
Electrophysiological Remodeling: Cardiac T-Tubules and ß-Adrenoceptors


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Electrophysiological Remodeling of the Heart and TATs
3. A Focus on the Ventricular Myocardium
4. The β-Adrenergic Receptors
5. cAMP Restriction and Compartmentation
6. The Molecular Modulation of Cardiac Electrophysiology by β-Adrenergic Receptors
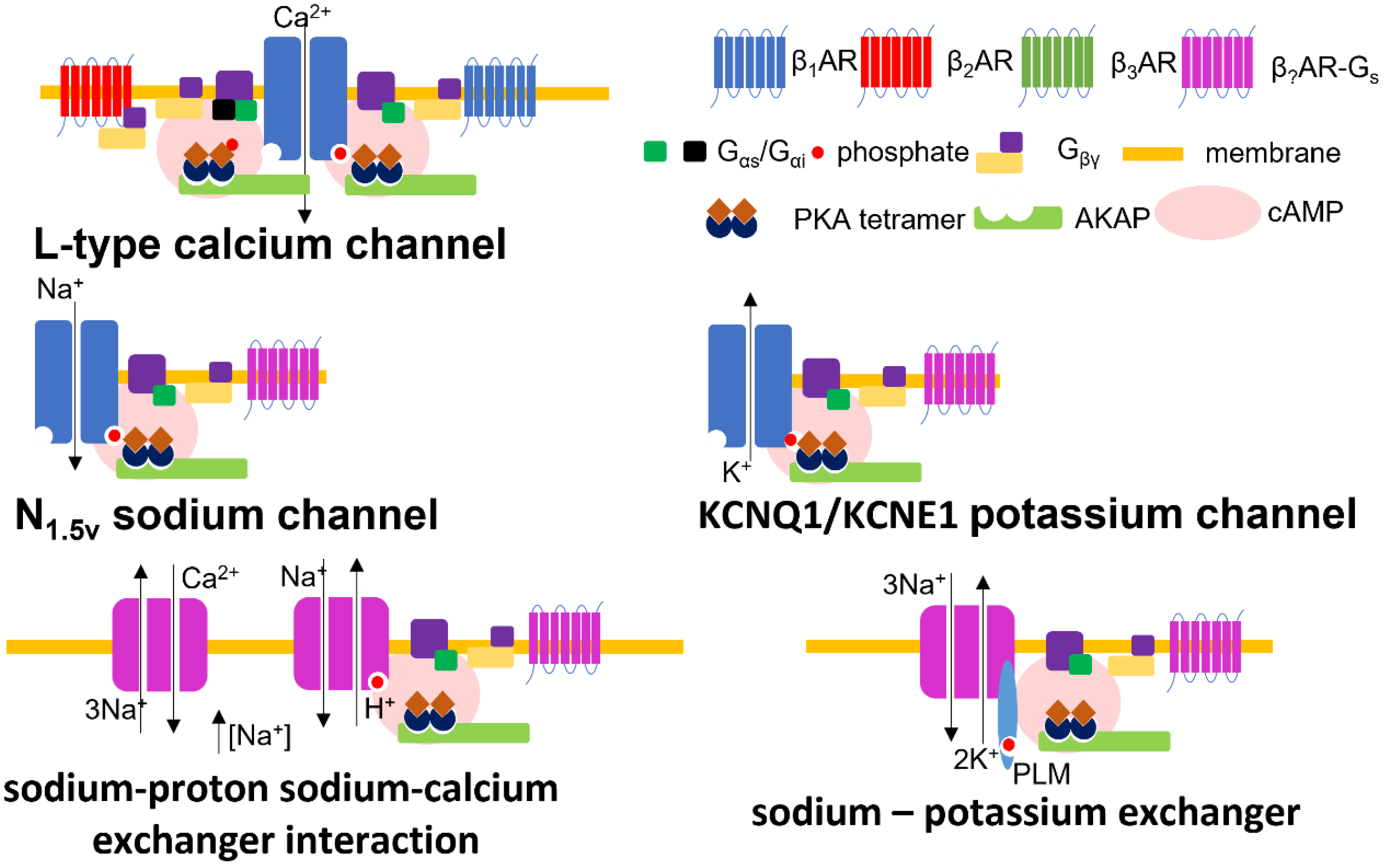
6.1. Calcium
6.2. Potassium
6.3. Sodium
6.4. Hyperpolarization Activated, Cyclic Nucleotide (HCN) Gated Channels and the Funny Current
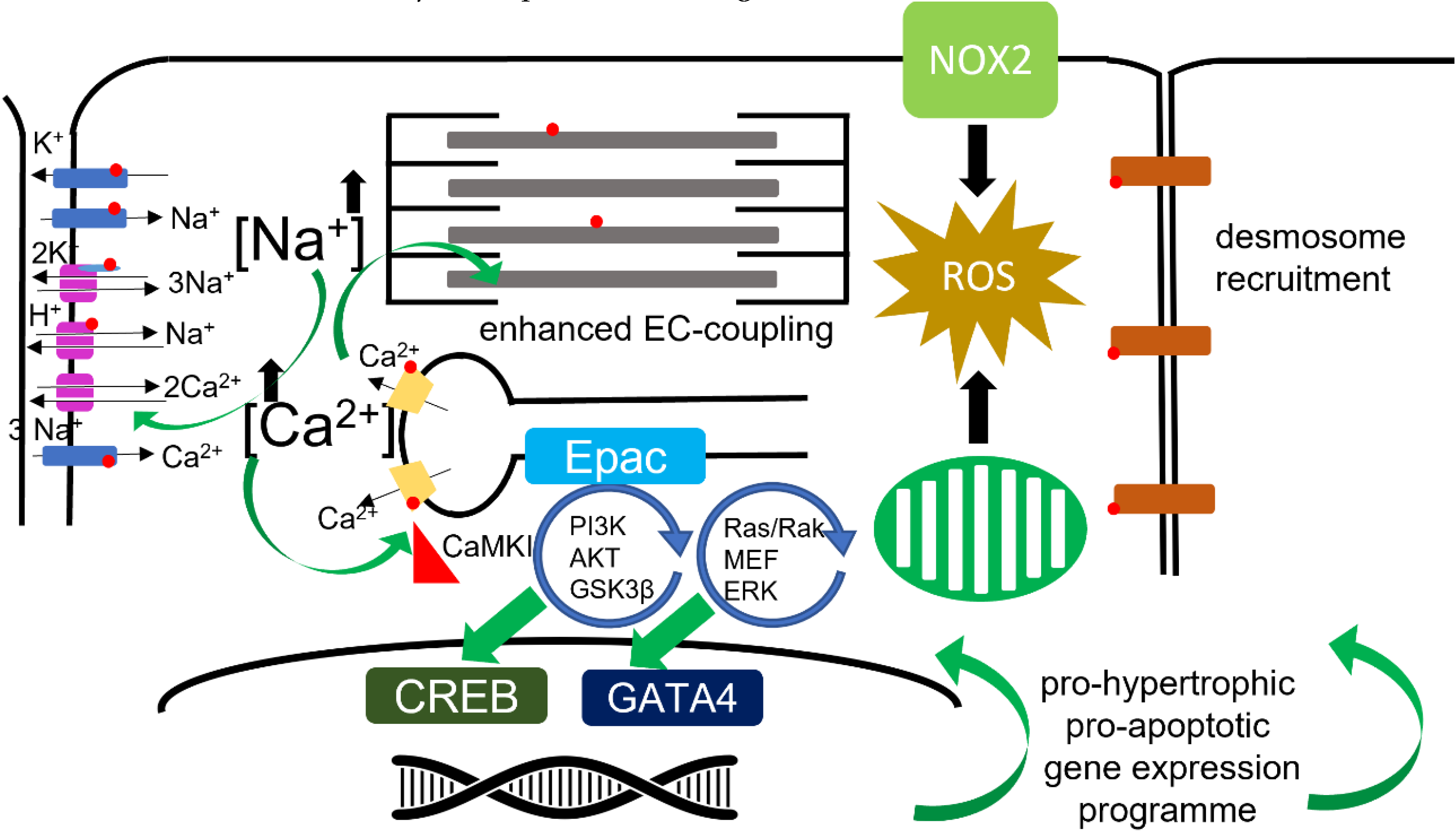
6.5. Sodium–Cation Exchangers
6.6. Reactive Oxygen Species
7. The ‘Structural’ Modulation of Cardiac Electrophysiology by β-Adrenergic Receptors
7.1. Cardiomyocyte Adhesion
7.2. Cardiomyocyte Hypertrophy
7.3. Extra-Cellular Matrix Remodelling
7.4. Mechano-Transduction
8. The Relevance of T-Tubules to the Modification of βAR Driven Changes in Cardiac Electrophysiology
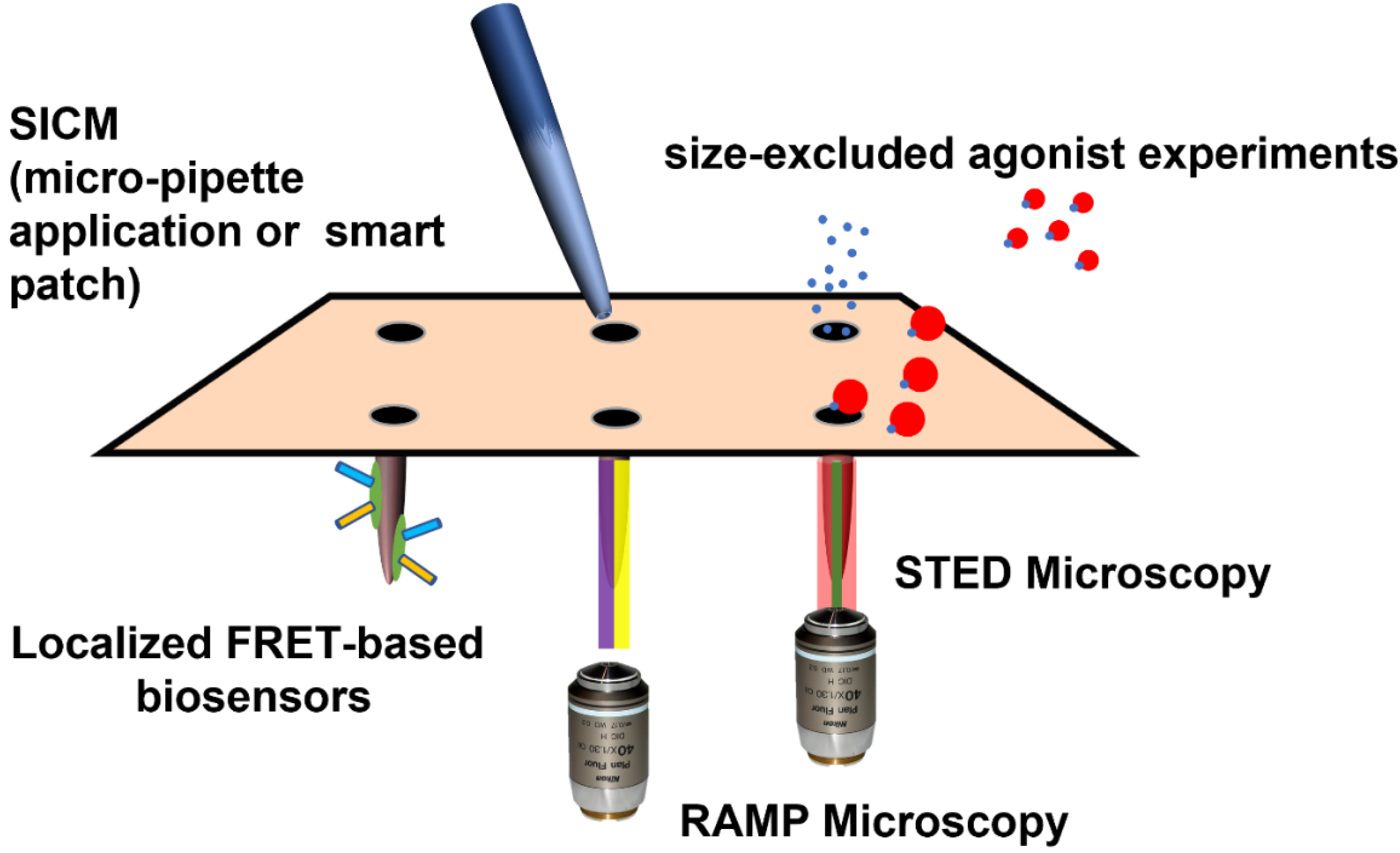
9. Experimental Techniques to Probe TAT/βAR Effects in Cardiomyocytes with ‘Tubular’ Resolution
9.1. Removing T-Tubules from Cardiomyocytes
9.2. Directly Applying Agonists to the T-Tubules
9.3. Localized Fluorescent Reporters
9.4. Polymer-Based Agonists
9.5. Measuring Activity within T-Tubules
9.6. Computational Models
9.7. Whole-Heart Models
10. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hüser, J.; Lipsius, S.L.; A’Blatter, L. Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J. Physiol. 1996, 494, 641–651. [Google Scholar] [CrossRef]
- Ayettey, A.S.; Navaratnam, V. The T-tubule system in the specialized and general myocardium of the rat. J. Anat. 1978, 127, 125–140. [Google Scholar]
- Glukhov, A.V.; Balycheva, M.; Sanchez-Alonso, J.L.; Ilkan, Z.; Alvarez-Laviada, A.; Bhogal, N.; Diakonov, I.; Schobesberger, S.; Sikkel, M.B.; Bhargava, A.; et al. Direct Evidence for Microdomain-Specific Localization and Remodeling of Functional L-Type Calcium Channels in Rat and Human Atrial Myocytes. Circulation 2015, 132, 2372–2384. [Google Scholar] [CrossRef] [Green Version]
- Forssmann, W.G.; Girardier, L. A study of the t system in rat heart. J. Cell Biol. 1970, 44, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Hibbs, R.G.; Ferrans, V.J. An ultrastructural and histochemical study of rat atrial myocardium. Am. J. Anat. 1969, 124, 251–279. [Google Scholar] [CrossRef]
- Brette, F.; Orchard, C. T-Tubule Function in Mammalian Cardiac Myocytes. Circ. Res. 2003, 92, 1182–1192. [Google Scholar] [CrossRef]
- Richards, M.A.; Clarke, J.D.; Saravanan, P.; Voigt, N.; Dobrev, D.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol. Circ. Physiol. 2011, 301, H1996–H2005. [Google Scholar] [CrossRef] [Green Version]
- Voigt, N.; Pearman, C.M.; Dobrev, D.; Dibb, K.M. Methods for isolating atrial cells from large mammals and humans. J. Mol. Cell. Cardiol. 2015, 86, 187–198. [Google Scholar] [CrossRef]
- Wright, P.; Bhogal, N.K.; Diakonov, I.; Pannell, L.M.; Perera, R.K.; Bork, N.I.; Schobesberger, S.; Lucarelli, C.; Faggian, G.; Alvarez-Laviada, A.; et al. Cardiomyocyte Membrane Structure and cAMP Compartmentation Produce Anatomical Variation in β2AR-cAMP Responsiveness in Murine Hearts. Cell Rep. 2018, 23, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.; Kukadia, P.; Siedlecka, U.; Cartledge, J.E.; Navaratnarajah, M.; Tokar, S.; Van Doorn, C.; Tsang, V.T.; Gorelik, J.; Yacoub, M.H.; et al. Cardiomyocyte Ca2+ handling and structure is regulated by degree and duration of mechanical load variation. J. Cell. Mol. Med. 2012, 16, 2910–2918. [Google Scholar] [CrossRef] [Green Version]
- Tse, G. Mechanisms of cardiac arrhythmias. J. Arrhythm. 2016, 32, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Anumonwo, J.M.; Pandit, S.V. Ionic mechanisms of arrhythmogenesis. Trends Cardiovasc. Med. 2015, 25, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.N.; Garfinkel, A.; Karagueuzian, H.S.; Nguyen, T.; Olcese, R.; Chen, P.-S.; Qu, Z. Perspective: A dynamics-based classification of ventricular arrhythmias. J. Mol. Cell. Cardiol. 2015, 82, 136–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antzelevitch, C.; Burashnikov, A. Overview of Basic Mechanisms of Cardiac Arrhythmia. Card. Electrophysiol. Clin. 2011, 3, 23–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, M.S.; Hawkey, L.A.; Sperelakis, N. The transverse-axial tubular system (tats) of mouse myocardium: Its morphology in the developing and adult animal. Am. J. Anat. 1984, 170, 143–162. [Google Scholar] [CrossRef]
