
Evolution of CD8+ T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer

 , , and
, , and
Abstract
:1. Introduction
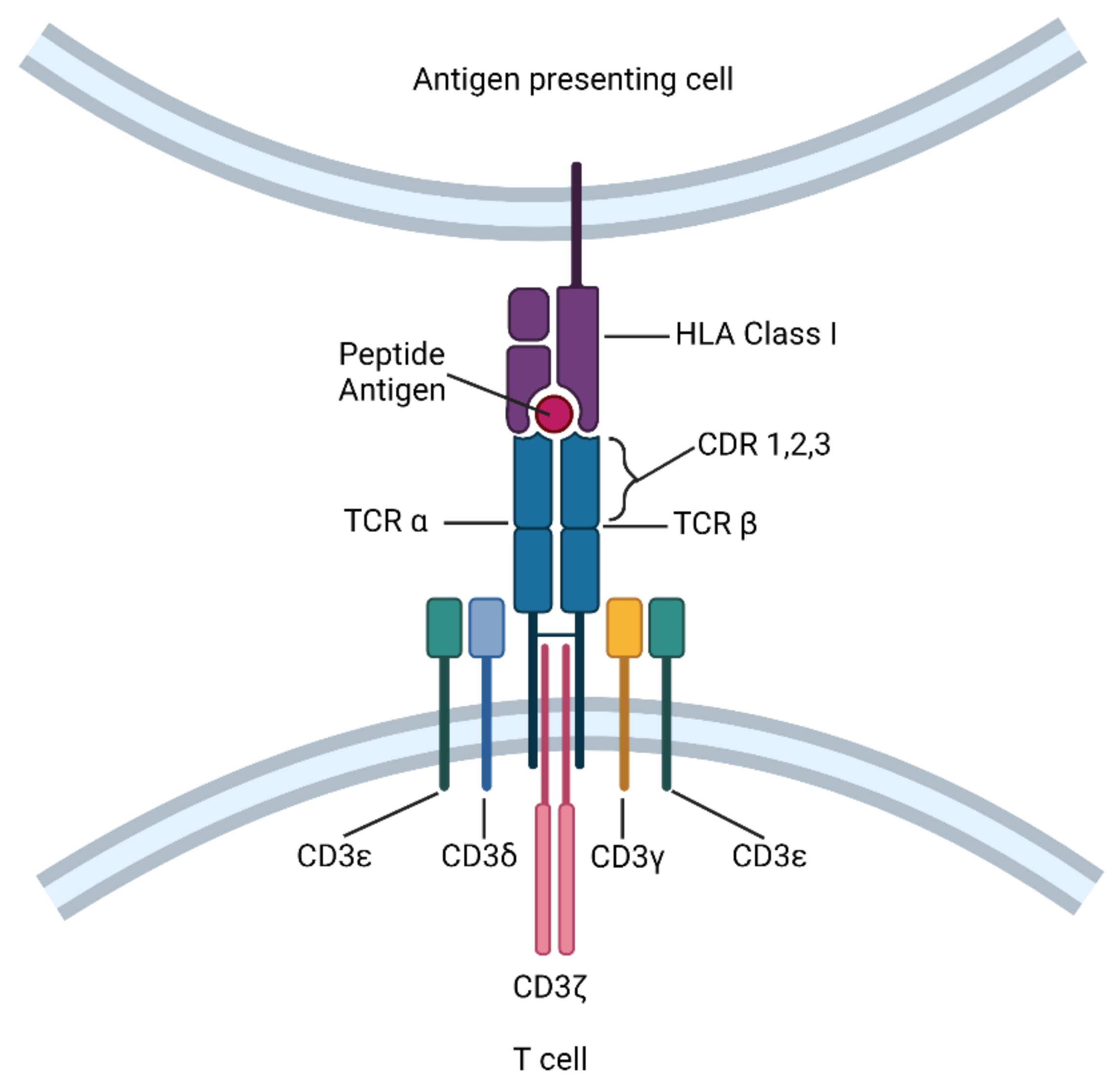
2. Definition and Mechanistic Overview of TCR-T Cell Immunotherapy
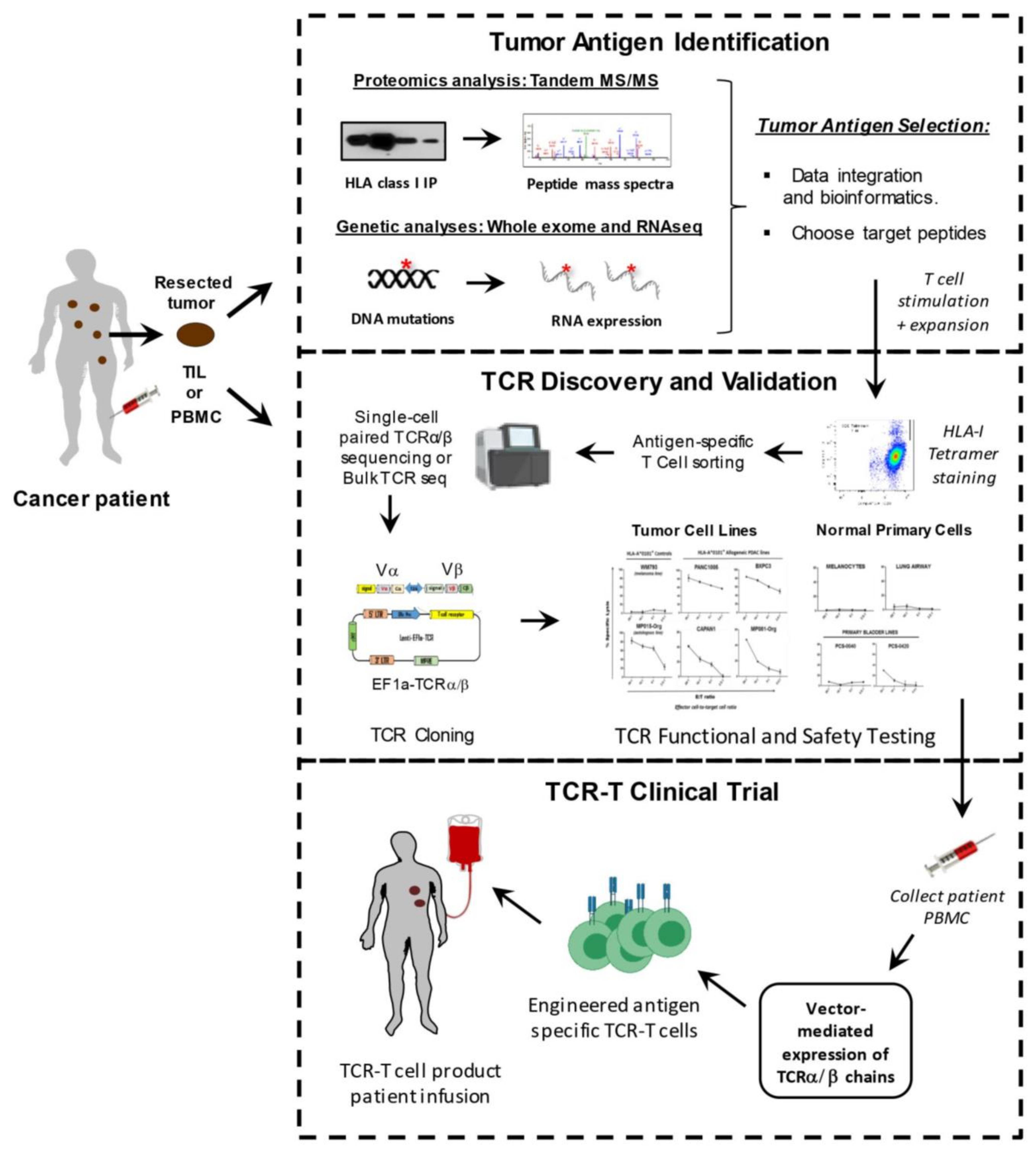
3. Tools and Techniques for TCR-T Cell Development
3.1. Target Antigens for TCR-T Therapy
3.2. Methods for Identifying Tumor-Associated Antigens
3.3. Approaches for Identifying Tumor Neoantigens
3.4. Isolation of Tumor-Specific T Cells and T Cell Receptors (TCR)
4. TCR Cloning and Validation Approaches
5. Clinical Trials of TCR-T Cell Therapy for Cancer
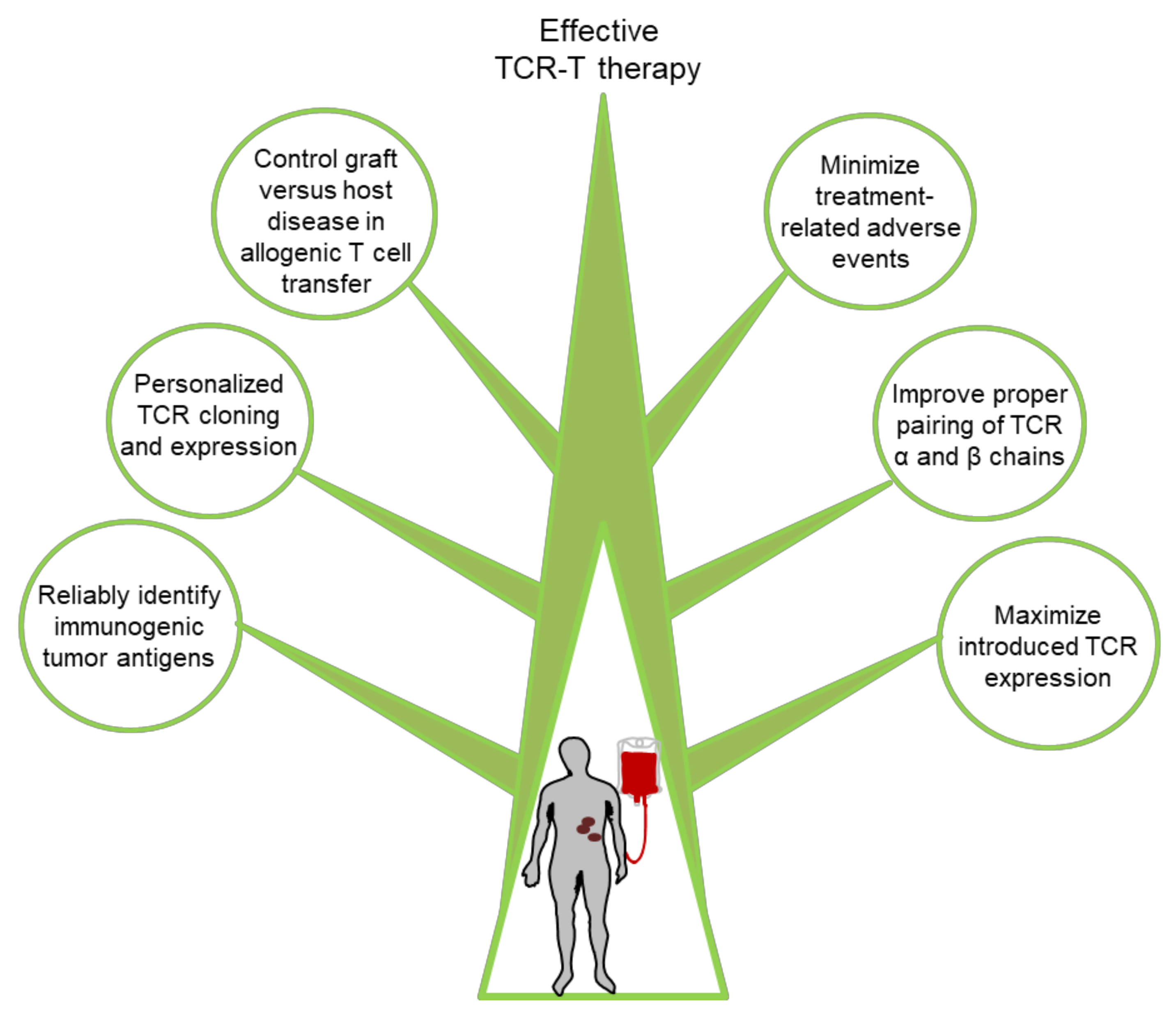
6. Challenges and Potential Solutions for Improving TCR-T Cell Therapy
6.1. Discovery of New Targets
6.2. Maximizing Therapeutic TCR Expression
6.3. Minimization of Adverse Events
6.4. Graft Versus Host Disease in Allogenic T Cell Transfer
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 2020, 11, 1689. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavunja, H.W.; Lang, S.; Sungsuwan, S.; Yin, Z.; Huang, X. Delivery of foreign cytotoxic T lymphocyte epitopes to tumor tissues for effective antitumor immunotherapy against pre-established solid tumors in mice. Cancer Immunol. Immunother. 2017, 66, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef]
- Kuhns, M.S.; Davis, M.M.; Garcia, K.C. Deconstructing the form and function of the TCR/CD3 complex. Immunity 2006, 24, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Samelson, L.E. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annu. Rev. Immunol. 2002, 20, 371–394. [Google Scholar] [CrossRef] [Green Version]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Clay, T.M.; Custer, M.C.; Sachs, J.; Hwu, P.; Rosenberg, S.A.; Nishimura, M.I. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J. Immunol. 1999, 163, 507–513. [Google Scholar]
- Dembic, Z.; Haas, W.; Weiss, S.; McCubrey, J.; Kiefer, H.; von Boehmer, H.; Steinmetz, M. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature 1986, 320, 232–238. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.; Hofmeister, R.; Quintas-Cardama, A. Synthetic T cell receptor-based lymphocytes for cancer therapy. Adv. Drug Deliv. Rev. 2019, 141, 47–54. [Google Scholar] [CrossRef]
