
GDF-15 Deficiency Reduces Autophagic Activity in Human Macrophages In Vitro and Decreases p62-Accumulation in Atherosclerotic Lesions in Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Transfection and Gene Silencing
2.2. Autophagic Activity Assay
2.3. Lysosomal Activity Assay
2.4. Transmission Electron Microscopy (TEM)
2.5. SDS-PAGE and Western Blot
2.6. GDF-15 ELISA
2.7. Animals
2.8. Genotyping
2.9. Dissection and Tissue Harvesting
2.10. Morphometry and Immunohistology
2.11. Statistical Analyses
3. Results
3.1. GDF-15 Silencing Reduces Autophagic Activity in Human THP-1 MΦ
3.2. rGDF-15 Enhances Autophagic Activity in Human THP-1 MΦ
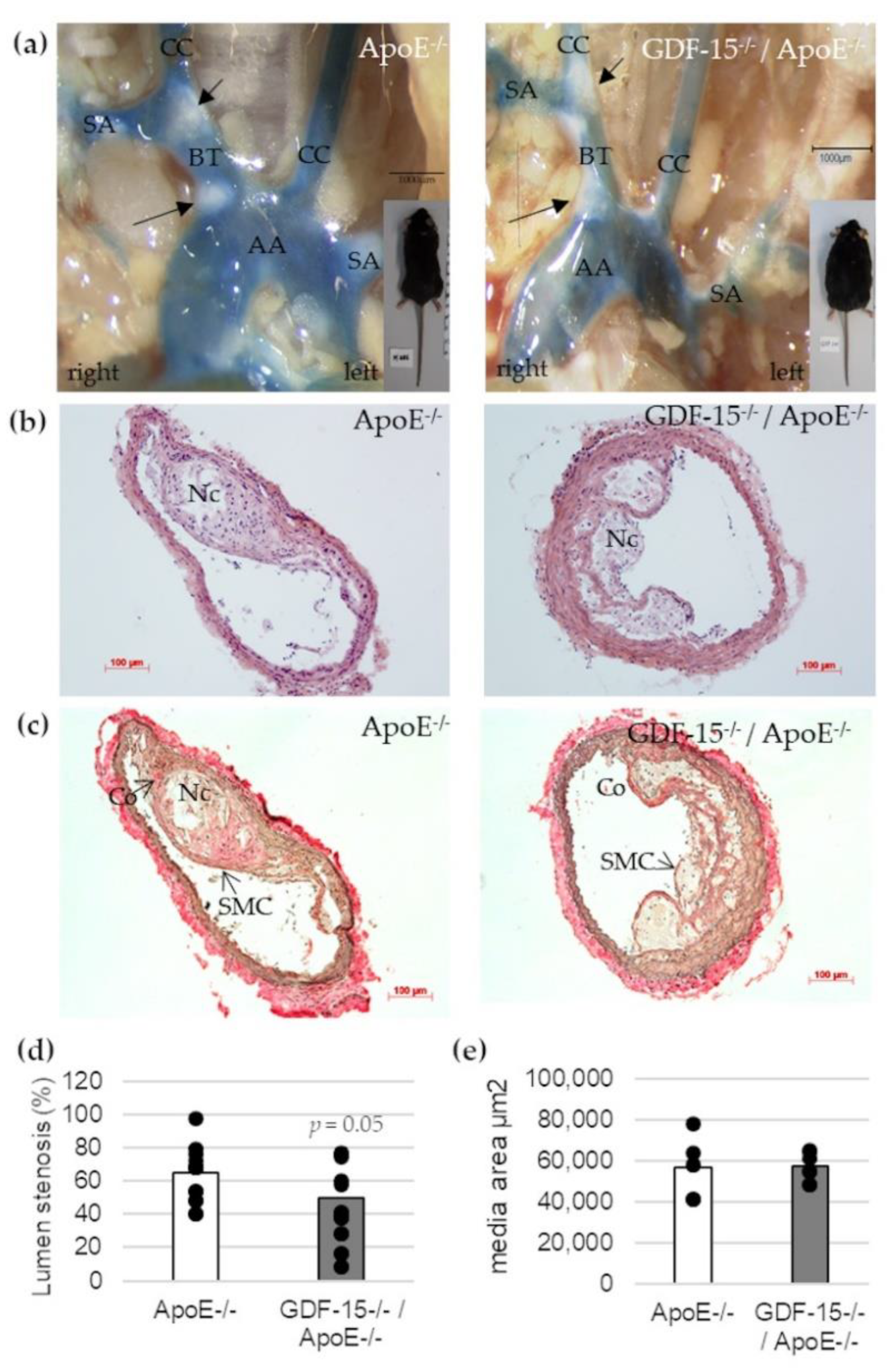
3.3. GDF-15−/− Reduces the Progression of Atherosclerotic Lesions in the BT of ApoE−/− Mice
3.4. GDF-15−/− Mice Characterized by Increased Body Weight, BMI and Increased Plasma TG Level
3.5. GDF-15 Deficiency Decreased the Accumulation of p62 in Atherosclerotic Lesions, Especially in ECs
3.6. GDF-15 Deficiency Increased the Survivin Expression in Atherosclerotic Lesions, Especially in ECs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ross, R. Atherosclerosis is an inflammatory disease. Am. Heart J. 1999, 138, S419–S420. [Google Scholar] [CrossRef]
- Kockx, M.; Herman, A.G. Apoptosis in atherosclerosis: Beneficial or detrimental? Cardiovasc. Res. 2000, 45, 736–746. [Google Scholar] [CrossRef] [Green Version]
- Bootcov, M.; Bauskin, A.R.; Valenzuela, S.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF- superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef] [Green Version]
- Tsai, V.W.; Husaini, Y.; Sainsbury, A.; Brown, D.A.; Breit, S.N. The MIC-1/GDF15-GFRAL Pathway in Energy Homeostasis: Implications for Obesity, Cachexia, and Other Associated Diseases. Cell Metab. 2018, 28, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochholzer, W.; Morrow, D.A.; Giugliano, R.P. Novel biomarkers in cardiovascular disease: Update 2010. Am. Heart J. 2010, 160, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Adela, R.; Banerjee, S.K. GDF-15 as a Target and Biomarker for Diabetes and Cardiovascular Diseases: A Translational Prospective. J. Diabetes Res. 2015, 2015, 490842. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.B.; Sapinoso, L.M.; Kern, S.G.; Brown, D.A.; Liu, T.; Bauskin, A.R.; Ward, R.L.; Hawkins, N.J.; Quinn, D.I.; Russell, P.J.; et al. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc. Natl. Acad. Sci. USA 2003, 100, 3410–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlittenhardt, D.; Schober, A.; Strelau, J.; Bonaterra, G.A.; Schmiedt, W.; Unsicker, K.; Kinscherf, R. Involvement of growth differentiation factor-15/macrophage inhibitory cytokine-1 (GDF-15/MIC-1) in oxLDL-induced apoptosis of human macrophages in vitro and in arteriosclerotic lesions. Cell Tissue Res. 2004, 318, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, K.; Bonaterra, G.A.; Kinscherf, R.; Schwarz, A. Growth differentiation factor-15 regulates oxLDL-induced lipid homeostasis and autophagy in human macrophages. Atherosclerosis 2019, 281, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A New Protein Conjugation System in Human. