
Perturbed Brain Glucose Metabolism Caused by Absent SIRT3 Activity


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Biochemicals
2.3. Administration of Labeled [1,6-13C]glucose
2.4. Tissue Extraction
2.5. NMR Spectroscopy
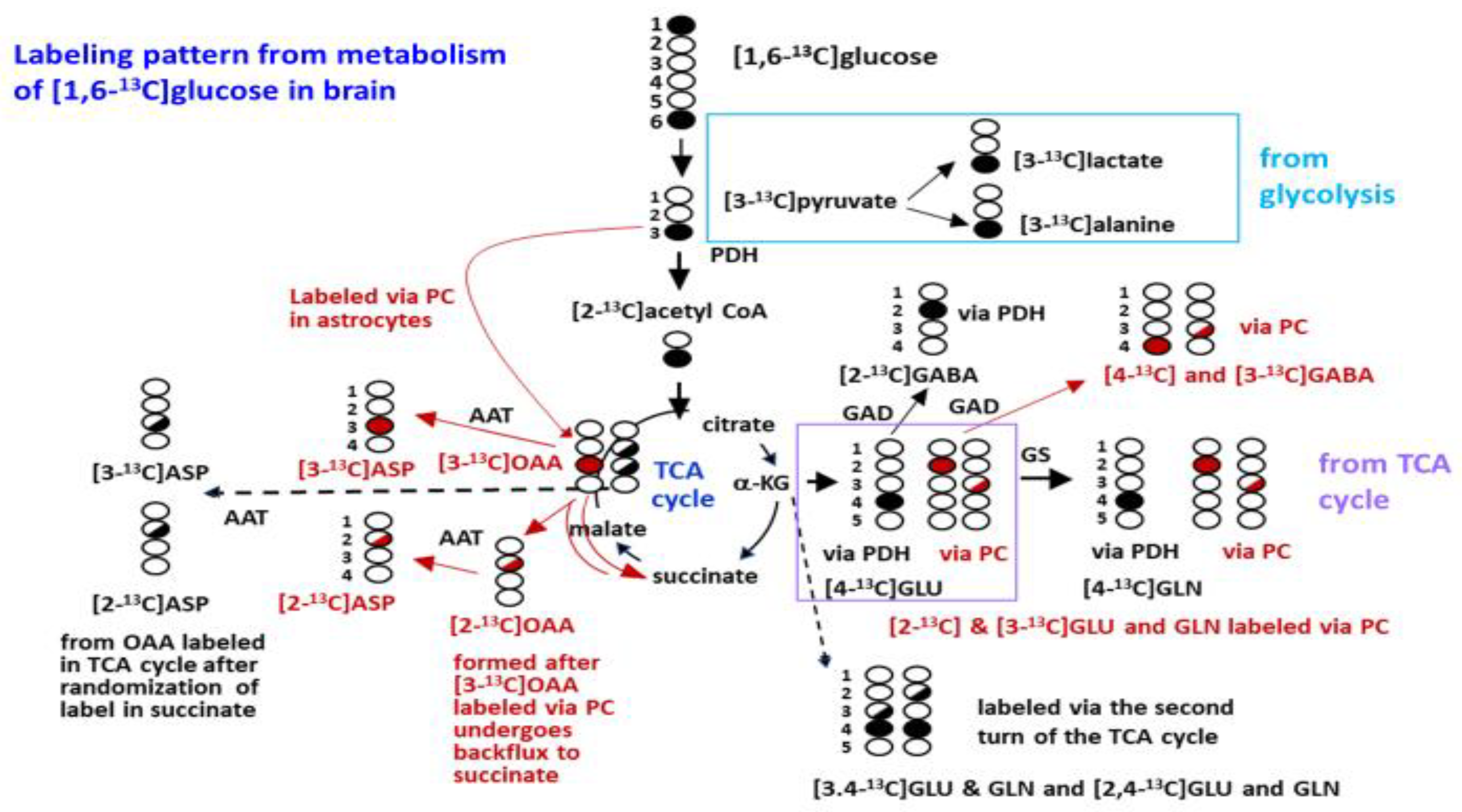
2.6. Labeling from the Metabolism of [1,6-13C]glucose in the Brain
2.7. Determination of Metabolite and Protein Concentration
2.8. Determination of Percent Enrichment of Labeled Metabolites
2.9. Statistical Analysis
3. Results
3.1. Concentration of Metabolites in the Brain
3.2. Metabolism Changes in SIRT3 KO Brain
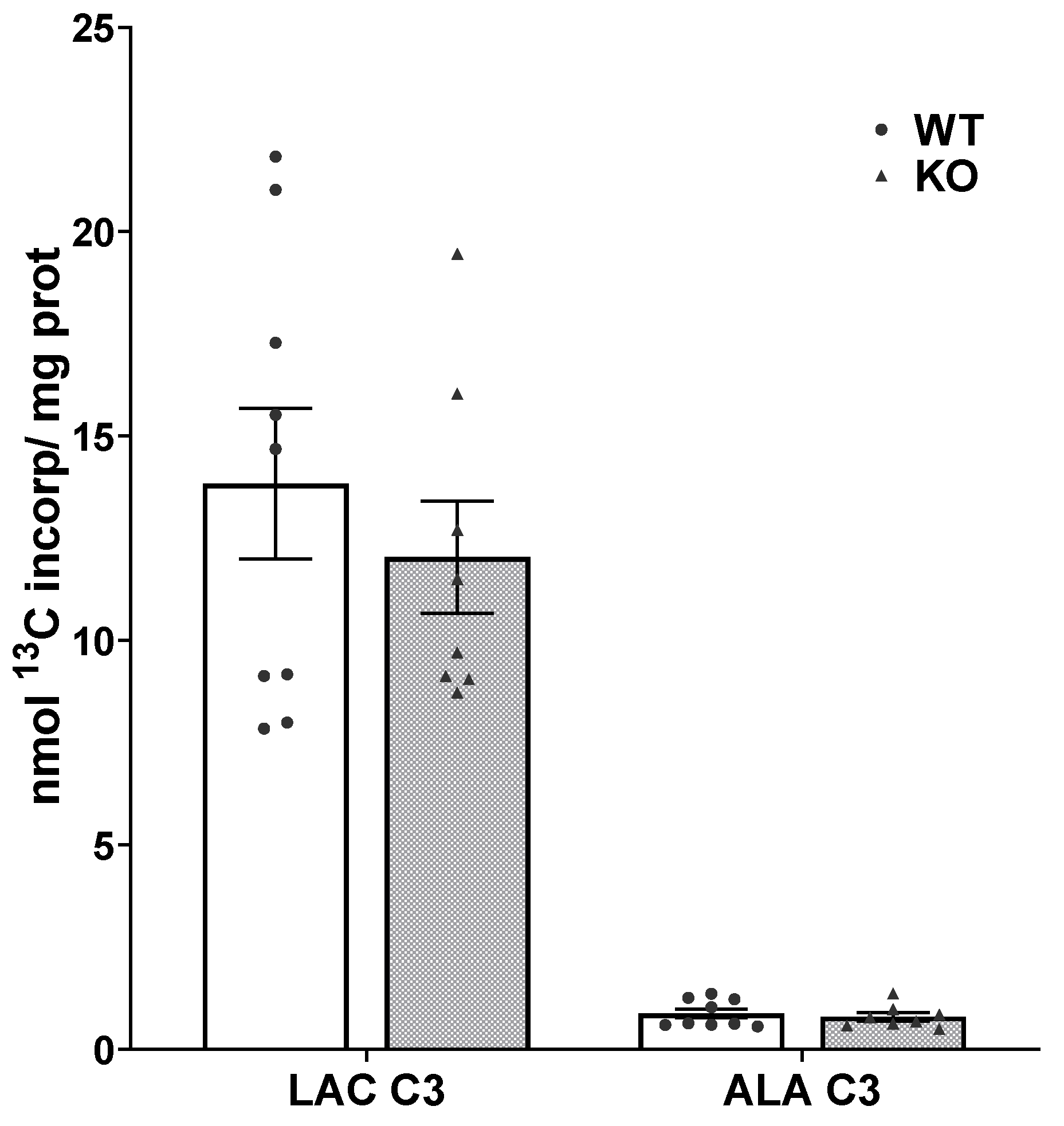
3.3. Glycolysis Is Not Impaired in SIRT3 KO Brain
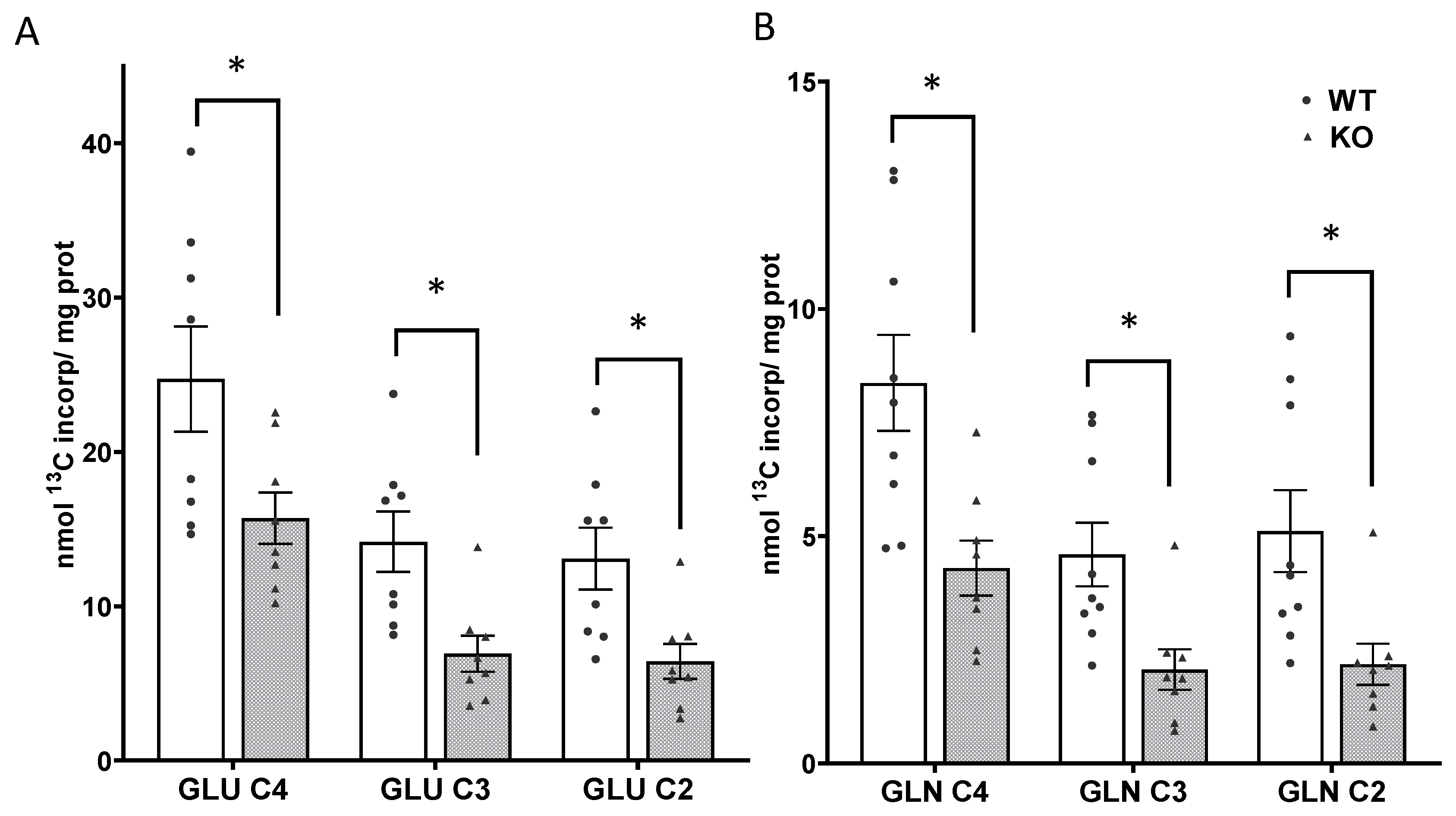
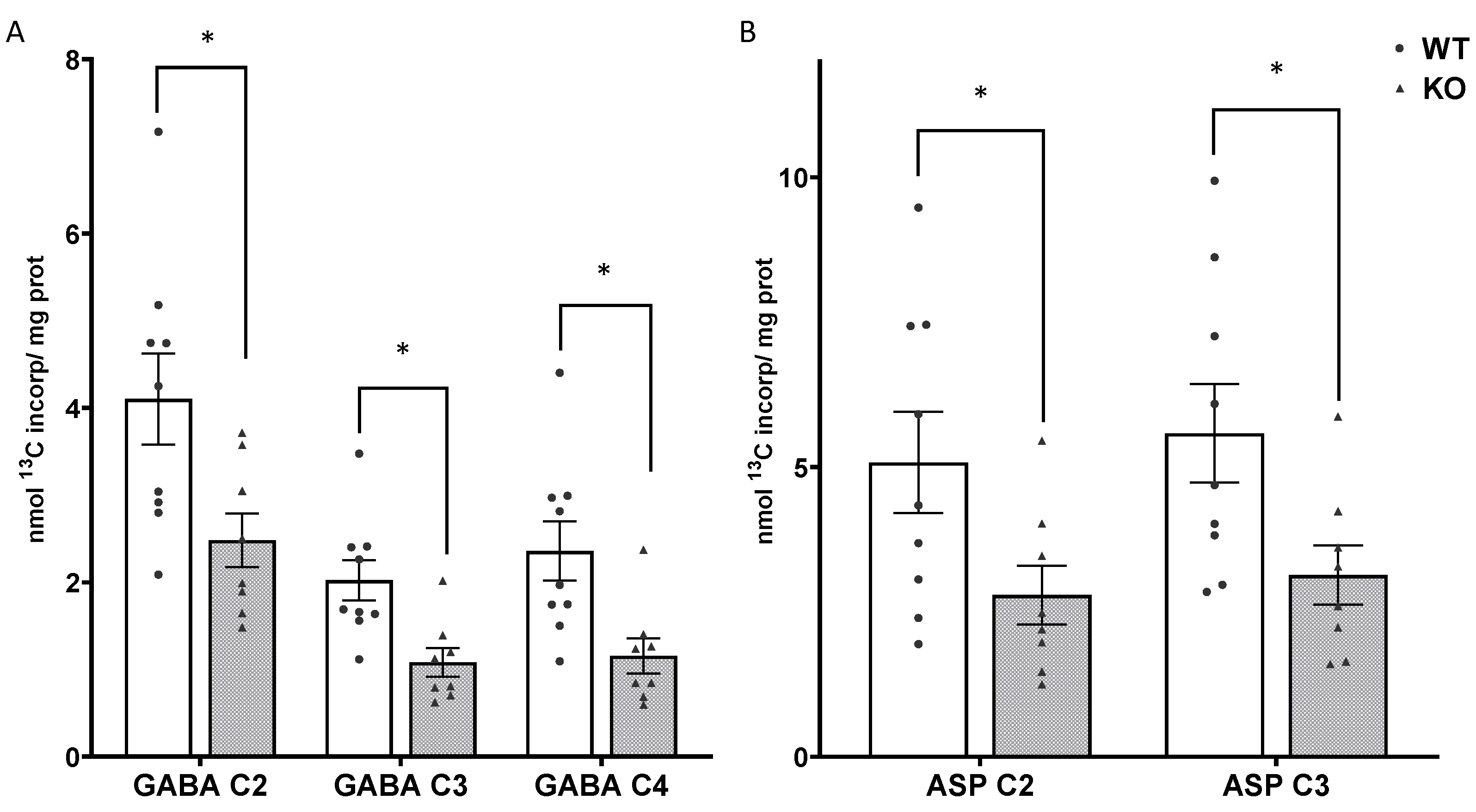
3.4. Mitochondrial Oxidative Metabolism Is Decreased in SIRT3 KO Brain
3.5. Cycling Ratios
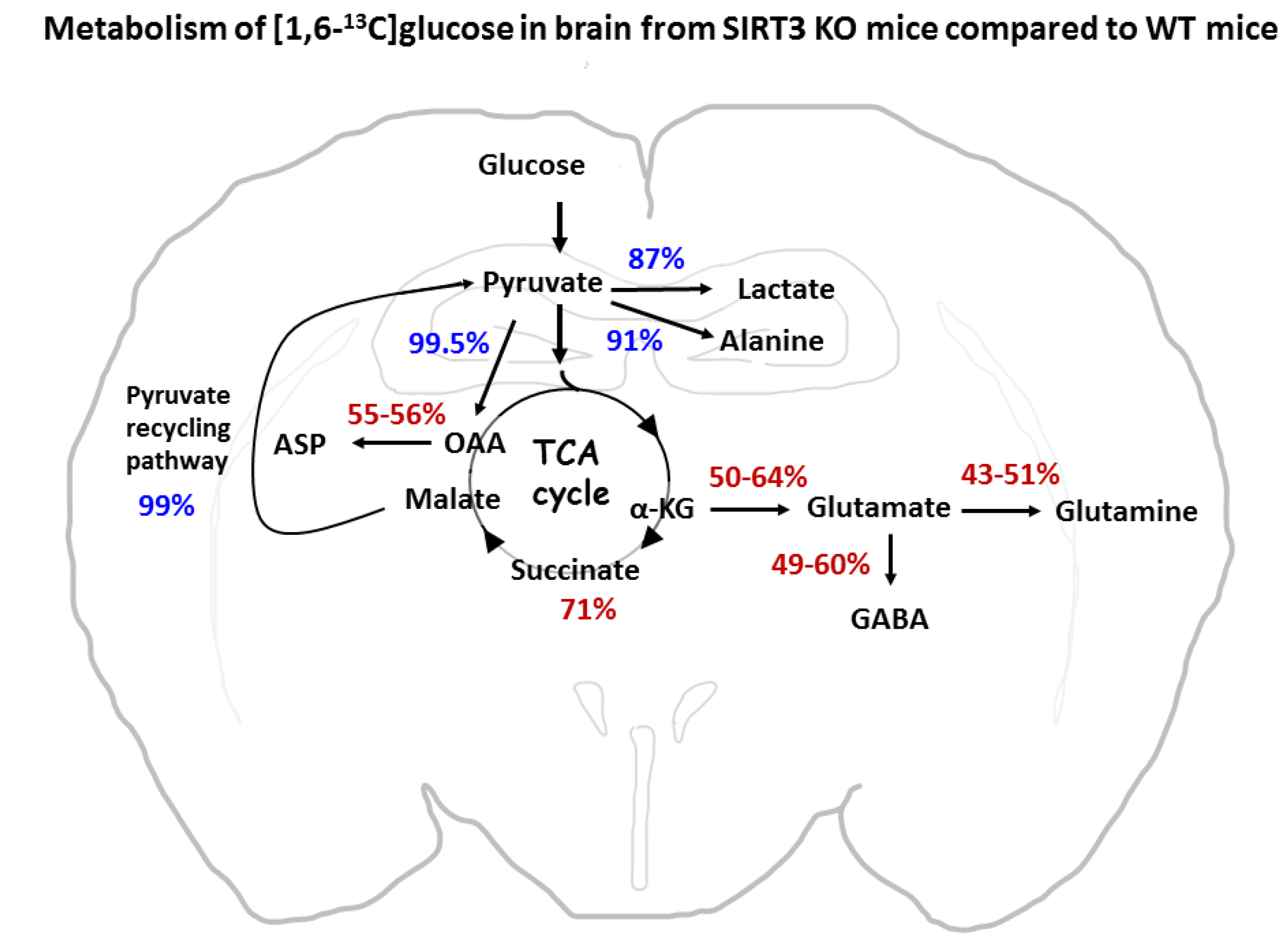
3.6. Metabolism via the TCA Cycle and the Pyruvate Recycling Pathway
3.7. Percent Enrichment of Metabolites
4. Discussion
4.1. Sirt3 Does Not Affect Glucose Uptake or Glycolytic Metabolism
4.2. Differential Incorporation of 13C into Metabolites in Wild Type and SIRT3 KO Mice
4.3. Perturbed Metabolic Coupling between Neurons and Astrocytes in SIRT3 KO Brain
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Close, P.; Creppe, C.; Gillard, M.; Ladang, A.; Chapelle, J.P.; Nguyen, L.; Chariot, A. The emerging role of lysine acetylation of non-nuclear proteins. Cell Mol. Life Sci. 2010, 67, 1255–1264. [Google Scholar] [CrossRef]