- Brandenburg, S.; Kohl, T.; Williams, G.S.B.; Gusev, K.; Wagner, E.; Rog-Zielinska, E.A.; Hebisch, E.; Dura, M.; Didié, M.; Gotthardt, M.; et al. Axial tubule junctions control rapid calcium signaling in atria. J. Clin. Investig. 2016, 126, 3999–4015. [Google Scholar] [CrossRef] [Green Version]
- Schobesberger, S.; Wright, P.; Tokar, S.; Bhargava, A.; Mansfield, C.; Glukhov, A.V.; Poulet, C.; Buzuk, A.; Monszpart, A.; Sikkel, M.; et al. T-tubule remodelling disturbs localized β2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc. Res. 2017, 113, 770–782. [Google Scholar] [CrossRef]
- Seidel, T.; Navankasattusas, S.; Ahmad, A.; Diakos, N.A.; Xu, W.D.; Tristani-Firouzi, M.; Bonios, M.J.; Taleb, I.; Li, D.Y.; Selzman, C.H.; et al. Sheet-Like Remodeling of the Transverse Tubular System in Human Heart Failure Impairs Excitation-Contraction Coupling and Functional Recovery by Mechanical Unloading. Circulation 2017, 135, 1632–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, F.R.; Bito, V.; Volders, P.G.; Antoons, G.; Mubagwa, K.; Sipido, K.R. Spatial and Temporal Inhomogeneities during Ca2+ Release from the Sarcoplasmic Reticulum in Pig Ventricular Myocytes. Circ. Res. 2002, 91, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.A.; Perbellini, F.; Terracciano, C.M. Cardiac t-tubules: Where structural plasticity meets functional adaptation. Cardiovasc. Res. 2016, 112, 423–425. [Google Scholar] [CrossRef]
- Wong, J.; Baddeley, D.; Bushong, E.A.; Yu, Z.; Ellisman, M.H.; Hoshijima, M.; Soeller, C. Nanoscale Distribution of Ryanodine Receptors and Caveolin-3 in Mouse Ventricular Myocytes: Dilation of T-Tubules near Junctions. Biophys. J. 2013, 104, L22–L24. [Google Scholar] [CrossRef] [Green Version]
- Burton, R.A.; Rog-Zielinska, E.A.; Corbett, A.; Peyronnet, R.; Bodi, I.; Fink, M.; Sheldon, J.; Hoenger, A.; Calaghan, S.; Bub, G.; et al. Caveolae in Rabbit Ventricular Myocytes: Distribution and Dynamic Diminution after Cell Isolation. Biophys. J. 2017, 113, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, H.; Hoshijima, M.; Song, L.-S. Ca2+ microdomains organized by junctophilins. Cell Calcium 2015, 58, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, T.-T.; Smyth, J.; Gao, D.; Chu, K.Y.; Vogan, J.M.; Fong, T.S.; Jensen, B.C.; Colecraft, H.M.; Shaw, R.M. BIN1 Localizes the L-Type Calcium Channel to Cardiac T-Tubules. PLoS Biol. 2010, 8, e1000312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani, B.; Woodman, S.E.; Lisanti, M.P. Caveolae: From Cell Biology to Animal Physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Wang, Y.; Chen, B.; Wang, Y.; Yuan, J.; Zhang, L.; Hall, D.; Wu, J.; Shi, Y.; Zhu, Q.; et al. E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 2018, 362, eaan3303. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.; Johnson, J.; Romero, C.M.; Eaton, D.M.; Poulet, C.; Sanchez-Alonso, J.; Lucarelli, C.; Ross, J.; Gibb, A.A.; Garbincius, J.F.; et al. Interaction of the Joining Region in Junctophilin-2 with the L-Type Ca2+ Channel Is Pivotal for Cardiac Dyad Assembly and Intracellular Ca2+ Dynamics. Circ. Res. 2021, 128, 92–114. [Google Scholar] [CrossRef]
- Poulet, C.; Sanchez-Alonso, J.; Swiatlowska, P.; Mouy, F.; Lucarelli, C.; Alvarez-Laviada, A.; Gross, P.; Terracciano, C.; Houser, S.; Gorelik, J. Junctophilin-2 tethers T-tubules and recruits functional L-type calcium channels to lipid rafts in adult cardiomyocytes. Cardiovasc. Res. 2021, 117, 149–161. [Google Scholar] [CrossRef]
- Hong, T.; Yang, H.; Zhang, S.-S.; Cho, H.C.; Kalashnikova, M.; Sun, B.; Zhang, H.; Bhargava, A.; Grabe, M.; E’Olgin, J.; et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 2014, 20, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Best, J.M.; Kamp, T.J. Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Masson-Pévet, M.; Gros, D.; Besselsen, E. The caveolae in rabbit sinus node and atrium. Cell Tissue Res. 1980, 208, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Sato, D.; Jiang, Y.; Ginsburg, K.S.; Ripplinger, C.; Bers, D.M. Calcium-Dependent Arrhythmogenic Foci Created by Weakly Coupled Myocytes in the Failing Heart. Circ. Res. 2017, 121, 1379–1391. [Google Scholar] [CrossRef]
- Brodde, O.-E. Beta-adrenoceptors in cardiac disease. Pharmacol. Ther. 1993, 60, 405–430. [Google Scholar] [CrossRef]
- Richer, L.-P.; Vinet, A.; Kus, T.; Cardinal, R.; Ardell, J.L.; Armour, J.A. Alpha-adrenoceptor blockade modifies neurally induced atrial arrhythmias. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1175–R1180. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Kobilka, B.K. Myocyte Adrenoceptor Signaling Pathways. Science 2003, 300, 1530–1532. [Google Scholar] [CrossRef]
