
All Roads Lead to Rome: Different Molecular Players Converge to Common Toxic Pathways in Neurodegeneration



Abstract
:1. Introduction
1.1. Mitochondrial Dysfunction and Oxidative Stress
1.2. Stress Granules
1.3. Disruption of Nucleocytoplasmic Transport (NCT)
1.4. Prion-Like Propagation
1.5. Non-Cell-Autonomous Toxicity
1.6. Disruption of Axonal Transport
1.7. Protein Misfolding
2. Proteostasis
2.1. Chaperones
2.2. ER-Associated Degradation (ERAD)
2.3. Ubiquitin-Proteasome System (UPS) and Autophagy
2.4. Stress Granule Formation
3. Alzheimer’s Disease
3.1. Etiology of AD
3.2. Pathophysiology of AD
3.3. Aβ Regulation in AD
3.4. Tau Regulation in AD
3.5. Mitochondrial Dysfunction in AD
3.6. Lysosomal Dysfunction in AD
3.7. Prion-Like Propagation in AD
3.8. Chaperones in AD
3.9. Current Treatments of AD
3.10. Clinical Trials in AD
4. Parkinson’s Disease
4.1. Etiology of PD
4.2. Pathophysiology of PD
4.3. α-Synuclein in PD
4.3.1. α-Syn PTMs
4.3.2. α-Syn Inclusions
4.3.3. α-Syn and Dopamine Toxicity
4.4. UCHL1 and Parkin in PD
4.5. Mitochondrial Dysfunction and Oxidative Stress in PD
4.6. Lysosomal Dysfunction in PD
4.7. Chaperones in PD
4.8. Current Treatments of PD
4.9. Clinical Trials in PD
5. Huntington’s Disease
5.1. Etiology and Clinical Manifestations of HD
5.2. Pathophysiology of HD
5.2.1. ER Stress in HD
5.2.2. Axonal Transport Disruption in HD
5.2.3. Mitochondrial Dysfunction in HD
5.3. Chaperones in HD
5.4. Current Treatments of HD
5.5. Clinical Trials in HD
6. Amyotrophic Lateral Sclerosis
6.1. Etiology of ALS
6.2. Cu/Zn Superoxide Dismutase (SOD1) in ALS
6.2.1. SOD1 and Mitochondrial Dysfunction
6.2.2. SOD1 and ER Stress
6.2.3. SOD1 Clearance
6.2.4. Prion-Like Propagation of SOD1
6.3. TAR DNA-Binding Protein 43 (TDP-43) in ALS
6.3.1. TDP-43 Mislocalization
6.3.2. TDP-43 Self-Regulation
6.3.3. TDP-43 and SG
6.3.4. TDP-43 C-Terminal Fragments and PTMs
6.3.5. TDP-43 Mitochondrial Dysfunction and Clearance
6.4. C9orf72 in ALS
6.5. Fused in Sarcoma (FUS)
6.6. Other Proteins
6.6.1. Ubiquilin2 in ALS
6.6.2. TANK Binding Kinase-1 (TBK1) in ALS
6.6.3. Profilin1 in ALS
6.7. Non-Cell Autonomous in ALS
6.8. Chaperones in ALS
6.9. Current Treatments of ALS
6.10. Clinical Trials in ALS
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heemels, M.T. Neurodegenerative diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Canter, R.G.; Penney, J.; Tsai, L.H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015; p. xxxviii. 905p. [Google Scholar]
- McQuillen, P.S.; Ferriero, D.M. Selective vulnerability in the developing central nervous system. Pediatr. Neurol. 2004, 30, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.H.; Greenamyre, J.T. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magri, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell. Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hamad, S.; Kahn, J.; Leyton-Jaimes, M.F.; Rosenblatt, J.; Israelson, A. Misfolded SOD1 Accumulation and Mitochondrial Association Contribute to the Selective Vulnerability of Motor Neurons in Familial ALS: Correlation to Human Disease. ACS Chem. Neurosci. 2017, 8, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef]
- Toombs, J.A.; Petri, M.; Paul, K.R.; Kan, G.Y.; Ben-Hur, A.; Ross, E.D. De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. USA 2012, 109, 6519–6524. [Google Scholar] [CrossRef] [Green Version]
- Babinchak, W.M.; Surewicz, W.K. Liquid-Liquid Phase Separation and Its Mechanistic Role in Pathological Protein Aggregation. J. Mol. Biol. 2020, 432, 1910–1925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958–971.e17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.; Chen, Y.H.; Duong, D.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steyaert, J.; Scheveneels, W.; Vanneste, J.; Van Damme, P.; Robberecht, W.; Callaerts, P.; Bogaert, E.; Bosch, L.V.D. FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum. Mol. Genet. 2018, 27, 4103–4116. [Google Scholar] [CrossRef] [Green Version]
- Giampetruzzi, A.; Danielson, E.W.; Gumina, V.; Jeon, M.; Boopathy, S.; Brown, R.H.; Ratti, A.; Landers, J.E.; Fallini, C. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 2019, 10, 3827. [Google Scholar] [CrossRef] [Green Version]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef] [Green Version]
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107.e6. [Google Scholar] [CrossRef] [Green Version]
- Veldman, M.B.; Yang, X.W. Huntington’s Disease: Nuclear Gatekeepers Under Attack. Neuron 2017, 94, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Jaunmuktane, Z.; Brandner, S. Invited Review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2020, 46, 522–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.L.; Lee, V.M.Y. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.-R.; Patani, R.; Sidle, K. In Vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, B.; Diamond, M.I. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 2010, 11, 155–159. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazawa, I.; Giasson, B.I.; Sasaki, R.; Zhang, B.; Joyce, S.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M.-Y. Mouse model of multiple system atrophy α-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 2005, 45, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.-Y.; Fang, Z.-H.; Yu, Z.-X.; Wang, C.-E.; Li, S.-H.; Li, X.-J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef]
- Rõlova, T.; Lehtonen, Š.; Goldsteins, G.; Kettunen, P.; Koistinaho, J. Metabolic and immune dysfunction of glia in neurodegenerative disorders: Focus on iPSC models. Stem Cells 2021, 39, 256–265. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Hinckelmann, M.-V.; Zala, D.; Saudou, F. Releasing the brake: Restoring fast axonal transport in neurodegenerative disorders. Trends Cell Biol. 2013, 23, 634–643. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.; Moughamian, A.J.; Holzbaur, E.L. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and Transport Deficits Early in the Pathogenesis of Alzheimer’s Disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Galvin, J.E.; Uryu, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [Green Version]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Lewis, A.J.; Genoud, C.; Pont, M.; Van De Berg, W.D.; Frank, S.; Stahlberg, H.; Shahmoradian, S.H.; Al-Amoudi, A. Imaging of post-mortem human brain tissue using electron and X-ray microscopy. Curr. Opin. Struct. Biol. 2019, 58, 138–148. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Smith, H.L.; Li, W.; Cheetham, M.E. Molecular chaperones and neuronal proteostasis. In Seminars in Cell and Developmental Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 40, pp. 142–152. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Shvil, N.; Banerjee, V.; Zoltsman, G.; Shani, T.; Kahn, J.; Abu-Hamad, S.; Papo, N.; Engel, S.; Bernhagen, J.; Israelson, A. MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: Implications for familial ALS. Cell Death Dis. 2018, 9, 107. [Google Scholar] [CrossRef]
- Knowles, T.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtis, N.; Tavernarakis, N. Small heat shock proteins and neurodegeneration: Recent developments. Biomol. Concepts 2018, 9, 94–102. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Shahheydari, H.; Ragagnin, A.; Walker, A.; Toth, R.P.; Vidal, M.; Jagaraj, C.J.; Perri, E.R.; Konopka, A.; Sultana, J.M.; Atkin, J.D. Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci. 2017, 10, 119. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Zattas, D.; Hochstrasser, M. Ubiquitin-dependent protein degradation at the yeast endoplasmic reticulum and nuclear envelope. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Galves, M.; Rathi, R.; Prag, G.; Ashkenazi, A. Ubiquitin Signaling and Degradation of Aggregate-Prone Proteins. Trends Biochem. Sci. 2019, 44, 872–884. [Google Scholar] [CrossRef]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef]
- Chitiprolu, M.; Jagow, C.; Tremblay, V.; Bondy-Chorney, E.; Paris, G.; Savard, A.; Palidwor, G.; Barry, F.A.; Zinman, L.; Keith, J.; et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 2018, 9, 2794. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Jin, X.; Liu, B. The involvement of stress granules in aging and aging-associated diseases. Aging Cell 2020, 19, e13136. [Google Scholar] [CrossRef] [Green Version]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Alzheimers Dementia. 2020 Alzheimer’s Disease Facts and Figures. 2020. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/full/10.1002/alz.12068 (accessed on 4 September 2021).