- Anderson, K.A.; Hirschey, M.D. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012, 52, 23–35. [Google Scholar] [CrossRef]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Newman, J.C.; Wang, M.Z.; Ho, L.; Verdin, E. Mitochondrial sirtuins: Regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 2012, 23, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; He, W.; Verdin, E. Mitochondrial protein acylation and intermediary metabolism: Regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 2012, 287, 42436–42443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.; Pugazhenthi, S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018, 8, 17547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidorova-Darmos, E.; Sommer, R.; Eubanks, J.H. The Role of SIRT3 in the Brain Under Physiological and Pathological Conditions. Front. Cell Neurosci. 2018, 12, 196. [Google Scholar] [CrossRef]
- Jesko, H.; Wencel, P.; Strosznajder, R.P.; Strosznajder, J.B. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem. Res. 2016, 42, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.; Benarroch, E.E. Mitochondrial dynamics: General concepts and clinical implications. Neurology 2012, 78, 1612–1619. [Google Scholar] [CrossRef]
- Scott, I.; Webster, B.R.; Li, J.H.; Sack, M.N. Identification of a molecular component of the mitochondrial acetyltransferase programme: A novel role for GCN5L1. Biochem. J. 2012, 443, 655–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Huang, J.Y.; Schwer, B.; Verdin, E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, H.B.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell. 2013, 50, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Tancopoulos, G.D.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Long, A.; Kristian, T. Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3 dependent mechanism in male mice. J. Neurosci. Res. 2019, 325, 113144. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C.; Scafidi, S.; Robertson, C.L. Metabolic Alterations in Developing Brain After Injury: Knowns and Unknowns. Neurochem. Res. 2015, 40, 2527–2543. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; O’Neill, B.T.; Rardin, M.J.; Kleinridders, A.; Ilkeyeva, O.R.; Ussar, S.; Bain, J.R.; Lee, K.Y.; Verdin, E.M.; Newgard, C.B.; et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013, 62, 3404–3417. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Nagasawa, K.; Munch, C.; Xu, Y.; Satterstrom, K.; Jeong, S.; Hayes, S.D.; Jedrychowski, P.; Vyas, F.S.; Zaganjor, E.; et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016, 167, 985–1000.e21. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddingr, S.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell. 2011, 44, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sol, E.M.; Wagner, S.A.; Weinert, B.T.; Kumar, A.; Kim, H.S.; Deng, C.X.; Choudhary, C. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase sirt3. PLoS ONE 2012, 7, e50545. [Google Scholar] [CrossRef]
- Ferreira, G.C.; Karimi, A.J.; Waddell, J.; McKenna, M.C. Metabolism of [1,6-13C]glucose in the cerebellum of 18-day-old rats: Comparison with cerebral metabolism. J. Neurochem. 2021. [Google Scholar] [CrossRef]
- Sibson, N.R.; Shen, J.; Mason, G.F.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. Functional energy metabolism: In vivo 13C-NMR spectroscopy evidence for coupling of cerebral glucose consumption and glutamatergic neuronal activity. Dev. Neurosci. 1998, 20, 321–330. [Google Scholar] [CrossRef]
- Richards, E.M.; Fiskum, G.; Rosenthal, R.E.; Hopkins, I.; McKenna, M.C. Hyperoxic reperfusion after global ischemia decreases hippocampal energy metabolism. Strokea 2007, 38, 1578–1584. [Google Scholar] [CrossRef] [Green Version]
