
Synapsin-Promoted Caveolin-1 Overexpression Maintains Mitochondrial Morphology and Function in PSAPP Alzheimer’s Disease Mice

 ,
,
Abstract
:
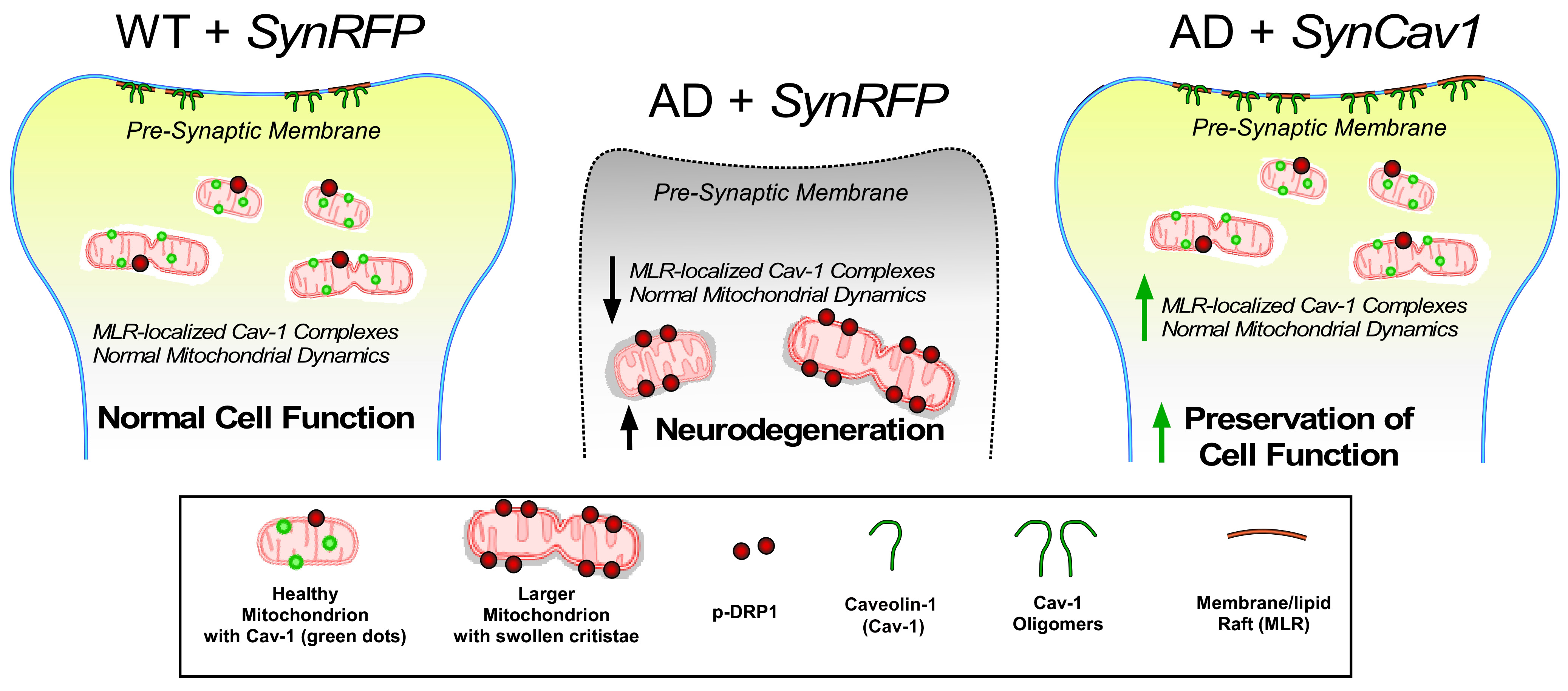{kind=link}
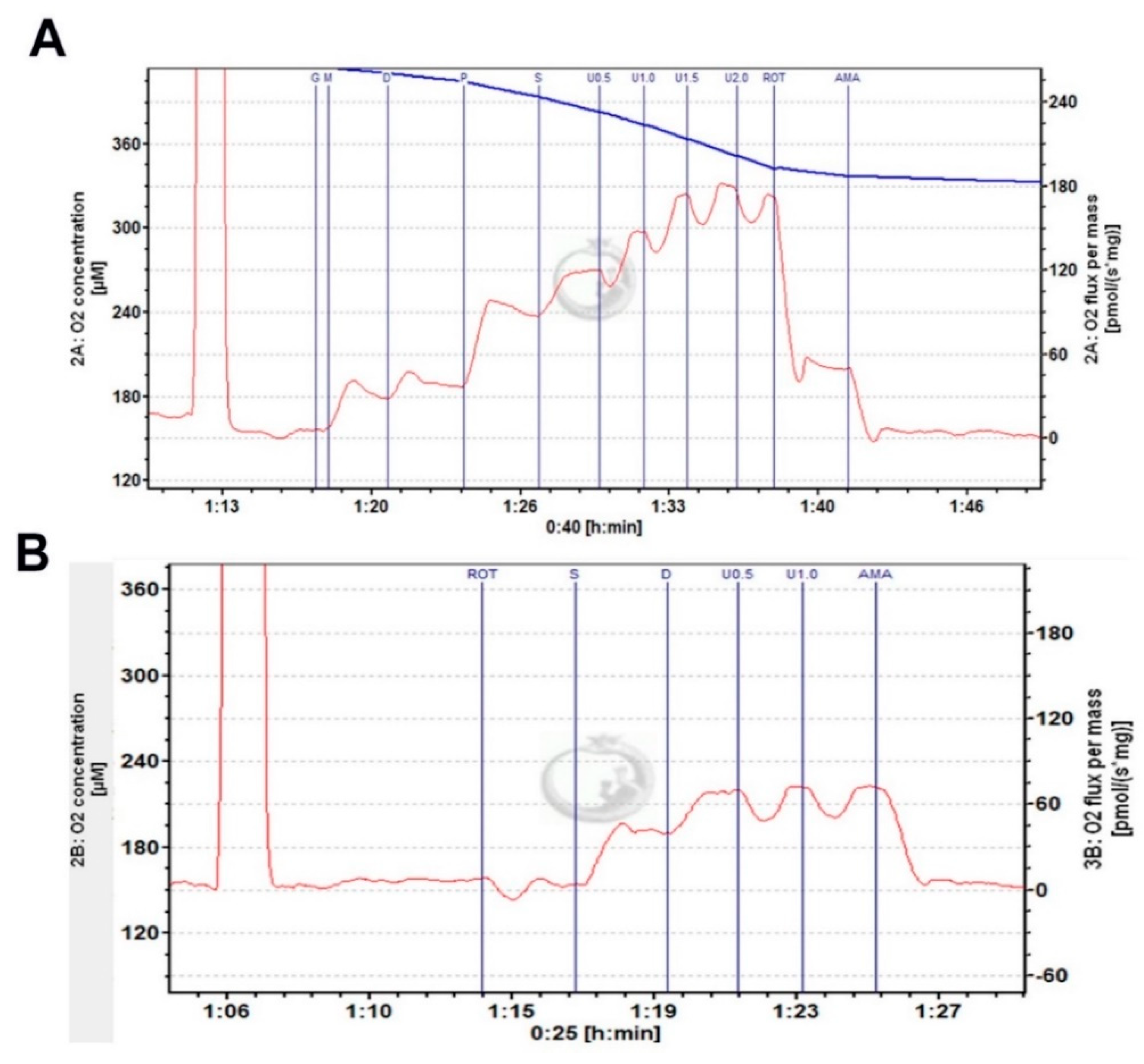{kind=link}
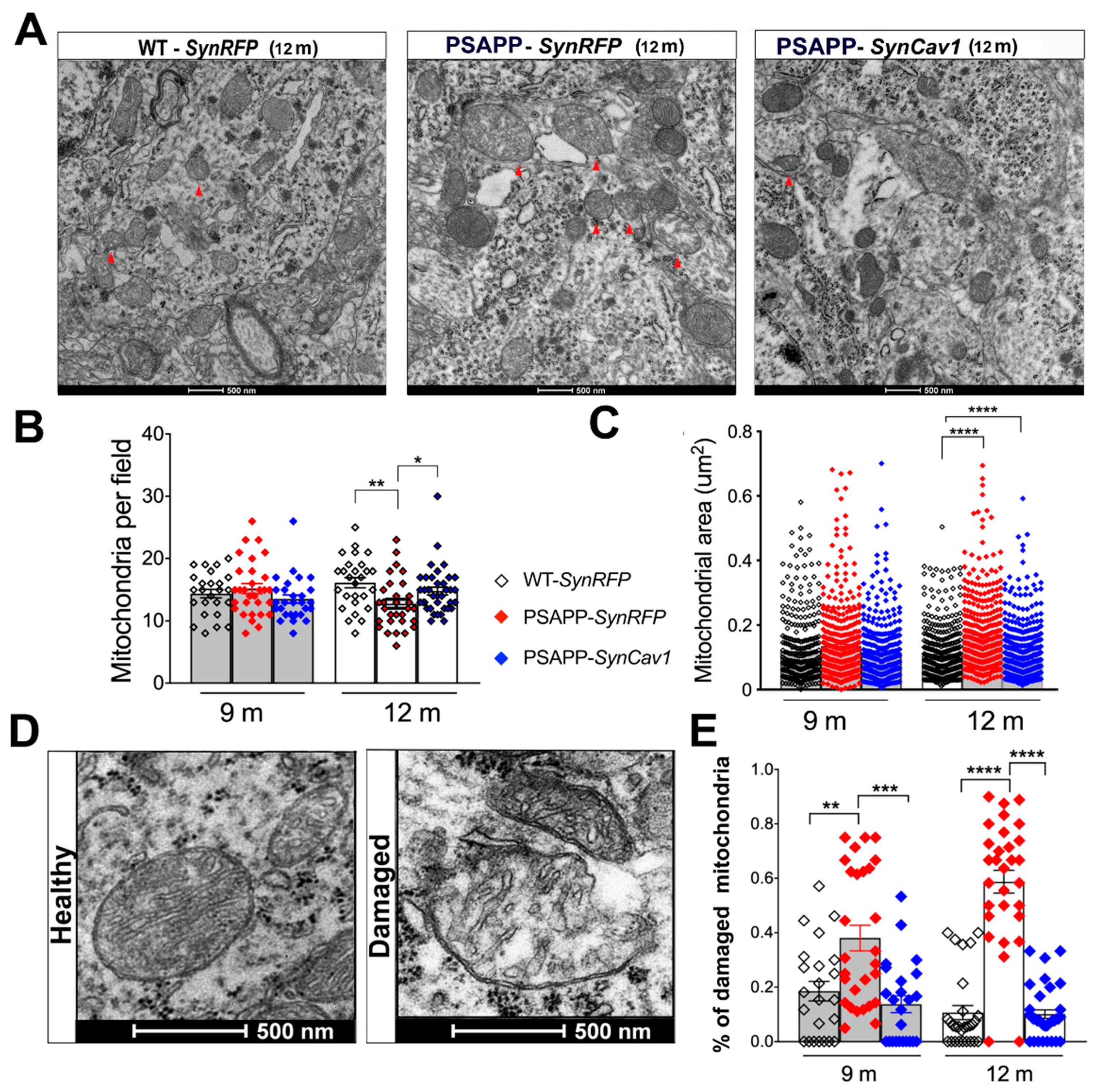{kind=link}
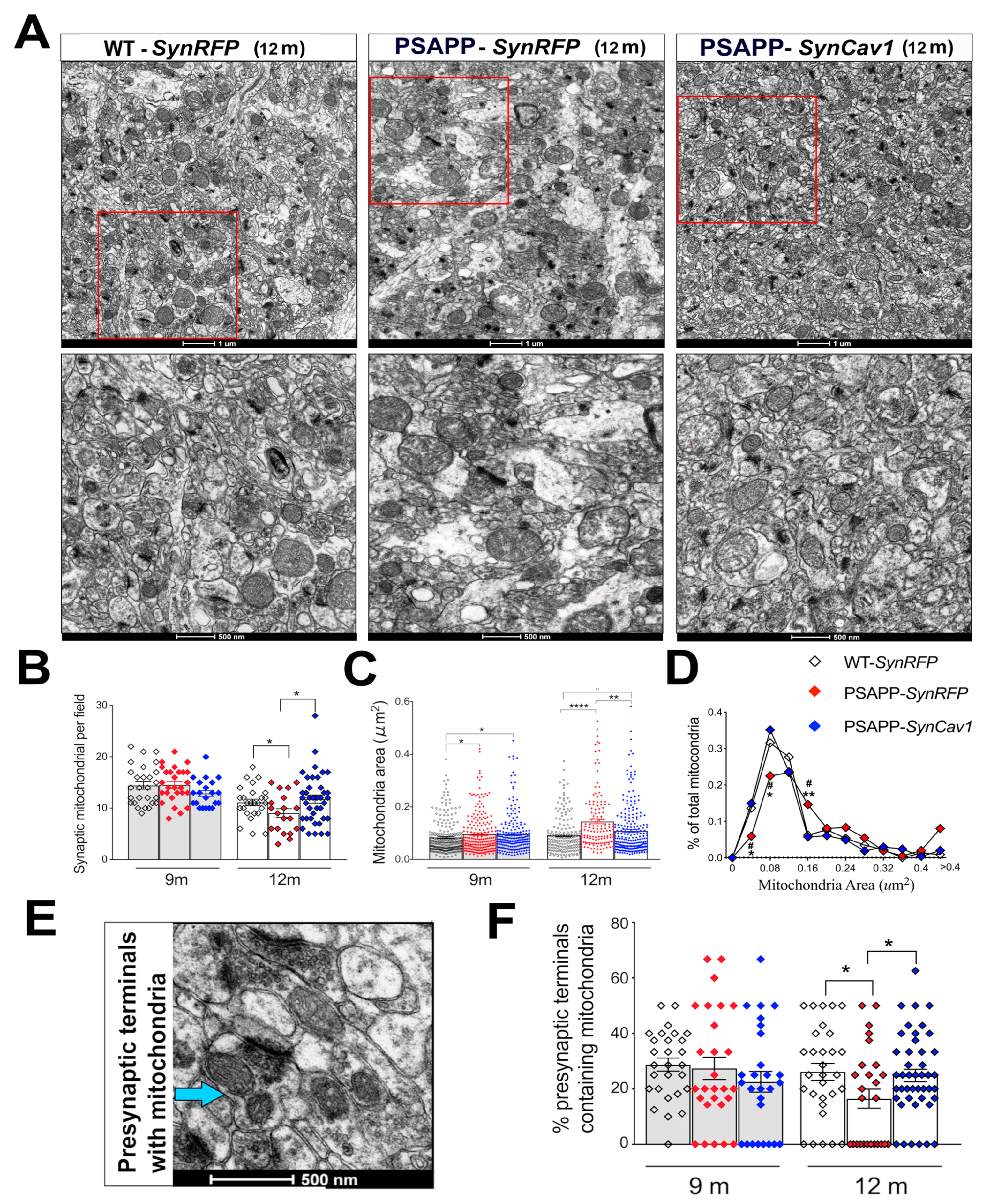{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Stereotactic Injection
2.3. Immunoblot Analysis
2.4. Electron Microscopy
2.5. High-Resolution Mitochondrial Respirometry
2.6. Statistical Analyses
3. Results
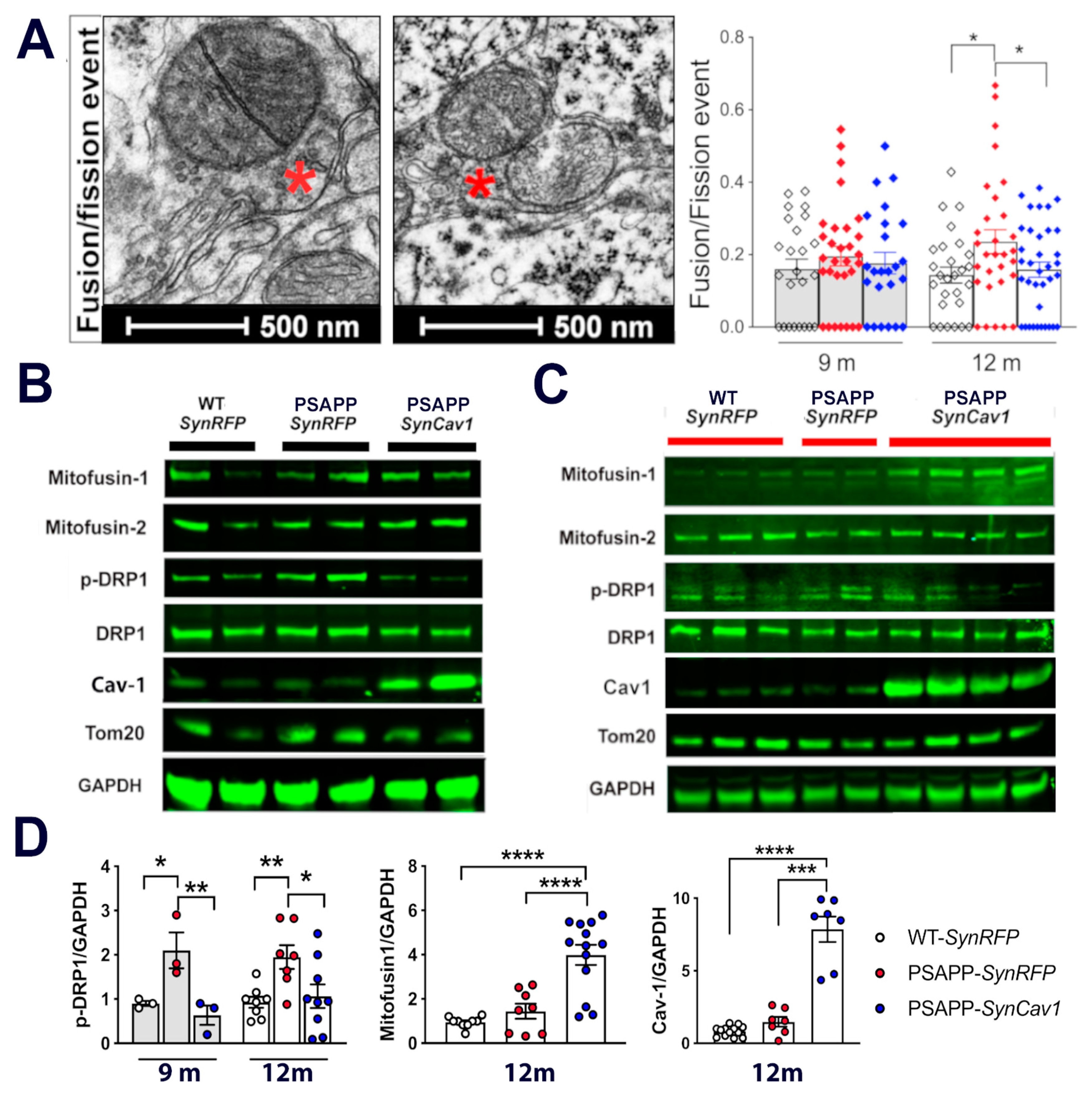
3.1. SynCav1 Preserves Soma Mitochondrial Morphology and Dynamics in Hippocampal Pyramidal Neurons of Symptomatic 12 m PSAPP Mice
3.2. SynCav1 Preserves Synaptic Mitochondrial Number and Morphology in Hippocampal Pyramidal Neurons of 12 m PSAPP Mice
3.3. Upregulated P-DRP1 in PSAPP Mice Is Inhibited by SynCav1