- Johnson, L.A.; Heemskerk, B.; Powell, D.J., Jr.; Cohen, C.J.; Morgan, R.A.; Dudley, M.E.; Robbins, P.F.; Rosenberg, S.A. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J. Immunol. 2006, 177, 6548–6559. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016, 22, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Peng, Y.; Wang, L.; Hong, Y.; Jiang, X.; Li, Q.; Liu, H.; Huang, L.; Wu, J.; Celis, E.; et al. Identification of alpha-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology 2018, 68, 574–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Bruggen, P.; Zhang, Y.; Chaux, P.; Stroobant, V.; Panichelli, C.; Schultz, E.S.; Chapiro, J.; Van Den Eynde, B.J.; Brasseur, F.; Boon, T. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol. Rev. 2002, 188, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Singh-Jasuja, H.; Emmerich, N.P.; Rammensee, H.G. The Tubingen approach: Identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol. Immunother. 2004, 53, 187–195. [Google Scholar] [CrossRef]
- Loffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Muhlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Ehx, G.; Perreault, C. Discovery and characterization of actionable tumor antigens. Genome Med. 2019, 11, 29. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019, 120, 109542. [Google Scholar] [CrossRef]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012, 135, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Rammensee, H.G.; Singh-Jasuja, H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev. Vaccines 2013, 12, 1211–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, S.D.; Talukder, A.H.; Lai, I.; Davis, R.; Alvarez, H.; Tiriac, H.; Zhang, M.; Chiu, Y.; Melendez, B.; Jackson, K.R.; et al. Vestigial-like 1 is a shared targetable cancer-placenta antigen expressed by pancreatic and basal-like breast cancers. Nat. Commun. 2020, 11, 5332. [Google Scholar] [CrossRef]
- Li, F.; Deng, L.; Jackson, K.R.; Talukder, A.H.; Katailiha, A.S.; Bradley, S.D.; Zou, Q.; Chen, C.; Huo, C.; Chiu, Y.; et al. Neoantigen vaccination induces clinical and immunologic responses in non-small cell lung cancer patients harboring EGFR mutations. J. Immunother. Cancer 2021, 9, e002531. [Google Scholar] [CrossRef]