- Melo, T.M.; Sonnewald, U.; Bastholm, I.A.; Nehlig, A. Astrocytes may play a role in the etiology of absence epilepsy: A comparison between immature GAERS not yet expressing seizures and adults. Neurobiol. Dis. 2007, 28, 227–235. [Google Scholar] [CrossRef]
- Sonnewald, U.; Kondziella, D. Neuronal glial interaction in different neurological diseases studied by ex vivo 13C NMR spectroscopy. NMR Biomed. 2003, 16, 424–429. [Google Scholar] [CrossRef]
- Rae, C.; Nasrallah, F.A.; Broer, S. Metabolic effects of blocking lactate transport in brain cortical tissue slices using an inhibitor specific to MCT1 and MCT2. Neurochem. Res. 2009, 34, 1783–1791. [Google Scholar] [CrossRef]
- Morken, T.S.; Brekke, E.; Haberg, A.; Wideroe, M.; Brubakk, A.M.; Sonnewald, U. Altered astrocyte-neuronal interactions after hypoxia-ischemia in the neonatal brain in female and male rats. Stroke 2014, 45, 2777–2785. [Google Scholar] [CrossRef] [Green Version]
- Haberg, A.K.; Qu, H.; Sonnewald, U. Acute changes in intermediary metabolism in cerebellum and contralateral hemisphere following middle cerebral artery occlusion in rat. J. Neurochem. 2009, 109 (Suppl. S1), 174–181. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Jeninga, E.H.; Canto, C.; Harach, T.; de Boer, V.C.; Andreux, P.; Moullan, N.; Pirinen, E.; Yamamoto, H.; Houten, S.M.; et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci. Rep. 2012, 2, 00425. [Google Scholar] [CrossRef] [PubMed]
- Scher, M.B.; Vaquero, A.; Reinberg, D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 2002, 158, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Lu, H.F.; Alano, C.C. Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS ONE 2011, 6, e14731. [Google Scholar] [CrossRef]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Fritz, K.S.; Galligan, J.J.; Hirschey, M.D.; Verdin, E.; Petersen, D.R. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 2012, 11, 1633–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittenhafer-Reed, K.E.; Richards, A.L.; Fan, J.; Smallegan, M.J.; Siahpirani, A.F.; Kemmerer, Z.A.; Prolla, T.A.; Roy, S.; Coon, J.J.; Denu, J.M. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 2015, 21, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, M.; Wang, Z.Q.; Namura, S.; Waeber, C.; Moskowitz, M.A. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 1997, 17, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Strosznajder, R.P.; Czubowicz, K.; Jesko, H.; Strosznajder, J.B. Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol. Neurobiol. 2010, 41, 187–196. [Google Scholar] [CrossRef]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Tang, L.; Wei, W.; Hong, Y.; Chen, H.; Ying, W.; Chen, S. Nicotinamide mononucleotide improves energy activity and survival rate in an in vitro model of Parkinson’s disease. Exp. Ther. Med. 2014, 8, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.N.; Owens, K.; Schlappal, A.E.; Kristian, T.; Fishman, P.S.; Schuh, R.A. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Buler, M.; Andersson, U.; Hakkola, J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. 2016, 30, 3942–3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Vigueira, P.A.; McCommis, K.S.; Schweitzer, G.G.; Remedi, M.S.; Chambers, K.T.; Fu, X.; McDonald, W.G.; Cole, S.L.; Colca, J.R.; Kletzien, R.F.; et al. Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep. 2014, 7, 2042–2053. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Liu, Q.; Li, Q.; Liu, B.; Yang, Y.; Zhang, N. Berberine Reduces Pyruvate-driven Hepatic Glucose Production by Limiting Mitochondrial Import of Pyruvate through Mitochondrial Pyruvate Carrier 1. EBioMedicine 2018, 34, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.X.; Li, X.; Dong, S.Y.; Guo, Y.J.; Liu, T.; Wu, Y.C. SIRT3 deacetylated and increased citrate synthase activity in PD model. Biochem. Biophys. Res. Commun. 2017, 484, 767–773. [Google Scholar] [CrossRef]