- Gong, H.; Adamson, D.L.; Ranu, H.K.; Koch, W.J.; Heubach, J.F.; Ravens, U.; Zolk, O.; E’Harding, S. The effect of Gi-protein inactivation on basal, and β1- and β2AR-stimulated contraction of myocytes from transgenic mice overexpressing the β2-adrenoceptor. Br. J. Pharmacol. 2000, 131, 594–600. [Google Scholar] [CrossRef] [Green Version]
- Belge, C.; Hammond, J.; Dubois-Deruy, E.; Manoury, B.; Hamelet, J.; Beauloye, C.; Markl, A.; Pouleur, A.-C.; Bertrand, L.; Esfahani, H.; et al. Enhanced Expression of β3-Adrenoceptors in Cardiac Myocytes Attenuates Neurohormone-Induced Hypertrophic Remodeling through Nitric Oxide Synthase. Circulation 2014, 129, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Baillie, G.S.; Sood, A.; McPhee, I.; Gall, I.; Perry, S.J.; Lefkowitz, R.J.; Houslay, M.D. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. USA 2003, 100, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.K.; Xiao, K.; Lefkowitz, R.J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-Q.; Wang, L.-P.; Gong, Y.-Y.; Fan, X.-X.; Zhu, S.-Y.; Wang, X.-T.; Wang, Y.-P.; Li, L.-L.; Xing, X.; Liu, X.-X.; et al. β2-Adrenergic Stimulation Compartmentalizes β1 Signaling into Nanoscale Local Domains by Targeting the C-Terminus of β1-Adrenoceptors. Circ. Res. 2019, 124, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Métrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.-L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac Mediates β-Adrenergic Receptor–Induced Cardiomyocyte Hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, T.C.; Fagan, K.A.; Nakata, H.; Schaack, J.; Cooper, D.M.; Karpen, J.W. Cyclic Nucleotide–Gated Channels Colocalize with Adenylyl Cyclase in Regions of Restricted Camp Diffusion. J. Gen. Physiol. 2000, 116, 147–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, T.; Schindler, R. New kids on the block: The Popeye domain containing (POPDC) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cell. Signal. 2017, 40, 156–165. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef]
- O’Nikolaev, V.; Gambaryan, S.; Lohse, M.J. Fluorescent sensors for rapid monitoring of intracellular cGMP. Nat. Chem. Biol. 2005, 3, 23–25. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel Single Chain cAMP Sensors for Receptor-induced Signal Propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [Green Version]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; MacHado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at b-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; Annibale, P.; Konrad, C.; Hannawacker, A.; Anton, S.E.; Maiellaro, I.; Zabel, U.; Sivaramakrishnan, S.; Falcke, M.; Lohse, M.J. Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 2020, 182, 1519–1530.e17. [Google Scholar] [CrossRef]
- Barthé, M.; Lefebvre, F.; Langlois, E.; Lefebvre, F.; Lechêne, P.; Iturrioz, X.; Ha-Duong, T.; Moine, L.; Tsapis, N.; Fischmeister, R. PEGylated isoprenaline reveals distinct functions of cardiac β-adrenergic receptors located in the T-tubule outer surface membrane. bioRxiv 2021. [CrossRef]
- Nivala, M.; Song, Z.; Weiss, J.N.; Qu, Z. T-tubule disruption promotes calcium alternans in failing ventricular myocytes: Mechanistic insights from computational modeling. J. Mol. Cell. Cardiol. 2015, 79, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Getz, M.; Rangamani, P.; Ghosh, P. Regulating cellular cyclic adenosine monophosphate: “Sources”, “sinks”, and now, “tunable valves”. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1490. [Google Scholar] [PubMed]
- Wright, P.T.; Nikolaev, V.O.; O’Hara, T.; Diakonov, I.; Bhargava, A.; Tokar, S.; Schobesberger, S.; Shevchuk, A.I.; Sikkel, M.B.; Wilkinson, R.; et al. Caveolin-3 regulates compartmentation of cardiomyocyte β2-adrenergic receptor-mediated cAMP signaling. J. Mol. Cell. Cardiol. 2014, 67, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDougall, D.A.; Agarwal, S.R.; Stopford, E.A.; Chu, H.; Collins, J.A.; Longster, A.L.; Colyer, J.; Harvey, R.D.; Calaghan, S. Caveolae compartmentalise β2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte. J. Mol. Cell. Cardiol. 2012, 52, 388–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.R.; MacDougall, D.A.; Tyser, R.; Pugh, S.D.; Calaghan, S.C.; Harvey, R.D. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 500–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, R.-P. β-Adrenergic Signaling in the Heart: Dual Coupling of the β2-Adrenergic Receptor to Gs and Gi; Proteins. Science 2001, 2001, re15. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.T.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac Ca1.2 channels during β1-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 16574–16579. [Google Scholar] [CrossRef] [Green Version]