- Li, F.; Chen, C.; Ju, T.; Gao, J.; Yan, J.; Wang, P.; Xu, Q.; Hwu, P.; Du, X.; Lizee, G. Rapid tumor regression in an Asian lung cancer patient following personalized neo-epitope peptide vaccination. Oncoimmunology 2016, 5, e1238539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.C.; Robbins, P.F. Cancer immunotherapy targeting neoantigens. Semin. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Cai, A.; Keskin, D.B.; DeLuca, D.S.; Alonso, A.; Zhang, W.; Zhang, G.L.; Hammond, N.N.; Nardi, V.; Stone, R.M.; Neuberg, D.; et al. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin. Cancer Res. 2012, 18, 5761–5772. [Google Scholar] [CrossRef] [Green Version]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Lower, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020, 38, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.K.; van Buuren, M.M.; Dang, K.K.; Hubbard-Lucey, V.M.; Sheehan, K.C.F.; Campbell, K.M.; Lamb, A.; Ward, J.P.; Sidney, J.; Blazquez, A.B.; et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183, 818–834. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef]
- Li, G.; Bethune, M.T.; Wong, S.; Joglekar, A.V.; Leonard, M.T.; Wang, J.K.; Kim, J.T.; Cheng, D.; Peng, S.; Zaretsky, J.M.; et al. T cell antigen discovery via trogocytosis. Nat. Methods 2019, 16, 183–190. [Google Scholar] [CrossRef]
- Gee, M.H.; Han, A.; Lofgren, S.M.; Beausang, J.F.; Mendoza, J.L.; Birnbaum, M.E.; Bethune, M.T.; Fischer, S.; Yang, X.; Gomez-Eerland, R.; et al. Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell 2018, 172, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Linnemann, C.; Heemskerk, B.; Kvistborg, P.; Kluin, R.J.; Bolotin, D.A.; Chen, X.; Bresser, K.; Nieuwland, M.; Schotte, R.; Michels, S.; et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat. Med. 2013, 19, 1534–1541. [Google Scholar] [CrossRef]
- Arber, C.; Feng, X.; Abhyankar, H.; Romero, E.; Wu, M.F.; Heslop, H.E.; Barth, P.; Dotti, G.; Savoldo, B. Survivin-specific T cell receptor targets tumor but not T cells. J. Clin. Investig. 2015, 125, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Obenaus, M.; Leitao, C.; Leisegang, M.; Chen, X.; Gavvovidis, I.; van der Bruggen, P.; Uckert, W.; Schendel, D.J.; Blankenstein, T. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat. Biotechnol. 2015, 33, 402–407. [Google Scholar] [CrossRef]