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Liu, D.; Ning, H.-F.; Yu, X.-C.; Guan, X.-R. The roles of p62/SQSTM1 on regulation of matrix metalloproteinase-9 gene expression in response to oxLDL in atherosclerosis. Biochem. Biophys. Res. Commun. 2016, 472, 451–458. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Triphan, J.; Aumüller, G.; Brandenburger, T.; Wilhelm, B. Localization and regulation of plasma membrane Ca2+-ATPase in bovine spermatozoa. Eur. J. Cell Biol. 2007, 86, 265–273. [Google Scholar] [CrossRef]
- Bonaterra, G.A.; Then, H.; Oezel, L.; Schwarzbach, H.; Ocker, M.; Thieme, K.; Di Fazio, P.; Kinscherf, R. Morphological Alterations in Gastrocnemius and Soleus Muscles in Male and Female Mice in a Fibromyalgia Model. PLoS ONE 2016, 11, e0151116. [Google Scholar] [CrossRef]
- Strelau, J.; Strzelczyk, A.; Rusu, P.; Bendner, G.; Wiese, S.; Diella, F.; Altick, A.L.; Von Bartheld, C.S.; Klein, R.; Sendtner, M.; et al. Progressive Postnatal Motoneuron Loss in Mice Lacking GDF-15. J. Neurosci. 2009, 29, 13640–13648. [Google Scholar] [CrossRef] [PubMed]
- Bonaterra, G.A.; Zügel, S.; Thogersen, J.; Walter, S.A.; Haberkorn, U.; Strelau, J.; Kinscherf, R. Growth Differentiation Factor-15 Deficiency Inhibits Atherosclerosis Progression by Regulating Interleukin-6–Dependent Inflammatory Response to Vascular Injury. J. Am. Heart Assoc. 2012, 1, e002550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargiulo, S.; Gramanzini, M.; Megna, R.; Greco, A.; Albanese, S.; Manfredi, C.; Brunetti, A. Evaluation of Growth Patterns and Body Composition in C57Bl/6J Mice Using Dual Energy X-Ray Absorptiometry. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Rasbach, E.; Splitthoff, P.; Bonaterra, G.A.; Schwarz, A.; Mey, L.; Schwarzbach, H.; Eiden, L.E.; Weihe, E.; Kinscherf, R. PACAP deficiency aggravates atherosclerosis in ApoE deficient mice. Immunobiology 2019, 224, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Vion, A.-C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; Breit, S.N.; Buring, J.; Fairlie, W.; Bauskin, A.R.; Liu, T.; Ridker, P.M. Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: A nested case-control study. Lancet 2002, 359, 2159–2163. [Google Scholar] [CrossRef]
- Lin, J.-F.; Wu, S.; Hsu, S.-Y.; Yeh, K.-H.; Chou, H.-H.; Cheng, S.-T.; Wu, T.-Y.; Hsu, W.-T.; Yang, C.-C.; Ko, Y.-L. Growth-Differentiation Factor-15 and Major Cardiac Events. Am. J. Med. Sci. 2014, 347, 305–311. [Google Scholar] [CrossRef]