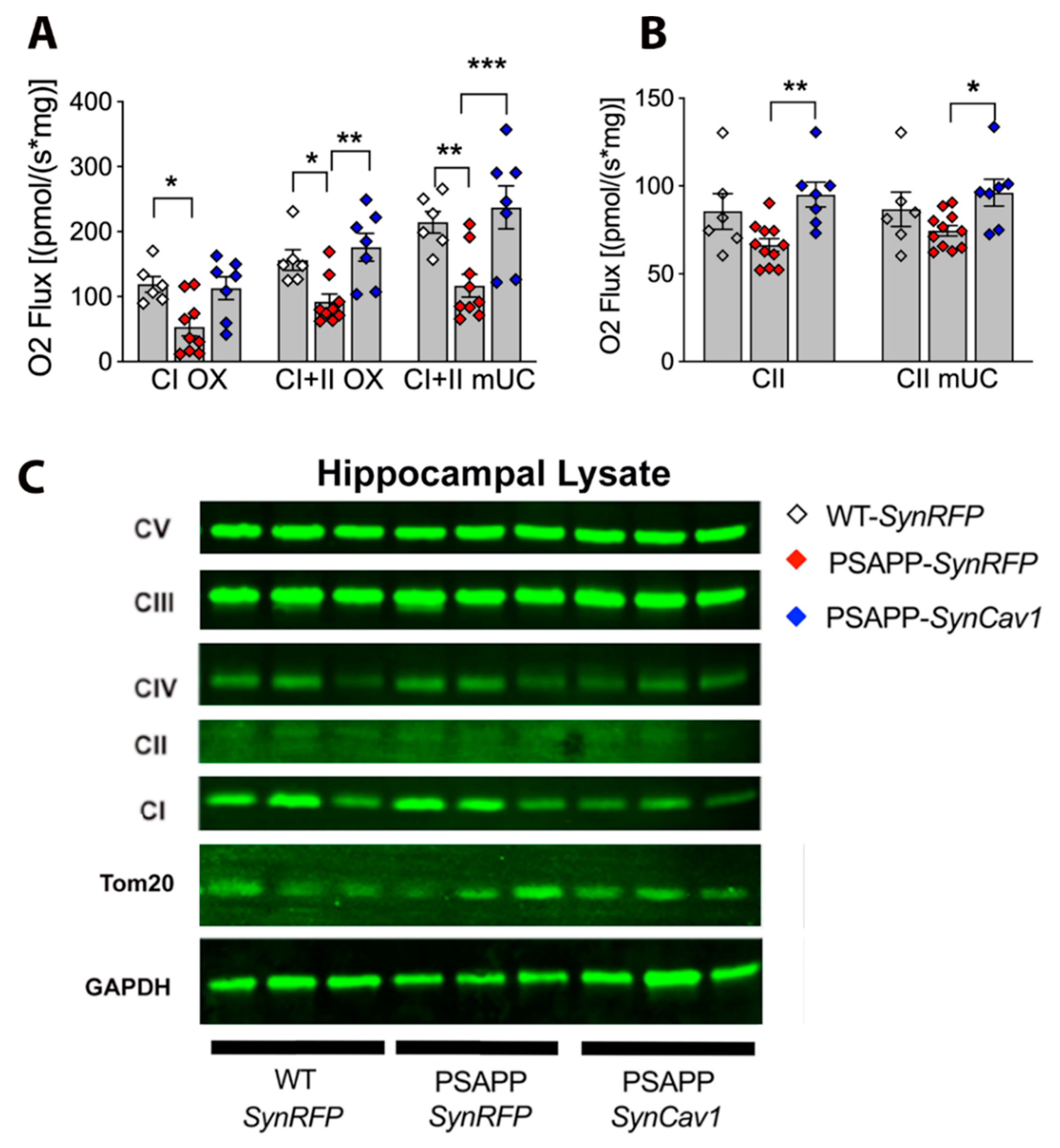
3.4. SynCav1 Augments Hippocampal Mitochondrial Respiration in Symptomatic PSAPP Mice
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.P.; Singh, B.K. Mitochondrial Dysfunction: A Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 3075–3088. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Mandyam, C.D.; Schilling, J.M.; Cui, W.; Egawa, J.; Niesman, I.R.; Kellerhals, S.E.; Staples, M.C.; Busija, A.R.; Risbrough, V.B.; Posadas, E.; et al. Neuron-Targeted Caveolin-1 Improves Molecular Signaling, Plasticity, and Behavior Dependent on the Hippocampus in Adult and Aged Mice. Biol. Psychiatry 2017, 81, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Head, B.P.; Hu, Y.; Finley, J.C.; Saldana, M.D.; Bonds, J.A.; Miyanohara, A.; Niesman, I.R.; Ali, S.S.; Murray, F.; Insel, P.A.; et al. Neuron-targeted caveolin-1 protein enhances signaling and promotes arborization of primary neurons. J. Biol. Chem. 2011, 286, 33310–33321. [Google Scholar] [CrossRef] [Green Version]