- Jaroudi, W.; Garami, J.; Garrido, S.; Hornberger, M.; Keri, S.; Moustafa, A.A. Factors underlying cognitive decline in old age and Alzheimer’s disease: The role of the hippocampus. Rev. Neurosci. 2017, 28, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454. [Google Scholar]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, M.S. In search of pathogenic amyloid β-peptide in familial Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2019, 168, 71–78. [Google Scholar] [CrossRef]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Mecocci, P.; Baroni, M.; Senin, U.; Boccardi, V. Brain Aging and Late-Onset Alzheimer’s Disease: A Matter of Increased Amyloid or Reduced Energy? J. Alzheimer’s Dis. 2018, 64, S397–S404. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell Rep. 2019, 29, 3592–3604.e5. [Google Scholar] [CrossRef] [Green Version]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.; Scheltens, P.; Verhey, F.R.J.; Visser, P.J.; Aalten, P.; Aarsland, D.; Alcolea, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Arbor, S.C.; Lafontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets—Protein processing, lipid rafts, and amyloid-beta pores. Yale J. Boil. Med. 2016, 89, 5–21. [Google Scholar]
- Zhang, C.; Browne, A.; Kim, D.; Tanzi, R. Familial Alzheimers Disease Mutations in Presenilin 1 Do Not Alter Levels of the Secreted Amyloid-β Protein Precursor Generated by β-Secretase Cleavage. Curr. Alzheimer Res. 2010, 7, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Strategies for disease modification in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 677–685. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.; Wang, Y.-J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Mandler, M.; Walker, L.; Santic, R.; Hanson, P.; Upadhaya, A.R.; Colloby, S.J.; Morris, C.M.; Thal, D.R.; Thomas, A.J.; Schneeberger, A.; et al. Pyroglutamylated amyloid-β is associated with hyperphosphorylated tau and severity of Alzheimer’s disease. Acta Neuropathol. 2014, 128, 67–79. [Google Scholar] [CrossRef]
- Leissring, M.A. The AβCs of Aβ-cleaving Proteases. J. Biol. Chem. 2008, 283, 29645–29649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Takaki, Y.; Iwata, N.; Trojanowski, J.; Saido, T.C. Alzheimer’s Disease, Neuropeptides, Neuropeptidase, and Amyloid- Peptide Metabolism. Sci. Aging Knowl. Environ. 2003, 2003, PE1. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Muñoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-β impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer’s disease. Glia 2021, 69, 997–1011. [Google Scholar] [CrossRef]
- Xin, S.-H.; Tan, L.; Cao, X.; Yu, J.-T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S. Microglia and Inflammation in Alzheimers Disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Weller, R.O.; Massey, A.; Newman, T.A.; Hutchings, M.; Kuo, Y.-M.; Roher, A.E. Cerebral Amyloid Angiopathy: Amyloid β Accumulates in Putative Interstitial Fluid Drainage Pathways in Alzheimer’s Disease. Am. J. Pathol. 1998, 153, 725–733. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139 (Suppl. 2), 237–252. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Takeuchi, H.; Tanaka, F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front. Neurol. 2019, 10, 980. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Armstrong, R.A. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009, 47, 289–299. [Google Scholar]
- Wang, J.-Z.; Xia, Y.-Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S123–S139. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Z.; Grundke-Iqbal, I.; Iqbal, K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007, 25, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shi, J.; Chu, D.; Hu, W.; Guan, Z.; Gong, C.-X.; Iqbal, K.; Liu, F. Relevance of Phosphorylation and Truncation of Tau to the Etiopathogenesis of Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.-X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef] [PubMed]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Flach, K.; Ramminger, E.; Hilbrich, I.; Arsalan-Werner, A.; Albrecht, F.; Herrmann, L.; Goedert, M.; Arendt, T.; Holzer, M. Axotrophin/MARCH7 acts as an E3 ubiquitin ligase and ubiquitinates tau protein In Vitro impairing microtubule binding. Biochim. Biophys. Acta 2014, 1842, 1527–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrucelli, L.; Dickson, D.W.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2020, 11, 595532. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Cao, J.; Zhong, M.B.; Toro, C.A.; Zhang, L.; Cai, D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci. Lett. 2019, 703, 68–78. [Google Scholar] [CrossRef]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, J.M.; Seward, M.E.; Bloom, G.S. Alzheimer disease: A tale of two prions. Prion 2013, 7, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, F.A.; Rüdiger, S.G.D. The Mitochondrial Hsp90 TRAP1 and Alzheimer’s Disease. Front. Mol. Biosci. 2021, 8, 697913. [Google Scholar] [CrossRef] [PubMed]
- Rowles, J.; Keane, K.; Heck, T.G.; Cruzat, V.; Verdile, G.; Newsholme, P. Are Heat Shock Proteins an Important Link between Type 2 Diabetes and Alzheimer Disease? Int. J. Mol. Sci. 2020, 21, 8204. [Google Scholar] [CrossRef] [PubMed]
- Arimon, M.; Grimminger, V.; Sanz, F.; Lashuel, H.A. Hsp104 Targets Multiple Intermediates on the Amyloid Pathway and Suppresses the Seeding Capacity of Aβ Fibrils and Protofibrils. J. Mol. Biol. 2008, 384, 1157–1173. [Google Scholar] [CrossRef] [Green Version]
- Zatsepina, O.; Evgen’Ev, M.; Garbuz, D. Role of a Heat Shock Transcription Factor and the Major Heat Shock Protein Hsp70 in Memory Formation and Neuroprotection. Cells 2021, 10, 1638. [Google Scholar] [CrossRef]
- Leak, R.K. Heat shock proteins in neurodegenerative disorders and aging. J. Cell Commun. Signal. 2014, 8, 293–310. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-analysis. J. Alzheimer’s Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Hoskin, J.L.; Sabbagh, M.N.; Al-Hasan, Y.; Decourt, B. Tau immunotherapies for Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kumar, R.; Pavlov, P.F.; Winblad, B. Small molecule therapeutics for tauopathy in Alzheimer’s disease: Walking on the path of most resistance. Eur. J. Med. Chem. 2021, 209, 112915. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. An Essay on the Shaking Palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236, Discussion 222. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Dick, F.D. Parkinson’s disease and pesticide exposures. Br. Med Bull. 2006, 79-80, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- Choi, B.-K.; Choi, M.-G.; Kim, J.-Y.; Yang, Y.; Lai, Y.; Kweon, D.-H.; Lee, N.K.; Shin, Y.-K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef] [Green Version]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of Toxic alpha-Synuclein Oligomer within Endoplasmic Reticulum Occurs in alpha-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef]