- Preusch, M.R.; Baeuerle, M.; Albrecht, C.; Blessing, E.; Bischof, M.; Katus, H.A.; Bea, F. GDF-15 protects from macrophage accumulation in a mousemodel of advanced atherosclerosis. Eur. J. Med. Res. 2013, 18, 19. [Google Scholar] [CrossRef] [Green Version]
- Johnen, H.; Kuffner, T.; Brown, D.A.; Wu, B.; Stocker, R.; Breit, S.N. Increased expression of the TGF-b superfamily cytokine MIC-1/GDF15 protects ApoE−/− mice from the development of atherosclerosis. Cardiovasc. Pathol. 2012, 21, 499–505. [Google Scholar] [CrossRef] [Green Version]
- De Jager, S.C.A.; Bermúdez, B.; Bot, I.; Koenen, R.; Bot, M.; Kavelaars, A.; De Waard, V.; Heijnen, C.J.; Muriana, F.J.G.; Weber, C.; et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J. Exp. Med. 2011, 208, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E. Chapter 10 Monitoring Autophagy by Electron Microscopy in Mammalian Cells. Methods Enzymol. 2009, 452, 143–164. [Google Scholar] [CrossRef]
- Eskelinen, E.-L. Fine Structure of the Autophagosome. In Autophagosome and Phagosome; Deretic, V., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 11–28. ISBN 978-1-59745-157-4. [Google Scholar]
- Kockx, M.M.; De Meyer, G.; Buyssens, N.; Knaapen, M.W.M.; Bult, H.; Herman, A.G. Cell Composition, Replication, and Apoptosis in Atherosclerotic Plaques After 6 Months of Cholesterol Withdrawal. Circ. Res. 1998, 83, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Martinet, W.; De Bie, M.; Schrijvers, D.M.; De Meyer, G.R.Y.; Herman, A.G.; Kockx, M.M. 7-Ketocholesterol Induces Protein Ubiquitination, Myelin Figure Formation, and Light Chain 3 Processing in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2296–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munafó, D.B.; Colombo, M.I. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J. Cell Sci. 2001, 114, 3619–3629. [Google Scholar] [CrossRef]
- Niemann, A.; Baltes, J.; Elsässer, H.-P. Fluorescence properties and staining behavior of monodansylpentane, a structural homologue of the lysosomotropic agent monodansylcadaverine. J. Histochem. Cytochem. 2001, 49, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Niemann, A.; Takatsuki, A.; Elsässer, H.-P. The Lysosomotropic Agent Monodansylcadaverine Also Acts as a Solvent Polarity Probe. J. Histochem. Cytochem. 2000, 48, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biederbick, A.; Kern, H.F.; Elsässer, H.P. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 1995, 66, 3–14. [Google Scholar] [PubMed]
- Bampton, E.T.W.; Goemans, C.G.; Niranjan, D.; Mizushima, N.; Tolkovsky, A.M. The Dynamics of Autophagy Visualised in Live Cells: From Autophagosome Formation to Fusion with Endo/lysosomes. Autophagy 2005, 1, 23–36. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S.; et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.-L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnen, H.; Lin, S.; Kuffner, T.; Brown, D.A.; Tsai, V.W.