- O’Marx, S.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA Phosphorylation Dissociates FKBP12.6 from the Calcium Release Channel (Ryanodine Receptor): Defective Regulation in Failing Hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef] [Green Version]
- MacDonnell, S.M.; García-Rivas, G.; Scherman, J.A.; Kubo, H.; Chen, X.; Valdivia, H.; Houser, S.R. Adrenergic Regulation of Cardiac Contractility Does Not Involve Phosphorylation of the Cardiac Ryanodine Receptor at Serine 2808. Circ. Res. 2008, 102, e65–e72. [Google Scholar] [CrossRef]
- Wehrens, X.H. CaMKII regulation of the cardiac ryanodine receptor and sarcoplasmic reticulum calcium release. Hear. Rhythm. 2011, 8, 323–325. [Google Scholar] [CrossRef] [Green Version]
- Dries, E.; Santiago, D.J.; Johnson, D.M.; Gilbert, G.; Holemans, P.; Korte, S.M.; Roderick, H.L.; Sipido, K.R. Calcium/calmodulin-dependent kinase II and nitric oxide synthase 1-dependent modulation of ryanodine receptors during β-adrenergic stimulation is restricted to the dyadic cleft. J. Physiol. 2016, 594, 5923–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulakhe, P.V.; Vo, X.T. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: Roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol. Cell. Biochem. 1995, 149–150, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, D.; Kuschel, M.; Kuramochi, T.; Zhu, W.; Cheng, H.; Xiao, R.-P. Frequency-encoding Thr17 phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. J. Biol. Chem. 2000, 275, 22532–22536. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Zhao, Y.-T.; Guo, Y.-B.; Xu, S.-M.; Bai, S.-H.; Lakatta, E.G.; Cheng, H.; Hao, X.-M.; Wang, S.-Q. β-Adrenergic signaling accelerates and synchronizes cardiac ryanodine receptor response to a single L-type Ca; channel. Proc. Natl. Acad. Sci. USA 2009, 106, 18028–18033. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Cheng, H.; Lao, D.H.; Na, L.; Van Oort, R.J.; Brown, J.H.; Wehrens, X.; Chen, J.; Bers, D.M. Epac2 Mediates Cardiac β1-Adrenergic–Dependent Sarcoplasmic Reticulum Ca2+ Leak and Arrhythmia. Circulation 2013, 127, 913–922. [Google Scholar] [CrossRef] [Green Version]
- McCrink, K.A.; Maning, J.; Vu, A.; Jafferjee, M.; Marrero, C.; Brill, A.; Bathgate-Siryk, A.; Dabul, S.; Koch, W.J.; Lymperopoulos, A. β-Arrestin2 Improves Post–Myocardial Infarction Heart Failure via Sarco(endo)plasmic Reticulum Ca2+-ATPase–Dependent Positive Inotropy in Cardiomyocytes. Hypertension 2017, 70, 972–981. [Google Scholar] [CrossRef]
- Mangmool, S.; Shukla, A.; Rockman, H.A. β-Arrestin–dependent activation of Ca2+/calmodulin kinase II after β1–adrenergic receptor stimulation. J. Cell Biol. 2010, 189, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, D.A.; Fernandez-Tenorio, M.; Ogrodnik, J.; Niggli, E. NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 2013, 100, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Moens, A.L.; Yang, R.; Watts, V.L.; Barouch, L.A. Beta 3-adrenoreceptor regulation of nitric oxide in the cardiovascular system. J. Mol. Cell. Cardiol. 2010, 48, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.M.; Antoons, G. Arrhythmogenic Mechanisms in Heart Failure: Linking β-Adrenergic Stimulation, Stretch, and Calcium. Front. Physiol. 2018, 9, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, M.; Noble, P.J.; Noble, D. Ca2+-induced delayed afterdepolarizations are triggered by dyadic subspace Ca2+ affirming that increasing SERCA reduces aftercontractions. Am. J. Physiol. Circ. Physiol. 2011, 301, H921–H935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, D.; Varghese, A. Modelling of sodium-overload arrhythmias and their suppression. Can. J. Cardiol. 1998, 14, 97–100. [Google Scholar]
- Zeng, J.; Rudy, Y. Early afterdepolarizations in cardiac myocytes: Mechanism and rate dependence. Biophys. J. 1995, 68, 949–964. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Larsson, H.P. Insights into Cardiac IKs (KCNQ1/KCNE1) Channels Regulation. Int. J. Mol. Sci. 2020, 21, 9440. [Google Scholar] [CrossRef] [PubMed]
- Trum, M.J.; Islam, M.M.T.; Lebek, S.; Baier, M.J.; Hegner, P.; Eaton, P.; Maier, L.S.; Wagner, S. Inhibition of cardiac potassium currents by oxidation-activated protein kinase A contributes to early afterdepolarizations in the heart. Am. J. Physiol. Circ. Physiol. 2020, 319, H1347–H1357. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, B.; Bossuyt, J.; Ginsburg, K.S.; Mendoza, L.M.; Talken, L.; Ferrier, W.T.; Pogwizd, S.M.; Izu, L.T.; Chen-Izu, Y.; Bers, D.M. Altered Repolarization Reserve in Failing Rabbit Ventricular Myocytes. Circ. Arrhythmia Electrophysiol. 2018, 11, e005852. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Shi, H.; Tonggu, L.; El-Din, T.M.G.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the Cardiac Sodium Channel. Cell 2020, 180, 122–134.e10. [Google Scholar] [CrossRef] [PubMed]