- Takahashi, H.; Wakabayashi, K. The cellular pathology of Parkinson’s disease. Neuropathology 2001, 21, 315–322. [Google Scholar] [CrossRef]
- Roberts, R.; Wade-Martins, R.; Alegre-Abarrategui, J. Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 2015, 138, 1642–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. [alpha]-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Eliezer, D. Biophysics of Parkinsons Disease: Structure and Aggregation of α-Synuclein. Curr. Protein Pept. Sci. 2009, 10, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Bodner, C.R.; Dobson, C.M.; Bax, A. Multiple Tight Phospholipid-Binding Modes of α-Synuclein Revealed by Solution NMR Spectroscopy. J. Mol. Biol. 2009, 390, 775–790. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Burré, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Südhof, T.C.; Brunger, A.T. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2013, 2, e00592. [Google Scholar] [CrossRef] [PubMed]
- George, J.M. The synucleins. Genome Biol. 2002, 3, REVIEWS3002. [Google Scholar] [CrossRef] [PubMed]
- Soper, J.H.; Roy, S.; Stieber, A.; Lee, E.; Wilson, R.B.; Trojanowski, J.Q.; Burd, C.G.; Lee, V.M.-Y. α-Synuclein–induced Aggregation of Cytoplasmic Vesicles in Saccharomyces cerevisiae. Mol. Biol. Cell 2008, 19, 1093–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Guerrero, E.; Vasudevaraju, P.; Hegde, M.; Britton, G.; Rao, K.S. Recent Advances in α-Synuclein Functions, Advanced Glycation, and Toxicity: Implications for Parkinson’s Disease. Mol. Neurobiol. 2013, 47, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; Von Saucken, V.; Sanderson, J.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [Green Version]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-Robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional Alterations to the Nigrostriatal System in Mice Lacking All Three Members of the Synuclein Family. J. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; De Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 Is Enhanced in Synucleinopathies, Inhibits -Synuclein Oligomerization, and Influences Synuclein-Membrane Interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Neumann, M.; Kahle, P.J.; Giasson, B.I.; Ozmen, L.; Borroni, E.; Spooren, W.; Muller, V.; Odoy, S.; Fujiwara, H.; Hasegawa, M.; et al. Misfolded proteinase K–resistant hyperphosphorylated α-synuclein in aged transgenic mice with locomotor deterioration and in human α-synucleinopathies. J. Clin. Investig. 2002, 110, 1429–1439. [Google Scholar] [CrossRef]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive Phosphorylation of the Parkinson’s Disease Associated α-Synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronin, A.N.; Morris, A.J.; Surguchov, A.; Benovic, J.L. Synucleins Are a Novel Class of Substrates for G Protein-coupled Receptor Kinases. J. Biol. Chem. 2000, 275, 26515–26522. [Google Scholar] [CrossRef] [Green Version]
- Inglis, K.J.; Chereau, D.; Brigham, E.F.; Chiou, S.-S.; Schöbel, S.; Frigon, N.L.; Yu, M.; Caccavello, R.J.; Nelson, S.; Motter, R.; et al. Polo-like Kinase 2 (PLK2) Phosphorylates α-Synuclein at Serine 129 in Central Nervous System. J. Biol. Chem. 2009, 284, 2598–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.W.; Margolis, R.L.; Li, X.; Troncoso, J.C.; Lee, M.; Dawson, V.; Dawson, T.M.; Iwatsubo, T.; Ross, C.A. Alpha-Synuclein Phosphorylation Enhances Eosinophilic Cytoplasmic Inclusion Formation in SH-SY5Y Cells. J. Neurosci. 2005, 25, 5544–5552. [Google Scholar] [CrossRef]
- Oueslati, A.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Polo-like kinase 2 regulates selective autophagic alpha-synuclein clearance and suppresses its toxicity In Vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E3945–E3954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Broe, M.; Huang, Y.; Anderson, J.P.; Gai, W.-P.; Milward, E.A.; Porritt, M.; Howells, D.; Hughes, A.J.; Wang, X.; et al. Changes in the solubility and phosphorylation of α-synuclein over the course of Parkinson’s disease. Acta Neuropathol. 2011, 121, 695–704. [Google Scholar] [CrossRef]
- Wu, J.; Lou, H.; Alerte, T.; Stachowski, E.; Chen, J.; Singleton, A.; Hamilton, R.; Perez, R. Lewy-like aggregation of α-synuclein reduces protein phosphatase 2A activity In Vitro and In Vivo. Neuroscience 2012, 207, 288–297. [Google Scholar] [CrossRef] [Green Version]
- Karpowicz, R.J.; Trojanowski, J.Q.; Lee, V.M.Y. Transmission of α-synuclein seeds in neurodegenerative disease: Recent developments. Lab. Investig. 2019, 99, 971–981. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Potential Pathways of Abnormal Tau and α-Synuclein Dissemination in Sporadic Alzheimer’s and Parkinson’s Diseases. Cold Spring Harb. Perspect. Biol. 2016, 8, a023630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-J.; Desplats, P.; Sigurdson, C.; Tsigelny, I.; Masliah, E. Cell-to-cell transmission of non-prion protein aggregates. Nat. Rev. Neurol. 2010, 6, 702–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pall, H.; Blake, D.; Gutteridge, J.; Williams, A.; Lunec, J.; Hall, M.; Taylor, A. Raised Cerebrospinal-Fluid Copper Concentration in Parkinson’s Disease. Lancet 1987, 330, 238–241. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.-D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B.H. Transition Metals, Ferritin, Glutathione, and Ascorbic Acid in Parkinsonian Brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, C.; Wang, S.; Yang, D.; Zhang, Y.; Xu, L.; Ma, L.; Zheng, J.; Petersen, R.B.; Zheng, L.; et al. Copper and iron ions accelerate the prion-like propagation of α-synuclein: A vicious cycle in Parkinson’s disease. Int. J. Biol. Macromol. 2020, 163, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643. [Google Scholar] [PubMed]
- Maker, H.S.; Weiss, C.; Silides, D.J.; Cohen, G. Coupling of Dopamine Oxidation (Monoamine Oxidase Activity) to Glutathione Oxidation Via the Generation of Hydrogen Peroxide in Rat Brain Homogenates. J. Neurochem. 1981, 36, 589–593. [Google Scholar] [CrossRef]