-W.; Bauskin, A.R.; Wu, L.; Pankhurst, G.; Jiang, L.; Junankar, S.; et al. Tumor-induced anorexia and weight loss are mediated by the TGF-β superfamily cytokine MIC-1. Nat. Med. 2007, 13, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage Autophagy Plays a Protective Role in Advanced Atherosclerosis. Cell Metab. 2012, 15, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Park, H.; Kim, C.-H.; Jeong, J.-H.; Park, M.; Kim, K.S. GDF15 contributes to radiation-induced senescence through the ROS-mediated p16 pathway in human endothelial cells. Oncotarget 2016, 7, 9634–9644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Jiang, D.; Matsuda, J.; Ternyak, K.; Zhang, B.; Chu, H.W. Cigarette Smoke Induces Human Airway Epithelial Senescence via Growth Differentiation Factor 15 Production. Am. J. Respir. Cell Mol. Biol. 2016, 55, 429–438. [Google Scholar] [CrossRef]
- Ha, G.; De Torres, F.; Arouche, N.; Benzoubir, N.; Ferratge, S.; Hatem, E.; Anginot, A.; Uzan, G. GDF15 secreted by senescent endothelial cells improves vascular progenitor cell functions. PLoS ONE 2019, 14, e0216602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altieri, D.C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar] [CrossRef] [Green Version]
- Connell, C.M.; Colnaghi, R.; Wheatley, S.P. Nuclear Survivin Has Reduced Stability and Is Not Cytoprotective. J. Biol. Chem. 2008, 283, 3289–3296. [Google Scholar] [CrossRef] [Green Version]
- Knauer, S.K.; Mann, W.; Stauber, R.H. Survivin’s Dual Role: An Export’s View. Cell Cycle 2007, 6, 518–521. [Google Scholar] [CrossRef]
- Blanc-Brude, O.; Yu, J.; Simosa, H.; Conte, M.S.; Sessa, W.C.; Altieri, D.C. Inhibitor of apoptosis protein survivin regulates vascular injury. Nat. Med. 2002, 8, 987–994. [Google Scholar] [CrossRef]
- Cheng, S.M.; Chang, Y.C.; Liu, C.Y.; Lee, J.Y.; Chan, H.H.; Kuo, C.W.; Lin, K.Y.; Tsai, S.L.; Chen, S.H.; Li, C.F.; et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br. J. Pharmacol. 2015, 172, 214–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.M.; Lin, T.-Y.; Chang, Y.-C.; Lin, I.-W.; Leung, E.; Cheung, C.H.A. YM155 and BIRC5 downregulation induce genomic instability via autophagy-mediated ROS production and inhibition in DNA repair. Pharmacol. Res. 2021, 166, 105474. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Chan, H.-H.; Chen, S.-H.; Sarvagalla, S.; Chen, P.-S.; Coumar, M.S.; Cheng, S.M.; Chang, Y.-C.; Lin, C.-H.; Leung, E.; et al. BIRC5/Survivin is a novel ATG12–ATG5 conjugate interactor and an autophagy-induced DNA damage suppressor in human cancer and mouse embryonic fibroblast cells. Autophagy 2020, 16, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.-Y.; Cheung, C.H.A.; Huang, C.-C.; Tsai, F.-Y.; Lee, J.Y.-C.; Cheng, S.M.; Chang, Y.-C.; Huang, Y.-C.; Chen, S.-H. Survivin—biology and potential as a therapeutic target in oncology. OncoTargets Ther. 2013, 6, 1453–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Cat Nr. | Company | Dilution |