- Weigand, L.U.; Liang, X.; Schmied, S.; Mall, S.; Klar, R.; Stotzer, O.J.; Salat, C.; Gotze, K.; Mautner, J.; Peschel, C.; et al. Isolation of human MHC class II-restricted T cell receptors from the autologous T-cell repertoire with potent anti-leukaemic reactivity. Immunology 2012, 137, 226–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossinger, G.; Bunse, M.; Bet, J.; Albrecht, J.; Paszkiewicz, P.J.; Weissbrich, B.; Schiedewitz, I.; Henkel, L.; Schiemann, M.; Neuenhahn, M.; et al. MHC multimer-guided and cell culture-independent isolation of functional T cell receptors from single cells facilitates TCR identification for immunotherapy. PLoS ONE 2013, 8, e61384. [Google Scholar] [CrossRef]
- Li, L.P.; Lampert, J.C.; Chen, X.; Leitao, C.; Popovic, J.; Muller, W.; Blankenstein, T. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat. Med. 2010, 16, 1029–1034. [Google Scholar] [CrossRef]
- Bonini, C.; Mondino, A. Adoptive T-cell therapy for cancer: The era of engineered T cells. Eur. J. Immunol. 2015, 45, 2457–2469. [Google Scholar] [CrossRef] [Green Version]
- D’Ippolito, E.; Wagner, K.I.; Busch, D.H. Needle in a Haystack: The Naive Repertoire as a Source of T Cell Receptors for Adoptive Therapy with Engineered T Cells. Int. J. Mol. Sci. 2020, 21, 8324. [Google Scholar] [CrossRef]
- Douglas, J.T. Adenoviral vectors for gene therapy. Mol. Biotechnol. 2007, 36, 71–80. [Google Scholar] [CrossRef]
- Cepko, C.; Pear, W. Overview of the retrovirus transduction system. Curr. Protoc. Mol. Biol. 2001, 36. [Google Scholar] [CrossRef]
- Doi, K.; Takeuchi, Y. Gene therapy using retrovirus vectors: Vector development and biosafety at clinical trials. Uirusu 2015, 65, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Verhoeyen, E.; Costa, C.; Cosset, F.L. Lentiviral vector gene transfer into human T cells. Methods Mol. Biol. 2009, 506, 97–114. [Google Scholar] [CrossRef]
- Zong, S.; Mi, T.; Flores, L.G., 2nd; Alpert, A.; Olivares, S.; Patel, K.; Maiti, S.; McNamara, G.; Cooper, L.J.N.; Torikai, H. Very rapid cloning, expression and identifying specificity of T-cell receptors for T-cell engineering. PLoS ONE 2020, 15, e0228112. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Albers, J.J.; Ammon, T.; Gosmann, D.; Audehm, S.; Thoene, S.; Winter, C.; Secci, R.; Wolf, A.; Stelzl, A.; Steiger, K.; et al. Gene editing enables T-cell engineering to redirect antigen specificity for potent tumor rejection. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Q.; Zhao, W.H.; Li, Y.; Zhu, B.; Yin, K.S. Adeno-associated virus mediated gene transfer into lung cancer cells promoting CD40 ligand-based immunotherapy. Virology 2007, 368, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Koeberl, D.D.; Pinto, C.; Sun, B.; Li, S.; Kozink, D.M.; Benjamin, D.K., Jr.; Demaster, A.K.; Kruse, M.A.; Vaughn, V.; Hillman, S.; et al. AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. Mol. Ther. 2008, 16, 665–672. [Google Scholar] [CrossRef]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Schambach, A. Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Birkholz, K.; Hombach, A.; Krug, C.; Reuter, S.; Kershaw, M.; Kampgen, E.; Schuler, G.; Abken, H.; Schaft, N.; Dorrie, J. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009, 16, 596–604. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.T.; Yang, N.; Lee Krishnamoorthy, T.; Oei, V.; Chua, A.; Zhao, X.; Tan, H.S.; Chia, A.; Le Bert, N.; Low, D.; et al. Use of Expression Profiles of HBV-DNA Integrated Into Genomes of Hepatocellular Carcinoma Cells to Select T Cells for Immunotherapy. Gastroenterology 2019, 156, 1862–1876. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Tran, E.; Parkhurst, M.R.; Cohen, C.J.; Robbins, P.F.; Cooper, L.J.; Rosenberg, S.A. Stable, Nonviral Expression of Mutated Tumor Neoantigen-specific T-cell Receptors Using the Sleeping Beauty Transposon/Transposase System. Mol. Ther. 2016, 24, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torikai, H.; Reik, A.; Liu, P.Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovich, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Hale, M.; Lee, B.; Honaker, Y.; Leung, W.H.; Grier, A.E.; Jacobs, H.M.; Sommer, K.; Sahni, J.; Jackson, S.W.; Scharenberg, A.M.; et al. Homology-Directed Recombination for Enhanced Engineering of Chimeric Antigen Receptor T Cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, S.N.; Huls, H.; Singh, H.; Dawson, M.; Figliola, M.; Olivares, S.; Rao, P.; Zhao, Y.J.; Multani, A.; Yang, G.; et al. Sleeping beauty system to redirect T-cell specificity for human applications. J. Immunother. 2013, 36, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarsheth, N.B.; Norberg, S.M.; Sinkoe, A.L.; Adhikary, S.; Meyer, T.J.; Lack, J.B.; Warner, A.C.; Schweitzer, C.; Doran, S.L.; Korrapati, S.; et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat. Med. 2021, 27, 419–425. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Perumal, D.; Imai, N.; Lagana, A.; Finnigan, J.; Melnekoff, D.; Leshchenko, V.V.; Solovyov, A.; Madduri, D.; Chari, A.; Cho, H.J.; et al. Mutation-derived Neoantigen-specific T-cell Responses in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 450–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Aggen, D.H.; Chervin, A.S.; Schmitt, T.M.; Engels, B.; Stone, J.D.; Richman, S.A.; Piepenbrink, K.H.; Baker, B.M.; Greenberg, P.D.; Schreiber, H.; et al. Single-chain ValphaVbeta T-cell receptors function without mispairing with endogenous TCR chains. Gene Ther. 2012, 19, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Knies, D.; Klobuch, S.; Xue, S.A.; Birtel, M.; Echchannaoui, H.; Yildiz, O.; Omokoko, T.; Guillaume, P.; Romero, P.; Stauss, H.; et al. An optimized single chain TCR scaffold relying on the assembly with the native CD3-complex prevents residual mispairing with endogenous TCRs in human T-cells. Oncotarget 2016, 7, 21199–21221. [Google Scholar] [CrossRef] [Green Version]
- Bendle, G.M.; Linnemann, C.; Hooijkaas, A.I.; Bies, L.; de Witte, M.A.; Jorritsma, A.; Kaiser, A.D.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010, 16, 565–570. [Google Scholar] [CrossRef]
- Thomas, S.; Stauss, H.J.; Morris, E.C. Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology 2010, 129, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Stanislawski, T.; Voss, R.H.; Lotz, C.; Sadovnikova, E.; Willemsen, R.A.; Kuball, J.; Ruppert, T.; Bolhuis, R.L.; Melief, C.J.; Huber, C.; et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat. Immunol. 2001, 2, 962–970. [Google Scholar] [CrossRef]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef] [Green Version]
- Kuball, J.; Dossett, M.L.; Wolfl, M.; Ho, W.Y.; Voss, R.H.; Fowler, C.; Greenberg, P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 2007, 109, 2331–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Xue, S.A.; Cesco-Gaspere, M.; San Jose, E.; Hart, D.P.; Wong, V.; Debets, R.; Alarcon, B.; Morris, E.; Stauss, H.J. Targeting the Wilms tumor antigen 1 by TCR gene transfer: TCR variants improve tetramer binding but not the function of gene modified human T cells. J. Immunol. 2007, 179, 5803–5810. [Google Scholar] [CrossRef] [Green Version]