- Smolkova, K.; Spackova, J.; Gotvaldova, K.; Dvorak, A.; Krenkova, A.; Hubalek, M.; Holendova, B.; Vitek, L.; Jezek, P. SIRT3 and GCN5L regulation of NADP+- and NADPH-driven reactions of mitochondrial isocitrate dehydrogenase IDH2. Sci. Rep. 2020, 10, 8677. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Weddle, A.; Kinter, C.S.; Humphries, K.M.; Mather, T.; Szweda, L.I.; Kinter, M. Lysine Acetylation Activates Mitochondrial Aconitase in the Heart. Biochemistry 2015, 54, 4008–4018. [Google Scholar] [CrossRef] [Green Version]
- Dessai, A.S.; Dominguez, M.P.; Chen, U.I.; Hasper, J.; Prechtl, C.; Yu, C.; Katsuta, E.; Dai, T.; Zhu, B.; Jung, S.Y.; et al. Transcriptional Repression of SIRT3 Potentiates Mitochondrial Aconitase Activation to Drive Aggressive Prostate Cancer to the Bone. Cancer Res. 2021, 81, 50–63. [Google Scholar] [CrossRef]
- Castro, L.; Tortora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron-Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 2019, 52, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; Cha, J.; Lee, J.; Roe, J.H. Glutathione reductase and a mitochondrial thioredoxin play overlapping roles in maintaining iron-sulfur enzymes in fission yeast. Eukaryot. Cell 2006, 5, 1857–1865. [Google Scholar] [CrossRef] [Green Version]
- Keyer, K.; Imlay, J.A. Inactivation of dehydratase [4Fe-4S] clusters and disruption of iron homeostasis upon cell exposure to peroxynitrite. J. Biol. Chem. 1997, 272, 27652–27659. [Google Scholar] [CrossRef] [Green Version]