- Ghassaban, K.; He, N.; Sethi, S.K.; Huang, P.; Chen, S.; Yan, F.; Haacke, E.M. Regional High Iron in the Substantia Nigra Differentiates Parkinson’s Disease Patients From Healthy Controls. Front. Aging Neurosci. 2019, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.-L.; Zhang, C.-W.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Parkinson’s Dis. 2018, 2018, 9163040. [Google Scholar] [CrossRef] [Green Version]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T.L., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lansbury, J.P.T. Vesicle Permeabilization by Protofibrillar α-Synuclein Is Sensitive to Parkinson’s Disease-Linked Mutations and Occurs by a Pore-like Mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- Volles, M.J.; Lee, S.-J.; Rochet, J.-C.; Shtilerman, M.D.; Ding, T.T.; Kessler, A.J.C.; Lansbury, J.P.T. Vesicle Permeabilization by Protofibrillar α-Synuclein: Implications for the Pathogenesis and Treatment of Parkinson’s Disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar] [CrossRef]
- Junn, E.; Mouradian, M. Human α-Synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 2002, 320, 146–150. [Google Scholar] [CrossRef]
- Liu, Y.; Fallon, L.; Lashuel, H.A.; Liu, Z.; Lansbury, P.T., Jr. The UCH-L1 Gene Encodes Two Opposing Enzymatic Activities that Affect α-Synuclein Degradation and Parkinson’s Disease Susceptibility. Cell 2002, 111, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Saigoh, K.; Wang, Y.-L.; Suh, J.-G.; Yamanishi, T.; Sakai, Y.; Kiyosawa, H.; Harada, T.; Ichihara, N.; Wakana, S.; Kikuchi, T.; et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999, 23, 47–51. [Google Scholar] [CrossRef]
- Setsuie, R.; Wang, Y.-L.; Mochizuki, H.; Osaka, H.; Hayakawa, H.; Ichihara, N.; Li, H.; Furuta, A.; Sano, Y.; Sun, Y.-J.; et al. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem. Int. 2007, 50, 119–129. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, J.; Chung, K.K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, T.M.; Dawson, V. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S32–S39. [Google Scholar] [CrossRef]
- Ganguly, U.; Banerjee, A.; Chakrabarti, S.S.; Kaur, U.; Sen, O.; Cappai, R.; Chakrabarti, S. Interaction of α-synuclein and Parkin in iron toxicity on SH-SY5Y cells: Implications in the pathogenesis of Parkinson’s disease. Biochem. J. 2020, 477, 1109–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Tan, J.M.M.; Ho, M.W.L.; Zaiden, N.; Wong, S.H.; Chew, C.L.C.; Eng, P.W.; Lim, T.M.; Dawson, T.M.; Lim, K.L. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson’s disease-linked point mutations. J. Neurochem. 2005, 93, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.; Henn, I.H.; Kay-Jackson, P.C.; Heller, U.; Tatzelt, J. Inactivation of Parkin by Oxidative Stress and C-terminal Truncations: A protective role of molecular chaperones. J. Biol. Chem. 2003, 278, 47199–47208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.-L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, T.M.; Dawson, V.L. Parkin Plays a Role in Sporadic Parkinson’s Disease. Neurodegener. Dis. 2014, 13, 69–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Jang, S.; Park, J.; Bang, S.; Choi, S.; Kwon, K.-Y.; Zhuang, X.; Kim, E.; Chung, J. Characterization of PINK1 (PTEN-induced Putative Kinase 1) Mutations Associated with Parkinson Disease in Mammalian Cells and Drosophila. J. Biol. Chem. 2013, 288, 5660–5672. [Google Scholar] [CrossRef] [Green Version]
- Van Der Vlag, M.; Havekes, R.; Heckman, P.R.A. The contribution of Parkin, PINK1 and DJ-1 genes to selective neuronal degeneration in Parkinson’s disease. Eur. J. Neurosci. 2020, 52, 3256–3268. [Google Scholar] [CrossRef] [Green Version]
- Lopert, P.; Patel, M. Brain mitochondria from DJ-1 knockout mice show increased respiration-dependent hydrogen peroxide consumption. Redox Biol. 2014, 2, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, M.K.; Cookson, M.R. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trempe, J.-F.; Fon, E.A. Structure and Function of Parkin, PINK1, and DJ-1, the Three Musketeers of Neuroprotection. Front. Neurol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Da Cruz, A.B.; Burbulla, L.F.; Lawrence, E.S.; Schuele, B.; Krainc, D.; Palmer, T.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.; Schapira, A.H. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol. Dis. 2016, 90, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Cleeter, M.W.; Chau, K.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.; Hardy, J.; Cooper, J.M.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Auluck, P.K.; Chan, H.Y.E.; Trojanowski, J.Q.; Lee, V.M.-Y.; Bonini, N.M. Chaperone Suppression of alpha-Synuclein Toxicity in a Drosophila Model for Parkinson’s Disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- McLean, P.; Kawamata, H.; Shariff, S.; Hewett, J.W.; Sharma, N.; Ueda, K.; Breakefield, X.O.; Hyman, B.T. TorsinA and heat shock proteins act as molecular chaperones: Suppression of α-synuclein aggregation. J. Neurochem. 2002, 83, 846–854. [Google Scholar] [CrossRef]
- Danzer, K.M.; Ruf, W.P.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.T.; McLean, P.J. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011, 25, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putcha, P.; Danzer, K.M.; Kranich, L.R.; Scott, A.; Silinski, M.; Mabbett, S.; Hicks, C.D.; Veal, J.M.; Steed, P.M.; Hyman, B.T.; et al. Brain-Permeable Small-Molecule Inhibitors of Hsp90 Prevent α-Synuclein Oligomer Formation and Rescue α-Synuclein-Induced Toxicity. J. Pharmacol. Exp. Ther. 2010, 332, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Raveendran, A.; Nagotu, S. Chaperones and Proteostasis: Role in Parkinson’s Disease. Diseases 2020, 8, 24. [Google Scholar] [CrossRef]
- Bose, S.; Cho, J. Targeting chaperones, heat shock factor-1, and unfolded protein response: Promising therapeutic approaches for neurodegenerative disorders. Ageing Res. Rev. 2017, 35, 155–175. [Google Scholar] [CrossRef]
- Faulkner, M.A. Safety overview of FDA-approved medications for the treatment of the motor symptoms of Parkinson’s disease. Expert Opin. Drug Saf. 2014, 13, 1055–1069. [Google Scholar] [CrossRef]
- Aradi, S.D.; Hauser, R.A. Medical Management and Prevention of Motor Complications in Parkinson’s Disease. Neurotherapeutics 2020, 17, 1339–1365. [Google Scholar] [CrossRef] [PubMed]