- Foley, K.C.; Spear, T.T.; Murray, D.C.; Nagato, K.; Garrett-Mayer, E.; Nishimura, M.I. HCV T Cell Receptor Chain Modifications to Enhance Expression, Pairing, and Antigen Recognition in T Cells for Adoptive Transfer. Mol. Ther. Oncolytics 2017, 5, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Voss, R.H.; Willemsen, R.A.; Kuball, J.; Grabowski, M.; Engel, R.; Intan, R.S.; Guillaume, P.; Romero, P.; Huber, C.; Theobald, M. Molecular design of the Calphabeta interface favors specific pairing of introduced TCRalphabeta in human T cells. J. Immunol. 2008, 180, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Hiasa, A.; Nishikawa, H.; Hirayama, M.; Kitano, S.; Okamoto, S.; Chono, H.; Yu, S.S.; Mineno, J.; Tanaka, Y.; Minato, N.; et al. Rapid alphabeta TCR-mediated responses in gammadelta T cells transduced with cancer-specific TCR genes. Gene Ther. 2009, 16, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Scholten, K.B.; Kramer, D.; Kueter, E.W.; Graf, M.; Schoedl, T.; Meijer, C.J.; Schreurs, M.W.; Hooijberg, E. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin. Immunol. 2006, 119, 135–145. [Google Scholar] [CrossRef]
- De Witte, M.A.; Jorritsma, A.; Kaiser, A.; van den Boom, M.D.; Dokter, M.; Bendle, G.M.; Haanen, J.B.; Schumacher, T.N. Requirements for effective antitumor responses of TCR transduced T cells. J. Immunol. 2008, 181, 5128–5136. [Google Scholar] [CrossRef]
- Yang, S.; Cohen, C.J.; Peng, P.D.; Zhao, Y.; Cassard, L.; Yu, Z.; Zheng, Z.; Jones, S.; Restifo, N.P.; Rosenberg, S.A.; et al. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther. 2008, 15, 1411–1423. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Arcangeli, S.; Mestermann, K.; Weber, J.; Bonini, C.; Casucci, M.; Hudecek, M. Overcoming key challenges in cancer immunotherapy with engineered T cells. Curr. Opin. Oncol. 2020, 32, 398–407. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef] [Green Version]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared p53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- Zamora, A.E.; Crawford, J.C.; Allen, E.K.; Guo, X.J.; Bakke, J.; Carter, R.A.; Abdelsamed, H.A.; Moustaki, A.; Li, Y.; Chang, T.C.; et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Wolff, R.A.; Wang-Gillam, A.; Alvarez, H.; Tiriac, H.; Engle, D.; Hou, S.; Groff, A.F.; San Lucas, A.; Bernard, V.; Allenson, K.; et al. Dynamic changes during the treatment of pancreatic cancer. Oncotarget 2018, 9, 14764–14790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. Int. Immunol. 2016, 28, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, 976. [Google Scholar] [CrossRef] [PubMed]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Okamoto, S.; Mineno, J.; Ikeda, H.; Fujiwara, H.; Yasukawa, M.; Shiku, H.; Kato, I. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009, 69, 9003–9011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Antigen | Number of Studies | Cancer Type | Trial Registration Number (ClinicalTrials.gov (Accessed on 9 August 2021)) |