- Panama, B.K.; Korogyi, A.S.; Aschar-Sobbi, R.; Oh, Y.; Gray, C.; Gang, H.; Brown, J.H.; Kirshenbaum, L.A.; Backx, P.H. Reductions in the Cardiac Transient Outward K+ Current Ito Caused by Chronic β-Adrenergic Receptor Stimulation Are Partly Rescued by Inhibition of Nuclear Factor κB. J. Biol. Chem. 2016, 291, 4156–4165. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, B.; Bányász, T.; Izu, L.T.; Belardinelli, L.; Bers, D.M.; Chen-Izu, Y. β-adrenergic regulation of late Na+ current during cardiac action potential is mediated by both PKA and CaMKII. J. Mol. Cell. Cardiol. 2018, 123, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Shuba, Y.M.; Morad, M. Regulation of cardiac sodium-calcium exchanger by β-adrenergic agonists. Proc. Natl. Acad. Sci. USA 1996, 93, 5527–5532. [Google Scholar] [CrossRef] [Green Version]
- Barman, P.; Choisy, S.C.M.; Hancox, J.C.; James, A.F. β-Adrenoceptor/PKA-stimulation, Na+-Ca2+ exchange and PKA-activated Cl− currents in rabbit cardiomyocytes: A conundrum. Cell Calcium 2011, 49, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Hinde, A.K.; Hancox, J.C. Anti-adrenergic effect of adenosine on Na+-Ca2+ exchange current recorded from guinea-pig ventricular myocytes. Cell Calcium 2001, 29, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, K.S.; Bers, D.M. Isoproterenol does not enhance Ca-dependent Na/Ca exchange current in intact rabbit ventricular myocytes. J. Mol. Cell. Cardiol. 2005, 39, 972–981. [Google Scholar] [CrossRef]
- Main, M.J.; Grantham, C.J.; Cannell, M.B. Changes in subsarcolemmal sodium concentration measured by Na-Ca exchanger activity during Na-pump inhibition and beta-adrenergic stimulation in guinea-pig ventricular myocytes. Pflugers Arch. 1997, 435, 112–118. [Google Scholar] [CrossRef]
- Ballard, C.; Schaffer, S. Stimulation of the Na+/Ca2+ exchanger by phenylephrine, angiotensin II and endothelin 1. J. Mol. Cell. Cardiol. 1996, 28, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Despa, S. Na/K-ATPase—An Integral Player in the Adrenergic Fight-or-Flight Response. Trends Cardiovasc. Med. 2009, 19, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Bundgaard, H.; Liu, C.-C.; Garcia, A.; Hamilton, E.J.; Huang, Y.; Chia, K.K.; Hunyor, S.N.; Figtree, G.A.; Rasmussen, H.H. β3 Adrenergic Stimulation of the Cardiac Na+-K+ Pump by Reversal of an Inhibitory Oxidative Modification. Circulation 2010, 122, 2699–2708. [Google Scholar] [CrossRef] [Green Version]
- Leineweber, K.; Heusch, G.; Schulz, R. Regulation and Role of the Presynaptic and Myocardial Na+/H+ Exchanger NHE1: Effects on the Sympathetic Nervous System in Heart Failure. Cardiovasc. Drug Rev. 2007, 25, 123–131. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+ in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [Green Version]
- Yeruva, S.; Kempf, E.; Egu, D.T.; Flaswinkel, H.; Kugelmann, D.; Waschke, J. Adrenergic Signaling-Induced Ultrastructural Strengthening of Intercalated Discs via Plakoglobin Is Crucial for Positive Adhesiotropy in Murine Cardiomyocytes. Front. Physiol. 2020, 11, 430. [Google Scholar] [CrossRef]
- Schinner, C.; Vielmuth, F.; Rötzer, V.; Hiermaier, M.; Radeva, M.Y.; Co, T.K.; Hartlieb, E.; Schmidt, A.; Imhof, A.; Messoudi, A.; et al. Adrenergic Signaling Strengthens Cardiac Myocyte Cohesion. Circ. Res. 2017, 120, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Shoykhet, M.; Trenz, S.; Kempf, E.; Williams, T.; Gerull, B.; Schinner, C.; Yeruva, S.; Waschke, J. Cardiomyocyte adhesion and hyperadhesion differentially require ERK1/2 and plakoglobin. JCI Insight 2020, 5, e140066. [Google Scholar] [CrossRef]
- Morisco, C.; Zebrowski, D.C.; Vatner, D.E.; Vatner, S.F.; Sadoshima, J. β -Adrenergic Cardiac Hypertrophy is Mediated Primarily by the β1-Subtype in the Rat Heart. J. Mol. Cell. Cardiol. 2001, 33, 561–573. [Google Scholar] [CrossRef]
- Kamide, T.; Okumura, S.; Ghosh, S.; Shinoda, Y.; Mototani, Y.; Ohnuki, Y.; Jin, H.; Cai, W.; Suita, K.; Sato, I.; et al. Oscillation of cAMP and Ca2+ in cardiac myocytes: A systems biology approach. J. Physiol. Sci. 2015, 65, 195–200. [Google Scholar] [CrossRef]
- Schäfer, M.; Frischkopf, K.; Taimor, G.; Piper, H.M.; Schlüter, K.-D. Hypertrophic effect of selective β1-adrenoceptor stimulation on ventricular cardiomyocytes from adult rat. Am. J. Physiol. Physiol. 2000, 279, C495–C503. [Google Scholar] [CrossRef]