- Dezsi, L.; Vecsei, L. Monoamine Oxidase B Inhibitors in Parkinson’s Disease. CNS Neurol. Disord.-Drug Targets 2017, 16, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Rascol, O. Efficacy and safety of amantadine for the treatment of l-DOPA-induced dyskinesia. J. Neural Transm. 2018, 125, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Pirtosek, Z. ’Bad guys’ among the antiparkinsonian drugs. Psychiatr. Danub. 2009, 21, 114–118. [Google Scholar] [PubMed]
- Stoker, T.B.; Barker, R.A. Recent developments in the treatment of Parkinson’s Disease. F1000Research 2020, 9, 862. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.A.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef]
- Mortiboys, H.; Aasly, J.; Bandmann, O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease. Brain 2013, 136, 3038–3050. [Google Scholar] [CrossRef] [Green Version]
- Mortiboys, H.; Furmston, R.; Bronstad, G.; Aasly, J.; Elliott, C.; Bandmann, O. UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2G2019S carriers and In Vivo. Neurology 2015, 85, 846–852. [Google Scholar] [CrossRef] [Green Version]
- Elsworth, J.D. Parkinson’s disease treatment: Past, present, and future. J. Neural Transm. 2020, 127, 785–791. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Chmielarz, P.; Saarma, M. Neurotrophic factors for disease-modifying treatments of Parkinson’s disease: Gaps between basic science and clinical studies. Pharmacol. Rep. 2020, 72, 1195–1217. [Google Scholar] [CrossRef]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.; Smeeth, L. The Prevalence of Huntington’s Disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Ha, A.D.; Fung, V.S. Huntington’s disease. Curr. Opin. Neurol. 2012, 25, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J. Juvenile Huntington’s Disease: Diagnostic and Treatment Considerations for the Psychiatrist. Curr. Psychiatry Rep. 2017, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Liu, J.-P.; Zeitlin, S.O. Is Huntingtin Dispensable in the Adult Brain? J. Huntington’s Dis. 2017, 6, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.; Finkbeiner, S. The Ubiquitin-Proteasome Pathway in Huntington’s Disease. Sci. World J. 2008, 8, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering Mutant Huntingtin Levels and Toxicity: Autophagy-Endolysosome Pathways in Huntington’s Disease. J. Mol. Biol. 2020, 432, 2673–2691. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Chillon-Marinas, C.; Goginashvili, A.; Atwal, R.S.; Artates, J.W.; Tabet, R.; Wheeler, V.C.; Bang, A.G.; Cleveland, D.W.; Lagier-Tourenne, C. Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron 2017, 94, 48–57.e4. [Google Scholar] [CrossRef] [Green Version]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Farshbaf, M.J.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Polyglutamine Disord. 2018, 1049, 59–83. [Google Scholar] [CrossRef]
- Koyuncu, S.; Fatima, A.; Gutierrez-Garcia, R.; Vilchez, D. Proteostasis of Huntingtin in Health and Disease. Int. J. Mol. Sci. 2017, 18, 1568. [Google Scholar] [CrossRef] [Green Version]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [Green Version]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease—Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef]
- Novak, M.J.; Tabrizi, S.J. Huntington’s Disease: Clinical Presentation and Treatment. Int. Rev. Neurobiol. 2011, 98, 297–323. [Google Scholar] [CrossRef] [PubMed]
- Bashir, H. Emerging therapies in Huntington’s disease. Expert Rev. Neurother. 2019, 19, 983–995. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: April 2020. J. Huntington’s Dis. 2020, 9, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.; Eisen, A.; Hardiman, O.; Burrell, J.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Marin, B.; Boumédiene, F.; Logroscino, G.; Couratier, P.; Babron, M.-C.; Leutenegger, A.-L.; Copetti, M.; Preux, P.-M.; Beghi, E. Variation in worldwide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Henden, L.; Twine, N.A.; Szul, P.; McCann, E.P.; Nicholson, G.A.; Rowe, D.B.; Kiernan, M.C.; Bauer, D.C.; Blair, I.; Williams, K.L. Identity by descent analysis identifies founder events and links SOD1 familial and sporadic ALS cases. NPJ Genom. Med. 2020, 5, 32. [Google Scholar] [CrossRef]
- Marogianni, C.; Rikos, D.; Provatas, A.; Dadouli, K.; Ntellas, P.; Tsitsi, P.; Patrinos, G.; Dardiotis, E.; Hadjigeorgiou, G.; Xiromerisiou, G. The role of C9orf72 in neurodegenerative disorders: A systematic review, an updated meta-analysis, and the creation of an online database. Neurobiol. Aging 2019, 84, 238.e25–238.e34. [Google Scholar] [CrossRef]
- Ferrari, R.; Kapogiannis, D.; Huey, E.D.; Momeni, P. FTD and ALS: A Tale of Two Diseases. Curr. Alzheimer Res. 2011, 8, 273–294. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.D.; Ullrich, M.S.; Calvo, J.S.; Behnia, F.; Meloni, G.; Winkler, D.D. Mutations in Superoxide Dismutase 1 (Sod1) Linked to Familial Amyotrophic Lateral Sclerosis Can Disrupt High-Affinity Zinc-Binding Promoted by the Copper Chaperone for Sod1 (Ccs). Molecules 2020, 25, 1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlary, L.; Aquilina, J.A.; Yerbury, J.J. Susceptibility of Mutant SOD1 to Form a Destabilized Monomer Predicts Cellular Aggregation and Toxicity but Not In Vitro Aggregation Propensity. Front. Neurosci. 2016, 10, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, C.; Phelan, J.P.; Hatzipetros, T.; Kidd, J.D.; Tassinari, V.R.; Levine, B.; Wang, M.Z.; Moreno, A.; Thompson, K.; Maier, M.; et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci. Rep. 2019, 9, 6724. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.; Chattopadhyay, M.; Whitelegge, J.; Valentine, J.S. SOD1 aggregation and ALS: Role of metallation states and disulfide status. Curr. Top. Med. Chem. 2012, 12, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Medinas, D.B.; Rozas, P.; Traub, F.M.; Woehlbier, U.; Brown, R.H.; Bosco, D.; Hetz, C. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 8209–8214. [Google Scholar] [CrossRef] [Green Version]
- Gal, J.; Kuang, L.; Barnett, K.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.; Hayward, L.; Kasarskis, E.J.; Zhu, H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016, 132, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Shaw, P.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Israelson, A.; Ditsworth, D.; Sun, S.; Song, S.; Liang, J.; Hruska-Plochan, M.; McAlonis-Downes, M.; Abu-Hamad, S.; Zoltsman, G.; Shani, T.; et al. Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron 2015, 86, 218–232. [Google Scholar] [CrossRef] [Green Version]