|---|---|---|---|
| HPV16-E7 | 6 |
|
|
| HPV16-E6 | 3 |
|
|
| KRAS | 3 |
|
|
| MAGE | 10 |
|
|
| LMP2 | 2 |
|
|
| TAC-1 | 1 |
|
|
| MLANA (MART-1) | 5 |
|
|
| EBV | 4 |
|
|
| PMEL(gp100) | 3 |
|
|
| HERV-E | 1 |
|
|
| HA-1H | 2 |
|
|
| TP53 | 2 |
|
|
| WT1 | 5 |
|
|
| PRAME | 1 |
|
|
| AFP | 4 |
|
|
| CTAG1A (NY-ESO-1) | 26 |
|
|
| Trial Registration Number (ClinicalTrials.gov (Accessed on 9 August 2021)) | Target Antigen | Cancer Type | Trial Stage | Clinical Response Results | Year |
|---|---|---|---|---|---|
| NCT00509288 | MART-1 | Metastatic melanoma | Phase 2 | PR 6/21 | 2012 |
| NCT00923195 | MART-1 Gp100 | Melanoma, skin cancer | Phase 2 | CR 0/2 PR 0/2 | 2015 |
| NCT02280811 | HPV16-E6 | Vaginal, cervical, anal, penile, oropharyngeal | Phase 1 and 2 | PR 2/6 | 2017 |
| NCT01352286 | NYESO | Multiple myeloma | Phase 2 | CR 3/25 PR 18/25 | 2019 |
| NCT01967823 | NYESO | Melanoma, meningioma, beast, non-small cell lung, hepatocellular | Phase 2 | CR 10% PR 50% | 2021 |
| NCT02858310 | HPV16-E7 | Virus-associated epithelial cancers | Phase 1 | PR 6/12 | 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Li, F.; Sonnemann, H.; Jackson, K.R.; Talukder, A.H.; Katailiha, A.S.; Lizee, G. Evolution of CD8+ T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer. Cells 2021, 10, 2379. https://doi.org/10.3390/cells10092379
Sun Y, Li F, Sonnemann H, Jackson KR, Talukder AH, Katailiha AS, Lizee G. Evolution of CD8+ T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer. Cells. 2021; 10(9):2379. https://doi.org/10.3390/cells10092379
Chicago/Turabian StyleSun, Yimo, Fenge Li, Heather Sonnemann, Kyle R. Jackson, Amjad H. Talukder, Arjun S. Katailiha, and Gregory Lizee. 2021. "Evolution of CD8+ T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer" Cells 10, no. 9: 2379. https://doi.org/10.3390/cells10092379
APA StyleSun, Y., Li, F., Sonnemann, H., Jackson, K. R., Talukder, A. H., Katailiha, A. S., & Lizee, G. (2021). Evolution of CD8+ T Cell Receptor (TCR) Engineered Therapies for the Treatment of Cancer. Cells, 10(9), 2379. https://doi.org/10.3390/cells10092379