- Khalilimeybodi, A.; Daneshmehr, A.; Sharif-Kashani, B. Investigating β-adrenergic-induced cardiac hypertrophy through computational approach: Classical and non-classical pathways. J. Physiol. Sci. 2018, 68, 503–520. [Google Scholar] [CrossRef] [PubMed]
- Morisco, C.; Seta, K.; Hardt, S.E.; Lee, Y.; Vatner, S.F.; Sadoshima, J. Glycogen Synthase Kinase 3β Regulates GATA4 in Cardiac Myocytes. J. Biol. Chem. 2001, 276, 28586–28597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saucerman, J.J.; McCulloch, A.D. Cardiac beta-Adrenergic Signaling: From Subcellular Microdomains to Heart Failure. Ann. N. Y. Acad. Sci. 2006, 1080, 348–361. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What Is the Role of β-Adrenergic Signaling in Heart Failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Siwik, D.A.; Kuster, G.M.; Brahmbhatt, J.V.; Zaidi, Z.; Malik, J.; Ooi, H.; Ghorayeb, G. EMMPRIN mediates β-adrenergic receptor-stimulated matrix metalloproteinase activity in cardiac myocytes. J. Mol. Cell. Cardiol. 2008, 44, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Burke, R.M. β-Adrenergic Blockade in Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2017, 70, 972–974. [Google Scholar] [CrossRef]
- Rona, G.; Chappel, C.I.; Balazs, T.; Gaudry, R. An infarct-like myocardial lesion and other toxic manifestations produced by isoproterenol in the rat. AMA Arch. Pathol. 1959, 67, 443–455. [Google Scholar]
- Schobesberger, S.; Wright, P.T.; Poulet, C.; Mardones, J.L.S.A.; Mansfield, C.; Friebe, A.; Harding, S.; Balligand, J.-L.; O’Nikolaev, V.; Gorelik, J. β3-Adrenoceptor redistribution impairs NO/cGMP/PDE2 signalling in failing cardiomyocytes. eLife 2020, 9, 52221. [Google Scholar] [CrossRef]
- Dries, E.; Santiago, D.J.; Gilbert, G.; Lenaerts, I.; Vandenberk, B.; Nagaraju, C.K.; Johnson, D.M.; Holemans, P.; Roderick, H.; Macquaide, N.; et al. Hyperactive ryanodine receptors in human heart failure and ischaemic cardiomyopathy reside outside of couplons. Cardiovasc. Res. 2018, 114, 1512–1524. [Google Scholar] [CrossRef] [Green Version]
- Boycott, H.E.; Nguyen, M.-N.; Vrellaku, B.; Gehmlich, K.; Robinson, P. Nitric Oxide and Mechano-Electrical Transduction in Cardiomyocytes. Front. Physiol. 2020, 11, 1629. [Google Scholar] [CrossRef]
- Curran, J.; Tang, L.; Roof, S.R.; Velmurugan, S.; Millard, A.; Shonts, S.; Wang, H.; Santiago, D.J.; Ahmad, U.; Perryman, M.; et al. Nitric Oxide-Dependent Activation of CaMKII Increases Diastolic Sarcoplasmic Reticulum Calcium Release in Cardiac Myocytes in Response to Adrenergic Stimulation. PLoS ONE 2014, 9, e87495. [Google Scholar] [CrossRef] [Green Version]
- Jian, Z.; Han, H.; Zhang, T.; Puglisi, J.; Izu, L.T.; Shaw, J.A.; Onofiok, E.; Erickson, J.R.; Chen, Y.-J.; Horvath, B.; et al. Mechanochemotransduction During Cardiomyocyte Contraction Is Mediated by Localized Nitric Oxide Signaling. Sci. Signal. 2014, 7, ra27. [Google Scholar] [CrossRef] [Green Version]
- Calaghan, S.; White, E.D. Activation of Na+-H+ exchange and stretch-activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart. J. Physiol. 2004, 559, 205–214. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS Signaling: Rapid Mechano-Chemo Transduction in Heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, B.; Shimkunas, R.; Jian, Z.; Izu, L.T.; Bers, D.M.; Chen-Izu, Y. Mechanoelectric coupling and arrhythmogenesis in cardiomyocytes contracting under mechanical afterload in a 3D viscoelastic hydrogel. Proc. Natl. Acad. Sci. USA 2021, 118, e2108484118. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Sejersted, O.M.; Swift, F. There Goes the Neighborhood: Pathological Alterations in T-Tubule Morphology and Consequences for Cardiomyocyte Ca2+ Handling. J. Biomed. Biotechnol. 2010, 2010, 503906. [Google Scholar] [CrossRef] [PubMed]
- Cros, C.; Brette, F. Functional subcellular distribution of β1- and β2-adrenergic receptors in rat ventricular cardiac myocytes. Physiol. Rep. 2013, 1, e00038. [Google Scholar] [CrossRef]
- Port, J.; Bristow, M.R. Altered Beta-adrenergic Receptor Gene Regulation and Signaling in Chronic Heart Failure. J. Mol. Cell. Cardiol. 2001, 33, 887–905. [Google Scholar] [CrossRef]