- Tak, Y.J.; Park, J.-H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef]
- Kanekura, K.; Suzuki, H.; Aiso, S.; Matsuoka, M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol. Neurobiol. 2009, 39, 81–89. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urushitani, M.; Kurisu, J.; Tsukita, K.; Takahashi, R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 83, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef] [Green Version]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [Green Version]
- Bidhendi, E.E.; Bergh, J.; Zetterström, P.; Forsberg, K.; Pakkenberg, B.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 136, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Mompeán, M.; Buratti, E.; Guarnaccia, C.; Brito, R.M.; Chakrabartty, A.; Baralle, F.E.; Laurents, D.V. Structural characterization of the minimal segment of TDP-43 competent for aggregation. Arch. Biochem. Biophys. 2014, 545, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Lee, V.M.; Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Ayala, Y.M.; Pantano, S.; D’Ambrogio, A.; Buratti, E.; Brindisi, A.; Marchetti, C.; Romano, M.; Baralle, F.E. Human, Drosophila, and C. elegans TDP43: Nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 2005, 348, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv. Genet. 2015, 91, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef] [Green Version]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Groote, C.C.-D.; Van Broeckhoven, C.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ray, P.; Rao, E.J.; Shi, C.; Guo, W.; Chen, X.; Woodruff, E.A., III; Fushimi, K.; Wu, J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 3169–3174. [Google Scholar] [CrossRef] [Green Version]
- Ash, P.E.; Zhang, Y.-J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef] [PubMed]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Velde, C.V. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nihei, Y.; Ito, D.; Suzuki, N. Roles of Ataxin-2 in Pathological Cascades Mediated by TAR DNA-binding Protein 43 (TDP-43) and Fused in Sarcoma (FUS). J. Biol. Chem. 2012, 287, 41310–41323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, L.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y.; et al. Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. Neuron 2019, 103, 802–819.e11. [Google Scholar] [CrossRef]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for Stress Granule Recruitment of Fused in Sarcoma (FUS) and TAR DNA-binding Protein of 43 kDa (TDP-43). J. Biol. Chem. 2012, 287, 23079–23094. [Google Scholar] [CrossRef] [Green Version]
- Babinchak, W.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.-K.; Surewicz, W.K. The role of liquid–liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inukai, Y.; Nonaka, T.; Arai, T.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.E.; Akiyama, H.; Hisanaga, S.-I.; et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008, 582, 2899–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M.-Y. Expression of TDP-43 C-terminal Fragments In Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef] [Green Version]
- Pesiridis, G.S.; Tripathy, K.; Tanik, S.; Trojanowski, J.Q.; Lee, V.M.-Y. A “Two-hit” Hypothesis for Inclusion Formation by Carboxyl-terminal Fragments of TDP-43 Protein Linked to RNA Depletion and Impaired Microtubule-dependent Transport. J. Biol. Chem. 2011, 286, 18845–18855. [Google Scholar] [CrossRef] [Green Version]
- Dayton, R.D.; Gitcho, M.A.; Orchard, E.A.; Wilson, J.D.; Wang, D.B.; Cain, C.D.; Johnson, J.A.; Zhang, Y.-J.; Petrucelli, L.; Mathis, J.M.; et al. Selective Forelimb Impairment in Rats Expressing a Pathological TDP-43 25 kDa C-terminal Fragment to Mimic Amyotrophic Lateral Sclerosis. Mol. Ther. 2013, 21, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Shaw, D.M.; Guarino, F.; Messina, A.; Walker, A.W.; Oddo, S. Reduced protein turnover mediates functional deficits in transgenic mice expressing the 25 kDa C-terminal fragment of TDP-43. Hum. Mol. Genet. 2015, 24, 4625–4635. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Majumder, S.; Oddo, S. Cognitive Decline Typical of Frontotemporal Lobar Degeneration in Transgenic Mice Expressing the 25-kDa C-Terminal Fragment of TDP-43. Am. J. Pathol. 2012, 180, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Berning, B.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotter, E.; Vance, C.; Nishimura, A.L.; Lee, Y.; Chen, H.-J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Sánchez, J.S.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, A.J.; Bäumer, D.; East, S.; Neal, J.; Morris, H.; Ansorge, O.; Blake, D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 2014, 35, 1779.e5–1779.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendron, T.F.; Petrucelli, L. Disease Mechanisms of C9ORF72 Repeat Expansions. Cold Spring Harb. Perspect. Med. 2018, 8, a024224. [Google Scholar] [CrossRef] [Green Version]
- Benussi, L.; Rossi, G.; Glionna, M.; Tonoli, E.; Piccoli, E.; Fostinelli, S.; Paterlini, A.; Flocco, R.; Albani, D.; Pantieri, R.; et al. C9ORF72 Hexanucleotide Repeat Number in Frontotemporal Lobar Degeneration: A Genotype-Phenotype Correlation Study. J. Alzheimer’s Dis. 2014, 38, 799–808. [Google Scholar] [CrossRef]
- Icardo, O.D.; García-Redondo, A.; Rojas-García, R.; Sanchez-Valle, R.; Noguera, A.; Gómez-Tortosa, E.; Pastor, P.; Hernández, I.; Esteban-Perez, J.; Suárez-Calvet, M.; et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2014, 23, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Suh, E.; Lee, E.B.; Neal, D.; Wood, E.M.; Toledo, J.; Rennert, L.; Irwin, D.; McMillan, C.; Krock, B.; Elman, L.B.; et al. Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol. 2015, 130, 363–372. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Highley, R.; Dickman, M.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Finch, N.A.; Wang, X.; Gendron, T.F.; Bieniek, K.; Heckman, M.G.; Vasilevich, A.; Murray, M.; Rousseau, L.; Weesner, R.; et al. In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol. 2017, 134, 255–269. [Google Scholar] [CrossRef]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobágyi, T.; Shaw, C. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef]