- Richardson, T.E.; Yu, A.E.; Wen, Y.; Yang, S.H.; Simpkins, J.W. Estrogen prevents oxidative damage to the mitochondria in Friedreich’s ataxia skin fibroblasts. PLoS ONE 2012, 7, e34600. [Google Scholar] [CrossRef]
- Klimova, N.; Fearnow, A.; Kristian, T. Role of NAD(+)-Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury. Brain Sci. 2020, 10, 449. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, C.; Gertz, M.; Papatheodorou, P.; Kachholz, B.; Becker, C.F.; Steegborn, C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 2008, 382, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C. The glutamate-glutamine cycle is not stoichiometric: Fates of glutamate in brain. J. Neurosci. Res. 2007, 85, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Lee, J.E.; Sweredoski, M.J.; Yang, S.J.; Jeon, S.J.; Harrison, J.S.; Yim, J.-H.; Lee, S.G.; Handa, H.; Kuhlman, B.; et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol. Cell 2016, 61, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, K.N.G.; Zaganjor, E.; Ishikawa, Y.; Spinelli, J.B.; Yoon, H.; Lin, J.R.; Satterstrom, F.K.; Rengel, A.; Mulei, S.; Souza, A.; et al. Small-Molecule Screen Identifies De Novo Nucleotide Synthesis as a Vulnerability of Cells Lacking SIRT3. Cell Rep. 2018, 22, 1945–1955. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhou, L.; Shi, Q.; Zhao, Y.; Lin, H.; Zhang, M.; Zhao, S.; Yang, Y.; Ling, Z.-Q.; Guan, K.-L.; et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015, 34, 1110–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | WT % Enrichment | SIRT3 KO % Enrichment | Significance |
|---|---|---|---|
| Glucose | 38.62 ± 7.23 | 37.98 ± 4.33 | t = −0.074, p = 0.942 |
| Lactate C3 | 28.25 ± 3.55 | 26.24 ± 3.45 | t = 0.423, p = 0.673 |
| Alanine C3 | 36.19 ± 5.16 | 33.26 ± 5.15 | t = 0.451, p = 0.659 |