- Lab, M.J.; Bhargava, A.; Wright, P.; Gorelik, J. The scanning ion conductance microscope for cellular physiology. Am. J. Physiol. Circ. Physiol. 2013, 304, H1–H11. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.; Sanchez-Alonso, J.L.; Lucarelli, C.; Alvarez-Laviada, A.; Poulet, C.E.; Bello, S.O.; Faggian, G.; Terracciano, C.M.; Gorelik, J. Partial Mechanical Unloading of the Heart Disrupts L-Type Calcium Channel and Beta-Adrenoceptor Signaling Microdomains. Front. Physiol. 2018, 9, 1302. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, A.; Lin, X.; Novak, P.; Mehta, K.; Korchev, Y.; Delmar, M.; Gorelik, J. Super-resolution Scanning Patch Clamp Reveals Clustering of Functional Ion Channels in Adult Ventricular Myocyte. Circ. Res. 2013, 112, 1112–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Alonso, J.L.; Bhargava, A.; O’Hara, T.; Glukhov, A.V.; Schobesberger, S.; Bhogal, N.; Sikkel, M.B.; Mansfield, C.; Korchev, Y.E.; Lyon, A.R.; et al. Microdomain-Specific Modulation of L-Type Calcium Channels Leads to Triggered Ventricular Arrhythmia in Heart Failure. Circ. Res. 2016, 119, 944–955. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M.; Xiang, Y.K.; Zaccolo, M. Whole-Cell cAMP and PKA Activity are Epiphenomena, Nanodomain Signaling Matters. Physiology 2019, 34, 240–249. [Google Scholar] [CrossRef]
- Bathe-Peters, M.; Gmach, P.; Boltz, H.-H.; Einsiedel, J.; Gotthardt, M.; Hübner, H.; Gmeiner, P.; Lohse, M.J.; Annibale, P. Visualization of β-adrenergic receptor dynamics and differential localization in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2101119118. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Scardigli, M.; Peyronnet, R.; Zgierski-Johnston, C.M.; Greiner, J.; Madl, J.; O’Toole, E.T.; Morphew, M.K.; Hoenger, A.; Sacconi, L.; et al. Beat-by-Beat Cardiomyocyte T-Tubule Deformation Drives Tubular Content Exchange. Circ. Res. 2021, 128, 203–215. [Google Scholar] [CrossRef]
- Rog-Zielinska, E.A.; Moss, R.; Kaltenbacher, W.; Greiner, J.; Verkade, P.; Seemann, G.; Kohl, P.; Cannell, M.B. Nano-scale morphology of cardiomyocyte t-tubule/sarcoplasmic reticulum junctions revealed by ultra-rapid high-pressure freezing and electron tomography. J. Mol. Cell. Cardiol. 2021, 153, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Scardigli, M.; Crocini, C.; Ferrantini, C.; Gabbrielli, T.; Silvestri, L.; Coppini, R.; Tesi, C.; Rog-Zielinska, E.A.; Kohl, P.; Cerbai, E.; et al. Quantitative assessment of passive electrical properties of the cardiac T-tubular system by FRAP microscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 5737–5742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, C.H.T.; Rog-Zielinska, E.A.; Kohl, P.; Orchard, C.H.; Cannell, M.B. Solute movement in the t-tubule system of rabbit and mouse cardiomyocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E7073–E7080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rog-Zielinska, E.A.; Kong, C.H.T.; Zgierski-Johnston, C.M.; Verkade, P.; Mantell, J.; Cannell, M.B.; Kohl, P. Species differences in the morphology of transverse tubule openings in cardiomyocytes. EP Eur. 2018, 20, iii120–iii124. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, L.; Ferrantini, C.; Lotti, J.; Coppini, R.; Yan, P.; Loew, L.; Tesi, C.; Cerbai, E.; Poggesi, C.; Pavone, F.S. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc. Natl. Acad. Sci. USA 2012, 109, 5815–5819. [Google Scholar] [CrossRef] [Green Version]
- Crocini, C.; Coppini, R.; Ferrantini, C.; Pavone, F.S.; Sacconi, L. Functional cardiac imaging by random access microscopy. Front. Physiol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocini, C.; Coppini, R.; Ferrantini, C.; Yan, P.; Loew, L.M.; Tesi, C.; Cerbai, E.; Poggesi, C.; Pavone, F.S.; Sacconi, S. Defects in T-tubular electrical activity underlies local alterations of calcium release in heart failure. Proc. Natl. Acad. Sci. USA 2014, 111, 15196–15201. [Google Scholar] [CrossRef] [Green Version]
- Crocini, C.; Coppini, R.; Ferrantini, C.; Yan, P.; Loew, L.M.; Poggesi, C.; Cerbai, E.; Pavone, F.S.; Sacconi, L. T-Tubular Electrical Defects Contribute to Blunted β-Adrenergic Response in Heart Failure. Int. J. Mol. Sci. 2016, 17, 1471. [Google Scholar] [CrossRef] [Green Version]
- Crocini, C.; Ferrantini, C.; Scardigli, M.; Coppini, R.; Mazzoni, L.; Lazzeri, E.; Pioner, J.M.; Scellini, B.; Guo, A.; Song, L.-S.; et al. Novel insights on the relationship between T-tubular defects and contractile dysfunction in a mouse model of hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 91, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Ter Keurs, H.E.D.J.; Boyden, P.A. Calcium and arrhythmogenesis. Physiol. Rev. 2007, 87, 457–506. [Google Scholar] [CrossRef]
- Doste, R.; Bueno-Orovio, A. Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function. Mathematics 2021, 9, 1785. [Google Scholar] [CrossRef]
- Vagos, M.; Van Herck, I.G.M.; Sundnes, J.; Arevalo, H.J.; Edwards, A.G.; Koivumäki, J.T. Computational Modeling of Electrophysiology and Pharmacotherapy of Atrial Fibrillation: Recent Advances and Future Challenges. Front. Physiol. 2018, 9, 1221. [Google Scholar] [CrossRef]