- Nwosu, Z.C.; Ebert, M.P.; Dooley, S.; Meyer, C. Caveolin-1 in the regulation of cell metabolism: A cancer perspective. Mol. Cancer 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, C.R.; Satomi, S.; Kato, Y.; Patel, H.H. The caveolar-mitochondrial interface: Regulation of cellular metabolism in physiology and pathophysiology. Biochem. Soc. Trans. 2020, 48, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Piorońska, W.; Wang, S.; Nwosu, Z.C.; Sticht, C.; Gao, Y.; Ebert, M.P.; Dooley, S.; Meyer, C. Hepatocyte caveolin-1 modulates metabolic gene profiles and functions in non-alcoholic fatty liver disease. Cell Death Dis. 2020, 11, 104. [Google Scholar] [CrossRef]
- Han, M.; Nwosu, Z.C.; Piorońska, W.; Ebert, M.P.; Dooley, S.; Meyer, C. Caveolin-1 Impacts on TGF-β Regulation of Metabolic Gene Signatures in Hepatocytes. Front. Physiol. 2019, 10, 1606. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Schilling, J.M.; Niesman, I.R.; Headrick, J.P.; Finley, J.C.; Kwan, E.; Patel, P.M.; Head, B.P.; Roth, D.M.; Yue, Y.; et al. Cardioprotective trafficking of caveolin to mitochondria is Gi-protein dependent. Anesthesiology 2014, 121, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridolfsson, H.N.; Kawaraguchi, Y.; Ali, S.S.; Panneerselvam, M.; Niesman, I.R.; Finley, J.C.; Kellerhals, S.E.; Migita, M.Y.; Okada, H.; Moreno, A.L.; et al. Mitochondria-localized caveolin in adaptation to cellular stress and injury. FASEB J. 2012, 26, 4637–4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.P.; Liu, P.; Pilcher, B.K.; Anderson, R.G. Cell-specific targeting of caveolin-1 to caveolae, secretory vesicles, cytoplasm or mitochondria. J. Cell Sci. 2001, 114, 1397–1408. [Google Scholar] [CrossRef]
