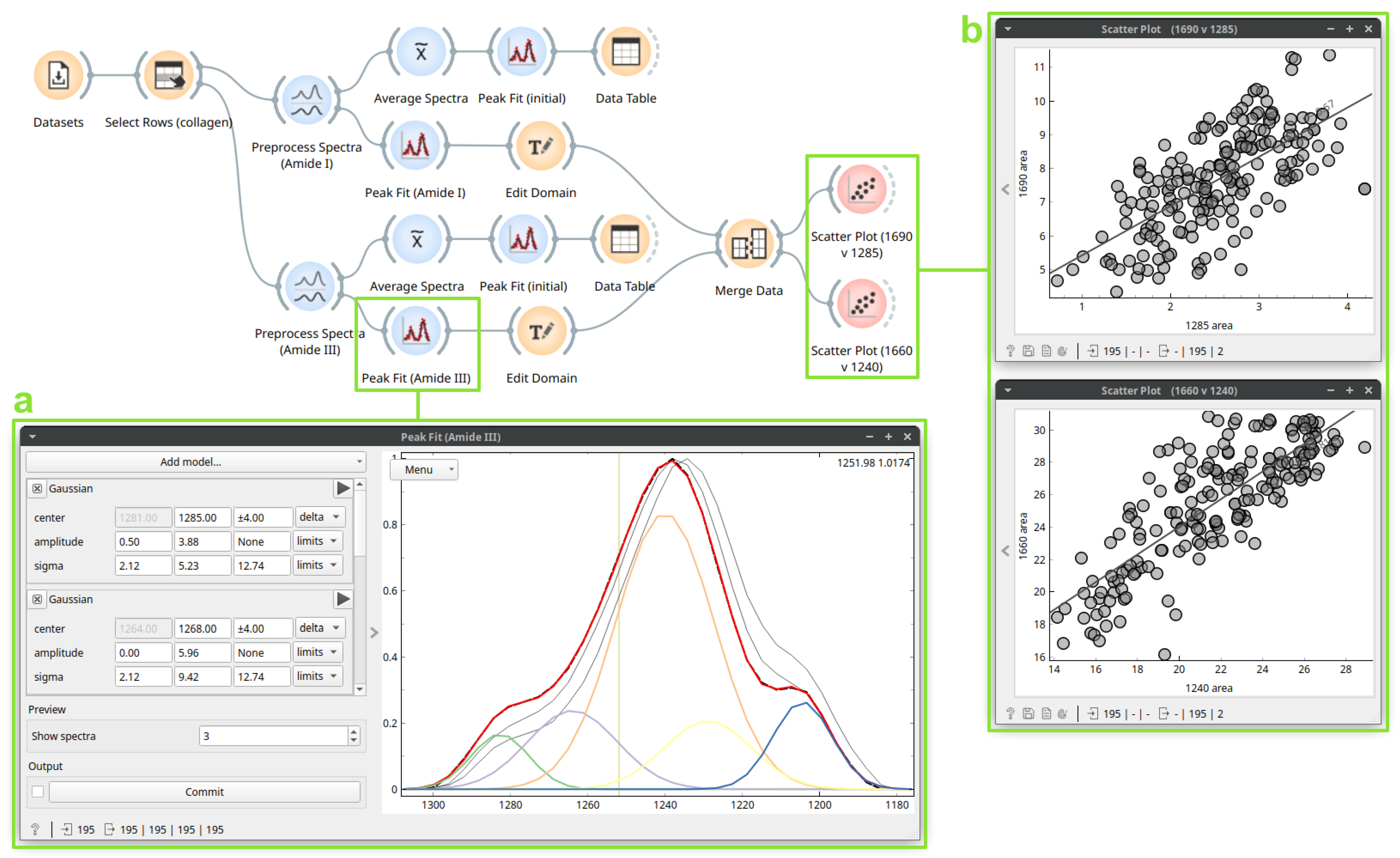
3.3. Case Study 3: Protein Secondary Structure Peak Fitting
3.4. Limitations and Further Development
3.5. Current Project Reach
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berisha, S.; Chang, S.; Saki, S.; Daeinejad, D.; He, Z.; Mankar, R.; Mayerich, D. SIproc: An open-source biomedical data processing platform for large hyperspectral images. Analyst 2017, 142, 1350–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.J.; Trevisan, J.; Bassan, P.; Bhargava, R.; Butler, H.J.; Dorling, K.M.; Fielden, P.R.; Fogarty, S.W.; Fullwood, N.J.; Heys, K.A.; et al. Using Fourier transform IR spectroscopy to analyze biological materials. Nat. Protoc. 2014, 9, 1771–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berisha, S.; Lotfollahi, M.; Jahanipour, J.; Gurcan, I.; Walsh, M.; Bhargava, R.; Van Nguyen, H.; Mayerich, D. Deep learning for FTIR histology: Leveraging spatial and spectral features with convolutional neural networks. Analyst 2019, 144, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, E.A.; Solheim, J.H.; Blazhko, U.; Tafintseva, V.; Tøndel, K.; Liland, K.H.; Dzurendova, S.; Shapaval, V.; Sandt, C.; Borondics, F.; et al. Deep convolutional neural network recovers pure absorbance spectra from highly scatter-distorted spectra of cells. J. Biophotonics 2020, 13, e202000204. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, A.; Xu, L.; Xie, H.; Qiao, H.; Lin, Q.; Cai, K. A deep learning CNN architecture applied in smart near-infrared analysis of water pollution for agricultural irrigation resources. Agric. Water Manag. 2020, 240, 106303. [Google Scholar] [CrossRef]
- Falahkheirkhah, K.; Yeh, K.; Mittal, S.; Pfister, L.; Bhargava, R. Deep learning-based protocols to enhance infrared imaging systems. Chemom. Intellig. Lab. Syst. 2021, 217, 104390. [Google Scholar] [CrossRef]
- Quasar Website. Available online: https://quasar.codes (accessed on 1 September 2021).