- Bigio, E.H.; Weintraub, S.; Rademakers, R.; Baker, M.; Ahmadian, S.S.; Rademaker, A.; Weitner, B.B.; Mao, Q.; Lee, K.-H.; Mishra, M.; et al. Frontotemporal lobar degeneration with TDP-43 proteinopathy and chromosome 9p repeat expansion in C9ORF72: Clinicopathologic correlation. Neuropathology 2013, 33, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellier, C.; Campanari, M.; Corbier, C.J.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef]
- Gijselinck, I.; Van Langenhove, T.; Van Der Zee, J.; Sleegers, K.; Philtjens, S.; Kleinberger, G.; Janssens, J.; Bettens, K.; Van Cauwenberghe, C.; Pereson, S.; et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65. [Google Scholar] [CrossRef]
- Zhu, Q.; Jiang, J.; Gendron, T.F.; McAlonis-Downes, M.; Jiang, L.; Taylor, A.; Garcia, S.D.; Dastidar, S.G.; Rodriguez, M.J.; King, P.; et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 2020, 23, 615–624. [Google Scholar] [CrossRef]
- Fratta, P.; Mizielinska, S.; Nicoll, A.J.; Zloh, M.; Fisher, E.; Parkinson, G.; Isaacs, A.M. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2012, 2, 1016. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.; Shaw, P.G.; Kim, M.-S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Almeida, S.; Moore, J.; Gendron, T.F.; Chalasani, U.; Lu, Y.; Du, X.; Nickerson, J.A.; Petrucelli, L.; Weng, Z.; et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 2015, 87, 1207–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, I.R.A.; Frick, P.; Grässer, F.A.; Gendron, T.F.; Petrucelli, L.; Cashman, N.R.; Edbauer, D.; Kremmer, E.; Prudlo, J.; Troost, D.; et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015, 130, 845–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-J.; Jansen-West, K.; Xu, Y.-F.; Gendron, T.F.; Bieniek, K.; Lin, W.-L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.M.; Rollinson, S.; Robinson, A.; Callister, J.B.; Thompson, J.C.; Snowden, J.S.; Gendron, T.; Petrucelli, L.; Masuda-Suzukake, M.; Hasegawa, M.; et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2013, 1, 68. [Google Scholar] [CrossRef]
- Chang, Y.-J.; Jeng, U.-S.; Chiang, Y.-L.; Hwang, I.-S.; Chen, Y.-R. The Glycine-Alanine Dipeptide Repeat from C9orf72 Hexanucleotide Expansions Forms Toxic Amyloids Possessing Cell-to-Cell Transmission Properties. J. Biol. Chem. 2016, 291, 4903–4911. [Google Scholar] [CrossRef] [Green Version]
- May, S.; Hornburg, D.; Schludi, M.H.; Arzberger, T.; Rentzsch, K.; Schwenk, B.M.; Grässer, F.A.; Mori, K.; Kremmer, E.; Banzhaf-Strathmann, J.; et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014, 128, 485–503. [Google Scholar] [CrossRef] [Green Version]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [Green Version]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., III; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [Green Version]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.-C.; Dastidar, S.G.; Tokunaga, S.; Ho, W.Y.; Lim, K.; Ilieva, H.; Parone, P.A.; Tyan, S.-H.; Tse, T.M.; Chang, J.-C.; et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. eLife 2019, 8, e40811. [Google Scholar] [CrossRef]
- Gao, F.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis–frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Julien, J.-P. Low expression of mutant Ubiquilin-2 exacerbates ALS/FTD features in a TDP-43 mouse model (P4.431). Neurology 2018, 90, P4.431. [Google Scholar]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491.e19. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 2019, 11, 2457–2476. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.P.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Smith, B.; Vance, C.; Scotter, E.; Troakes, C.; Wong, C.H.; Topp, S.; Maekawa, S.; King, A.; Mitchell, J.C.; Lund, K.; et al. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 2015, 36, 1602.e17–1602.e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Danielson, E.W.; Qiao, T.; Metterville, J.; Brown, R.H.; Landers, J.E.; Xu, Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc. Natl. Acad. Sci. USA 2016, 113, E6209–E6218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nonaka, T.; Suzuki, G.; Kametani, F.; Hasegawa, M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum. Mol. Genet. 2016, 25, 1420–1433. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Sharma, K.; Grisotti, G.; Roos, R.P. The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2009, 35, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Bogaert, E.; Dewil, M.; Hersmus, N.; Kiraly, D.; Scheveneels, W.; Bockx, I.; Braeken, D.; Verpoorten, N.; Verhoeven, K.; et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 14825–14830. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.-G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef]
- Chen, H.; Kankel, M.W.; Su, S.C.; Han, S.W.S.; Ofengeim, D. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018, 25, 648–662. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; DEL Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11, 592851. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Wang, L.; Li, D.; Zhao, C.; Li, J.; Liu, T. Exploring the Extended Biological Functions of the Human Copper Chaperone of Superoxide Dismutase 1. Protein J. 2019, 38, 463–471. [Google Scholar] [CrossRef]
- Wright, G.S. Molecular and pharmacological chaperones for SOD1. Biochem. Soc. Trans. 2020, 48, 1795–1806. [Google Scholar] [CrossRef]
- Farhan, S.M.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef]
- Rusmini, P.; Cristofani, R.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Messi, E.; Piccolella, M.; et al. The Role of the Heat Shock Protein B8 (HSPB8) in Motoneuron Diseases. Front. Mol. Neurosci. 2017, 10, 176. [Google Scholar] [CrossRef] [Green Version]
- Leyton-Jaimes, M.F.; Kahn, J.; Israelson, A. Macrophage migration inhibitory factor: A multifaceted cytokine implicated in multiple neurological diseases. Exp. Neurol. 2018, 301, 83–91. [Google Scholar] [CrossRef]
- Leyton-Jaimes, M.F.; Benaim, C.; Abu-Hamad, S.; Kahn, J.; Guetta, A.; Bucala, R.; Israelson, A. Endogenous macrophage migration inhibitory factor reduces the accumulation and toxicity of misfolded SOD1 in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2016, 113, 10198–10203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyton-Jaimes, M.F.; Kahn, J.; Israelson, A. AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc. Natl. Acad. Sci. USA 2019, 116, 14755–14760. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Ginsburg, S.; Trobiani, L.; Setini, A.; Favaloro, F.L.; Giorda, E.; Jacob, A.; Hauschner, H.; Levy, L.; Cestra, G.; DE Jaco, A.; et al. A Lipophilic 4-Phenylbutyric Acid Derivative That Prevents Aggregation and Retention of Misfolded Proteins. Chemistry 2020, 26, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Ginsburg, S.; Di Salvio, M.; Weitman, M.; Afri, M.; Ribeiro, S.; Ebbinghaus, S.; Cestra, G.; Gruzman, A. Chemical chaperones targeted to the endoplasmic reticulum (ER) and lysosome prevented neurodegeneration in a C9orf72 repeat expansion Drosophila amyotrophic lateral sclerosis (ALS) model. Pharmacol. Rep. 2021, 73, 536–550. [Google Scholar] [CrossRef]