- Suchaoin, W.; Chanvorachote, P. Caveolin-1 attenuates hydrogen peroxide-induced oxidative damage to lung carcinoma cells. Anticancer. Res. 2012, 32, 483–490. [Google Scholar] [PubMed]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; Pestell, R.G.; et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: A transcriptional informatics analysis with validation. Cell Cycle 2010, 9, 2201–2219. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, N.; Zheng, Y.; Zhang, J.; Zhang, F.; Wang, Z. Caveolin-1: An Oxidative Stress-Related Target for Cancer Prevention. Oxid. Med. Cell. Longev. 2017, 2017, 7454031. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volonte, D.; Galbiati, F. Polymerase I and transcript release factor (PTRF)/cavin-1 is a novel regulator of stress-induced premature senescence. J. Biol. Chem. 2011, 286, 28657–28661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, A.; Bartholomew, J.N.; Volonte, D.; Galbiati, F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006, 66, 10805–10814. [Google Scholar] [CrossRef] [Green Version]
- Powter, E.E.; Coleman, P.R.; Tran, M.H.; Lay, A.J.; Bertolino, P.; Parton, R.G.; Vadas, M.A.; Gamble, J.R. Caveolae control the anti-inflammatory phenotype of senescent endothelial cells. Aging Cell 2015, 14, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Volonte, D.; Liu, Z.; Shiva, S.; Galbiati, F. Caveolin-1 controls mitochondrial function through regulation of m-AAA mitochondrial protease. Aging 2016, 8, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Mufson, E.J.; He, B.; Ginsberg, S.D.; Carper, B.A.; Bieler, G.S.; Crawford, F.; Alvarez, V.E.; Huber, B.R.; Stein, T.D.; McKee, A.C.; et al. Gene Profiling of Nucleus Basalis Tau Containing Neurons in Chronic Traumatic Encephalopathy: A Chronic Effects of Neurotrauma Consortium Study. J. Neurotrauma 2018, 35, 1260–1271. [Google Scholar] [CrossRef]