- The GitHub Project Page for Orange Spectroscopy. Available online: https://github.com/Quasars/orange-spectroscopy (accessed on 1 September 2021).
- Toplak, M.; Read, S.; Borondics, F. Quasar. Zenodo 2021. [Google Scholar] [CrossRef]
- Orange Website. Available online: https://orangedatamining.com/ (accessed on 1 September 2021).
- The Github Project Page for Orange. Available online: https://github.com/biolab/orange3 (accessed on 1 September 2021).
- Curk, T.; Demsar, J.; Xu, Q.; Leban, G.; Petrovic, U.; Bratko, I.; Shaulsky, G.; Zupan, B. Microarray data mining with visual programming. Bioinformatics 2005, 21, 396–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Marko Toplak, M.P.; Starič, A.; Štajdohar, M.; et al. Orange: Data Mining Toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Godec, P.; Pančur, M.; Ilenič, N.; Čopar, A.; Stražar, M.; Erjavec, A.; Pretnar, A.; Demšar, J.; Starič, A.; Toplak, M.; et al. Democratized image analytics by visual programming through integration of deep models and small-scale machine learning. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stražar, M.; Žagar, L.; Kokošar, J.; Tanko, V.; Erjavec, A.; Poličar, P.G.; Starič, A.; Demšar, J.; Shaulsky, G.; Menon, V.; et al. scOrange—A tool for hands-on training of concepts from single-cell data analytics. Bioinformatics 2019, 35, i4–i12. [Google Scholar] [CrossRef] [PubMed]
- Toplak, M.; Birarda, G.; Read, S.; Sandt, C.; Rosendahl, S.; Vaccari, L.; Demšar, J.; Borondics, F. Infrared orange: Connecting hyperspectral data with machine learning. Synchrotron Radiat. News 2017, 30, 40–45. [Google Scholar] [CrossRef]
- Sandt, C.; Borondics, F. A new typology of human hair medullas based on lipid composition analysis by synchrotron FTIR microspectroscopy. Analyst 2021, 146, 3942–3954. [Google Scholar] [CrossRef] [PubMed]
- Sandt, C.; Feraud, O.; Bonnet, M.L.; Desterke, C.; Khedhir, R.; Flamant, S.; Bailey, C.G.; Rasko, J.E.; Dumas, P.; Bennaceur-Griscelli, A.; et al. Direct and rapid identification of T315I-Mutated BCR-ABL expressing leukemic cells using infrared microspectroscopy. Biochem. Biophys. Res. Commun. 2018, 503, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Newville, M.; Otten, R.; Nelson, A.; Ingargiola, A.; Stensitzki, T.; Allan, D.; Fox, A.; Carter, F.; Michał; Pustakhod, D.; et al. lmfit/lmfit-py 1.0.2. Zenodo 2021. [Google Scholar] [CrossRef]
- Le Naour, F.; Sandt, C.; Peng, C.; Trcera, N.; Chiappini, F.; Flank, A.M.; Guettier, C.; Dumas, P. In situ chemical composition analysis of cirrhosis by combining synchrotron fourier transform infrared and synchrotron X-ray fluorescence microspectroscopies on the same tissue section. Anal. Chem. 2012, 84, 10260–10266. [Google Scholar] [CrossRef]
- Stani, C.; Vaccari, L.; Mitri, E.; Birarda, G. FTIR investigation of the secondary structure of type I collagen: New insight into the amide III band. Spectrochim. Acta Part Mol. Biomol. Spectrosc. 2020, 229, 118006. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toplak, M.; Read, S.T.; Sandt, C.; Borondics, F. Quasar: Easy Machine Learning for Biospectroscopy. Cells 2021, 10, 2300. https://doi.org/10.3390/cells10092300
Toplak M, Read ST, Sandt C, Borondics F. Quasar: Easy Machine Learning for Biospectroscopy. Cells. 2021; 10(9):2300. https://doi.org/10.3390/cells10092300
Chicago/Turabian StyleToplak, Marko, Stuart T. Read, Christophe Sandt, and Ferenc Borondics. 2021. "Quasar: Easy Machine Learning for Biospectroscopy" Cells 10, no. 9: 2300. https://doi.org/10.3390/cells10092300
APA StyleToplak, M., Read, S. T., Sandt, C., & Borondics, F. (2021). Quasar: Easy Machine Learning for Biospectroscopy. Cells, 10(9), 2300. https://doi.org/10.3390/cells10092300