- Bryson, H.M.; Fulton, B.; Benfield, P. Riluzole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs 1996, 52, 549–563. [Google Scholar] [CrossRef]
- Kanai, K.; Kuwabara, S.; Misawa, S.; Tamura, N.; Ogawara, K.; Nakata, M.; Sawai, S.; Hattori, T.; Bostock, H. Altered axonal excitability properties in amyotrophic lateral sclerosis: Impaired potassium channel function related to disease stage. Brain 2006, 129, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, K.-M.; Hwang, J.-Y.; Shin, H.-C.; Koh, J.-Y. A Novel Neuroprotective Mechanism of Riluzole: Direct Inhibition of Protein Kinase C. Neurobiol. Dis. 2000, 7, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amyotrophic Lateral Sclerosis/Riluzole Study Group II; Lacomblez, L.; Bensimon, G.; Meininger, V.; Leigh, P.; Guillet, P. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef]
- Jami, M.-S.; Salehi-Najafabadi, Z.; Ahmadinejad, F.; Hoedt, E.; Chaleshtori, M.H.; Ghatrehsamani, M.; Neubert, T.A.; Larsen, J.P.; Møller, S.G. Edaravone leads to proteome changes indicative of neuronal cell protection in response to oxidative stress. Neurochem. Int. 2015, 90, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Goutman, S.A.; Brown, M.B.; Glass, J.D.; Boulis, N.; Johe, K.; Hazel, T.; Cudkowicz, M.; Atassi, N.; Borges, L.; Patil, P.G.; et al. Long-term Phase 1/2 intraspinal stem cell transplantation outcomes in ALS. Ann. Clin. Transl. Neurol. 2018, 5, 730–740. [Google Scholar] [CrossRef]
- Petrou, P.; Gothelf, Y.; Argov, Z.; Gotkine, M.; Levy, Y.S.; Kassis, I.; Vaknin-Dembinsky, A.; Ben-Hur, T.; Offen, D.; Abramsky, O.; et al. Safety and Clinical Effects of Mesenchymal Stem Cells Secreting Neurotrophic Factor Transplantation in Patients with Amyotrophic Lateral Sclerosis: Results of Phase 1/2 and 2a Clinical Trials. JAMA Neurol. 2016, 73, 337–344. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, H. A promising new therapy for ALS. Nat. Rev. Neurol. 2021, 17, 327. [Google Scholar] [CrossRef]
- Ludolph, A.C. The TUDCA trial—Innovative trial designs for amyotrophic lateral sclerosis drugs? Eur. J. Neurol. 2015, 23, 11–12. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Atassi, N.; David, W.; Cudkowicz, M.; Schoenfeld, D. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology 2018, 90, e565–e574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filosto, M.; Scarpelli, M.; Cotelli, M.S.; Vielmi, V.; Todeschini, A.; Gregorelli, V.; Tonin, P.; Tomelleri, G.; Padovani, A. The role of mitochondria in neurodegenerative diseases. J. Neurol. 2011, 258, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef]
- Cabral-Miranda, F.; Hetz, C. ER Stress and Neurodegenerative Disease: A Cause or Effect Relationship? Curr. Top. Microbiol. Immunol. 2018, 414, 131–157. [Google Scholar] [CrossRef]
- Borza, L.R. A review on the cause-effect relationship between oxidative stress and toxic proteins in the pathogenesis of neurodegenerative diseases. Med -Surg. J. 2014, 118, 19–27. [Google Scholar]
- Kumar, V.; Sami, N.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, I. Protein aggregation and neurodegenerative diseases: From theory to therapy. Eur. J. Med. Chem. 2016, 124, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]


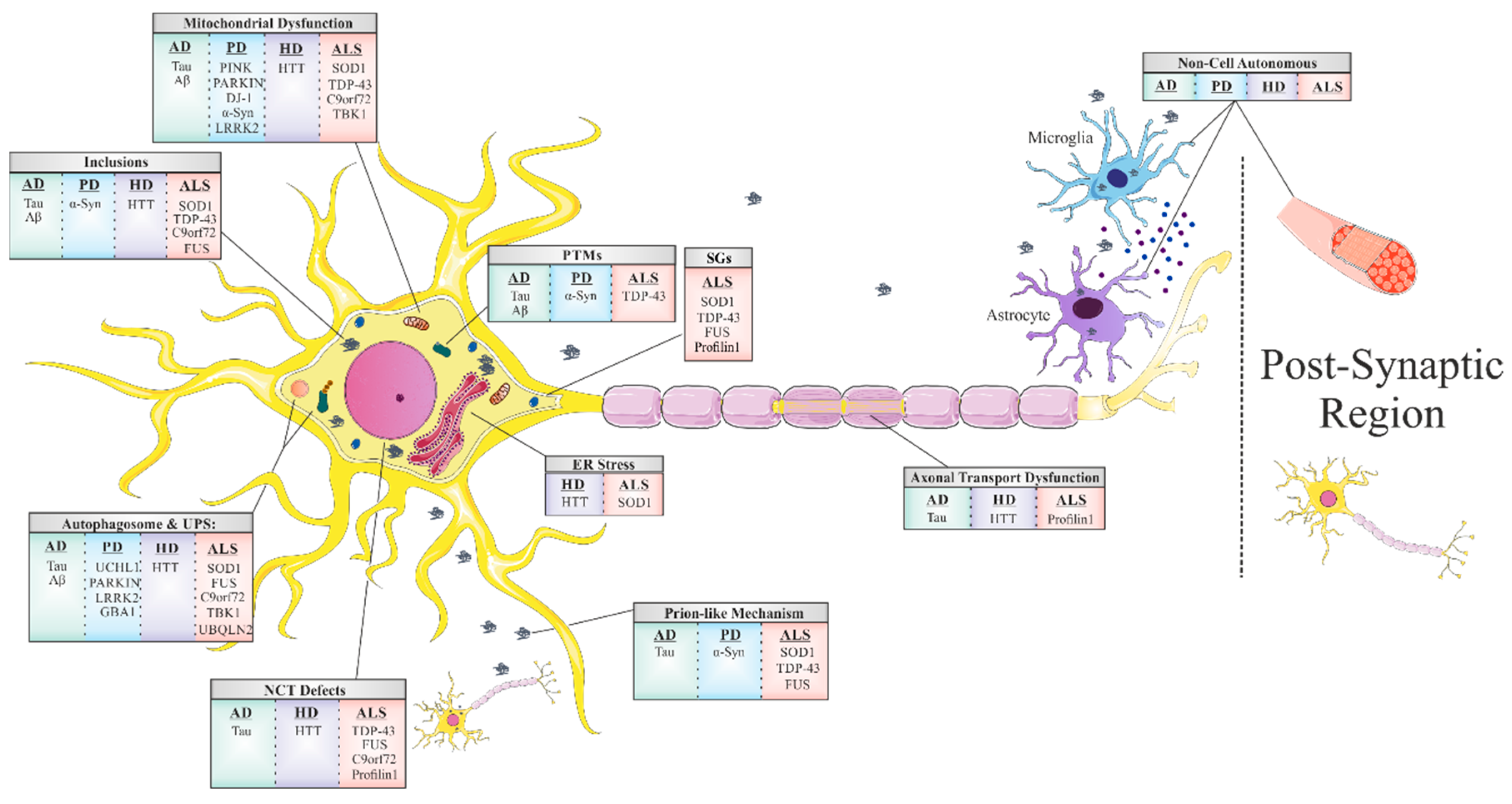{kind=link}
| Disease | Associated Genes | Associated/Pathogenic Protein | Normal Function | Toxicity Mechanism | Related Chaperones | Current Treatments |
|---|---|---|---|---|---|---|
| AD | MAPT | Tau | Polymerizes tubulin into microtubules | Gain and loss of function | HSP90 HSP104 HSP70 HSP60 HSC70 | AChE inhibitors:
|
| APP PSEN1 PSEN2 | Amyloid β | There are evidence for its precursor participation in:
| Gain of toxic function | |||
| PD | SNCA | α-Synuclein |
| Gain of toxic function | HSP70 HSP40 HSP90 | Precursor of dopamine
|
| UCHL1 | UCHL1 | Neuron-specific deubiquitinating enzyme | Gain of toxic function | |||
| PARK2 | Parkin |
| Loss of function | |||
| PINK1 | PINK1 |
function of the mitochondria and mitophagy function of the mitochondria and mitophagy
| Loss of function | |||
| PARK7 | DJ-1 |
| Loss of function | |||
| LRRK2 | LRRK2 | Mitochondrial clearance | Gain of toxic function | |||
| GBA | GCase | Lysosomal enzyme | Gain and loss of function | |||
| HD | HTT | HTT |
| Gain of toxic function | HSP70 HSP40 HSP90 | Only supportive treatments are available |
| ALS | SOD1 | SOD1 | Catalyzes the conversion of superoxide radical into peroxide and oxygen | Gain of toxic function | CCS HSP70 HSP40 HSP110 HSPB8 HSP27 MIF 4-PBA | Glutamate antagonist:
|
| TARDBP | TDP-43 | RNA metabolism | Gain and loss of function | |||
| C9orf72 | C9orf72 | Yet to be established | Gain and loss of function | |||
| FUS | FUS |
| Gain and loss of function | |||
| UBQLN2 | Ubiquilin2 | Degradation of ubiquitinated proteins | Yet to be established | |||
| TBK1 | TBK1 |
| Loss of function | |||
| PFN1 | Profilin1 | Actin polymerization | Gain and loss of function |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Argueti-Ostrovsky, S.; Alfahel, L.; Kahn, J.; Israelson, A. All Roads Lead to Rome: Different Molecular Players Converge to Common Toxic Pathways in Neurodegeneration. Cells 2021, 10, 2438. https://doi.org/10.3390/cells10092438
Argueti-Ostrovsky S, Alfahel L, Kahn J, Israelson A. All Roads Lead to Rome: Different Molecular Players Converge to Common Toxic Pathways in Neurodegeneration. Cells. 2021; 10(9):2438. https://doi.org/10.3390/cells10092438
Chicago/Turabian StyleArgueti-Ostrovsky, Shirel, Leenor Alfahel, Joy Kahn, and Adrian Israelson. 2021. "All Roads Lead to Rome: Different Molecular Players Converge to Common Toxic Pathways in Neurodegeneration" Cells 10, no. 9: 2438. https://doi.org/10.3390/cells10092438
APA StyleArgueti-Ostrovsky, S., Alfahel, L., Kahn, J., & Israelson, A. (2021). All Roads Lead to Rome: Different Molecular Players Converge to Common Toxic Pathways in Neurodegeneration. Cells, 10(9), 2438. https://doi.org/10.3390/cells10092438