| Glutamate C4 | 32.40 ± 3.88 | 23.02 ± 3.15 | t = 1.847, p = 0.084 |
| Glutamate C3 | 18.15 ± 2.20 | 10.23 ± 1.93 | t = 2.669, p = 0.017 |
| Glutamate C2 | 16.69 ± 2.23 | 9.53 ± 1.88 | t = 2.419, p = 0.028 |
| Glutamine C4 | 23.18 ± 2.22 | 16.26 ± 2.38 | t = 2.12, p = 0.050 |
| Glutamine C3 | 12.67 ± 1.56 | 7.87 ± 1.68 | t = 2.09, p = 0.053 |
| Glutamine C2 | 14.00 ± 2.14 | 8.32 ± 1.73 | t = 2.02, p = 0.061 |
| GABA C2 | 25.90 ± 2.49 | 21.55 ± 3.26 | t = 1.076, p = 0.299 |
| GABA C3 | 12.92 ± 1.24 | 9.40 ± 1.61 | t = 1.752, p = 0.100 |
| GABA C4 | 14.75 ± 1.56 | 10.06 ± 1.93 | t = 1.901, p = 0.076 |
| Lactate C2 | 1.37 ± 0.14 | 1.34 ± 0.17 | t = 0.15, p = 0.882 |
| Aspartate C2 | 27.51 ± 3.99 | 18.14 ± 3.75 | t = 1.69, p = 0.110 |
| Aspartate C3 | 30.62 ± 3.73 | 20.15 ± 3.76 | t = 1.97, p = 0.067 |
| Succinate C2/3 | 26.64 ± 3.32 | 20.68 ± 3.39 | t = 1.253, p = 0.229 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kristian, T.; Karimi, A.J.; Fearnow, A.; Waddell, J.; McKenna, M.C. Perturbed Brain Glucose Metabolism Caused by Absent SIRT3 Activity. Cells 2021, 10, 2348. https://doi.org/10.3390/cells10092348
Kristian T, Karimi AJ, Fearnow A, Waddell J, McKenna MC. Perturbed Brain Glucose Metabolism Caused by Absent SIRT3 Activity. Cells. 2021; 10(9):2348. https://doi.org/10.3390/cells10092348
Chicago/Turabian StyleKristian, Tibor, Arman J. Karimi, Adam Fearnow, Jaylyn Waddell, and Mary C. McKenna. 2021. "Perturbed Brain Glucose Metabolism Caused by Absent SIRT3 Activity" Cells 10, no. 9: 2348. https://doi.org/10.3390/cells10092348
APA StyleKristian, T., Karimi, A. J., Fearnow, A., Waddell, J., & McKenna, M. C. (2021). Perturbed Brain Glucose Metabolism Caused by Absent SIRT3 Activity. Cells, 10(9), 2348. https://doi.org/10.3390/cells10092348