- McCabe, K.J.; Rangamani, P. Computational modeling approaches to cAMP/PKA signaling in cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 154, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Loucks, A.D.; O’Hara, T.; Trayanova, N.A. Degradation of T-Tubular Microdomains and Altered cAMP Compartmentation Lead to Emergence of Arrhythmogenic Triggers in Heart Failure Myocytes: An in silico Study. Front. Physiol. 2018, 9, 1737. [Google Scholar] [CrossRef]
- Sanchez-Alonso, J.L.; Loucks, A.; Schobesberger, S.; van Cromvoirt, A.M.; Poulet, C.; Chowdhury, R.A.; Trayanova, N.; Gorelik, J. Nanoscale regulation of L-type calcium channels differentiates between ischemic and dilated cardiomyopathies. EBioMedicine 2020, 57, 102845. [Google Scholar] [CrossRef]
- Mora, M.T.; Gong, J.Q.; Sobie, E.A.; Trenor, B. The role of β-adrenergic system remodeling in human heart failure: A mechanistic investigation. J. Mol. Cell. Cardiol. 2021, 153, 14–25. [Google Scholar] [CrossRef]
- Pueyo, E.; Orini, M.; Rodríguez, J.F.; Taggart, P. Interactive effect of beta-adrenergic stimulation and mechanical stretch on low-frequency oscillations of ventricular action potential duration in humans. J. Mol. Cell. Cardiol. 2016, 97, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Lyon, A.; Dupuis, L.J.; Arts, T.; Crijns, H.J.G.M.; Prinzen, F.W.; Delhaas, T.; Heijman, J.; Lumens, J. Differentiating the effects of β-adrenergic stimulation and stretch on calcium and force dynamics using a novel electromechanical cardiomyocyte model. Am. J. Physiol. Circ. Physiol. 2020, 319, H519–H530. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, C.; Guo, A.; Song, L.-S. In situ single photon confocal imaging of cardiomyocyte T-tubule system from Langendorff-perfused hearts. Front. Physiol. 2015, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Lang, D.; Holzem, K.; Kang, C.; Xiao, M.; Hwang, H.J.; Ewald, G.A.; Yamada, K.A.; Efimov, I.R. Arrhythmogenic Remodeling of β2 Versus β1 Adrenergic Signaling in the Human Failing Heart. Circ. Arrhythm. Electrophysiol. 2015, 8, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomek, J.; Hao, G.; Tomkova, M.; Lewis, A.; Carr, C.; Paterson, D.J.; Rodriguez, B.; Bub, G.; Herring, N. β-Adrenergic Receptor Stimulation and Alternans in the Border Zone of a Healed Infarct: An ex vivo Study and Computational Investigation of Arrhythmogenesis. Front. Physiol. 2019, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Badiceanu, A.; Brennan, J.A.; Gloschat, C.; Qiao, Y.; Trayanova, N.; Efimov, I.R. β-adrenergic stimulation augments transmural dispersion of repolarization via modulation of delayed rectifier currents IKs and IKr in the human ventricle. Sci. Rep. 2017, 7, 15922. [Google Scholar] [CrossRef]
- Kamada, R.; Yokoshiki, H.; Mitsuyama, H.; Watanabe, M.; Mizukami, K.; Tenma, T.; Takahashi, M.; Takada, S.; Anzai, T. Arrhythmogenic β-adrenergic signaling in cardiac hypertrophy: The role of small-conductance calcium-activated potassium channels via activation of CaMKII. Eur. J. Pharmacol. 2019, 844, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.D.; Montaigne, D.; Tinker, A. Tissue-Level Cardiac Electrophysiology Studied in Murine Myocardium Using a Microelectrode Array: Autonomic and Thermal Modulation. J. Membr. Biol. 2017, 250, 471–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brook, J.; Kim, M.-Y.; Koutsoftidis, S.; Pitcher, D.; Agha-Jaffar, D.; Sufi, A.; Jenkins, C.; Tzortzis, K.; Ma, S.; Jabbour, R.; et al. Development of a pro-arrhythmic ex vivo intact human and porcine model: Cardiac electrophysiological changes associated with cellular uncoupling. Pflüg. Arch.-Eur. J. Physiol. 2020, 472, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.; Tsui, S.F.; Francis, A.J.; MacLeod, K.T.; Marston, S.B. Approaches to High-Throughput Analysis of Cardiomyocyte Contractility. Front. Physiol. 2020, 11, 612. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, P.T.; Gorelik, J.; Harding, S.E. Electrophysiological Remodeling: Cardiac T-Tubules and ß-Adrenoceptors. Cells 2021, 10, 2456. https://doi.org/10.3390/cells10092456
Wright PT, Gorelik J, Harding SE. Electrophysiological Remodeling: Cardiac T-Tubules and ß-Adrenoceptors. Cells. 2021; 10(9):2456. https://doi.org/10.3390/cells10092456
Chicago/Turabian StyleWright, Peter T., Julia Gorelik, and Sian E. Harding. 2021. "Electrophysiological Remodeling: Cardiac T-Tubules and ß-Adrenoceptors" Cells 10, no. 9: 2456. https://doi.org/10.3390/cells10092456
APA StyleWright, P. T., Gorelik, J., & Harding, S. E. (2021). Electrophysiological Remodeling: Cardiac T-Tubules and ß-Adrenoceptors. Cells, 10(9), 2456. https://doi.org/10.3390/cells10092456