|---|---|---|---|
| Primary Antibodies | |||
| goat anti-mouse Smooth Muscle Actin (sm-α-actin) | ABIN185271 | Antibodies online | 1:10 |
| rabbit anti-mouse APG5L/ATG5 | ab108327 | ABCAM, Cambridge, UK | 1:10 |
| rat anti-mouse CD68 | MCA1957T | Bio-Rad Laboratories Inc., Hercules, CA, USA | 1:10 |
| rabbit anti-mouse p62/SQSTM1 | P0068 | Sigma-Aldrich Chemie GmbH, Munich, Germany | 1:50 |
| rat anti-mouse CD31 | ab73888 | ABCAM, Cambridge, UK | 1:10 |
| Survivin Alexa Fluor674 | sc-17779 AF647 | Santa Cruz Biotechnology, Heidelberg, Germany | 1:50 |
| p53 Alexa Fluor674 | sc-126 AF647 | Santa Cruz Biotechnology, Heidelberg, Germany | 1:50 |
| Secondary Antibodies | |||
| goat Fab anti-rat IgG (H + L)-Cy3 | 112-167-003 | Jackson ImmunoResearch, Ely, UK | 1:100 |
| rabbit IgG anti-goat IgG (H + L)-Cy3 | 305-165-003 | Jackson ImmunoResearch, Ely, UK | 1:100 |
| goat anti-rabbit IgG (H + L)-Alexa Fluor 488 | A-11008 | Thermo Fisher Scientific, Rockford, IL, USA | 1:200 |
| ApoE−/− (n) | GDF-15−/−/ApoE−/− (n) | |
|---|---|---|
| Body Size (cm) | 9.77 ± 0.19 (6) | 9.50 ± 0.32 (14) |
| Body Weight (g) | 30.91 ± 2.21 (6) | 44.44 ± 3.35 *** (14) |
| BMI (g/cm2) | 3.61 ± 0.05 (6) | 4.42 ± 0.06 *** (14) |
| TG (mg/dL) | 135.66 ± 53.42 (6) | 207.07 ± 78,81 (10) |
| TC (mg/dL) | 440.37 ± 70.37 (6) | 514.36 ± 92.11 (10) |
| FC (mg/dL) | 126.42 ± 61.20 (6) | 112.34 ± 36.32 (10) |
| CE (mg/dL) | 313.94 ± 40.71 (6) | 402.02 ± 106.99 (10) |
| HDL (mg/dL) | 0.51 ± 0.58 (6) | 1.58 ± 1.61 (10) |
| LDL/VLDL (mg/dL) | 12.99 ± 4.23 (6) | 13.67 ± 6.58 (10) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heduschke, A.; Ackermann, K.; Wilhelm, B.; Mey, L.; Bonaterra, G.A.; Kinscherf, R.; Schwarz, A. GDF-15 Deficiency Reduces Autophagic Activity in Human Macrophages In Vitro and Decreases p62-Accumulation in Atherosclerotic Lesions in Mice. Cells 2021, 10, 2346. https://doi.org/10.3390/cells10092346
Heduschke A, Ackermann K, Wilhelm B, Mey L, Bonaterra GA, Kinscherf R, Schwarz A. GDF-15 Deficiency Reduces Autophagic Activity in Human Macrophages In Vitro and Decreases p62-Accumulation in Atherosclerotic Lesions in Mice. Cells. 2021; 10(9):2346. https://doi.org/10.3390/cells10092346
Chicago/Turabian StyleHeduschke, Aline, Kathrin Ackermann, Beate Wilhelm, Lilli Mey, Gabriel Alejandro Bonaterra, Ralf Kinscherf, and Anja Schwarz. 2021. "GDF-15 Deficiency Reduces Autophagic Activity in Human Macrophages In Vitro and Decreases p62-Accumulation in Atherosclerotic Lesions in Mice" Cells 10, no. 9: 2346. https://doi.org/10.3390/cells10092346
APA StyleHeduschke, A., Ackermann, K., Wilhelm, B., Mey, L., Bonaterra, G. A., Kinscherf, R., & Schwarz, A. (2021). GDF-15 Deficiency Reduces Autophagic Activity in Human Macrophages In Vitro and Decreases p62-Accumulation in Atherosclerotic Lesions in Mice. Cells, 10(9), 2346. https://doi.org/10.3390/cells10092346