- Sawada, A.; Wang, S.; Jian, M.; Leem, J.; Wackerbarth, J.; Egawa, J.; Schilling, J.M.; Platoshyn, O.; Zemljic-Harpf, A.; Roth, D.M.; et al. Neuron-targeted caveolin-1 improves neuromuscular function and extends survival in SOD1(G93A) mice. FASEB J. 2019, 33, 7545–7554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Leem, J.S.; Podvin, S.; Hook, V.; Kleschevnikov, N.; Savchenko, P.; Dhanani, M.; Zhou, K.; Kelly, I.C.; Zhang, T.; et al. Synapsin-caveolin-1 gene therapy preserves neuronal and synaptic morphology and prevents neurodegeneration in a mouse model of AD. Mol. Ther. Methods Clin. Dev. 2021, 21, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G.; Palacios, H.H.; Gasimov, E.; Obrenovich, M.E.; Morales, L.; Leszek, J.; Bragin, V.; Solis Herrera, A.; Gokhman, D. Oxidative Stress Induced Mitochondrial Failure and Vascular Hypoperfusion as a Key Initiator for the Development of Alzheimer Disease. Pharmaceuticals 2010, 3, 158–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonda, D.J.; Wang, X.; Perry, G.; Nunomura, A.; Tabaton, M.; Zhu, X.; Smith, M.A. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 2010, 59, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Egawa, J.; Zemljic-Harpf, A.; Mandyam, C.D.; Niesman, I.R.; Lysenko, L.V.; Kleschevnikov, A.M.; Roth, D.M.; Patel, H.H.; Patel, P.M.; Head, B.P. Neuron-Targeted Caveolin-1 Promotes Ultrastructural and Functional Hippocampal Synaptic Plasticity. Cereb. Cortex 2018, 28, 3255–3266. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, G.; Chen, L.; Zhang, W.; Feng, D.; Liu, L.; Chen, Q. Monitoring mitophagy in mammalian cells. Methods Enzym. 2014, 547, 39–55. [Google Scholar] [CrossRef]
- Chen, L.; Ma, K.; Han, J.; Chen, Q.; Zhu, Y. Monitoring Mitophagy in Mammalian Cells. Methods Enzym. 2017, 588, 187–208. [Google Scholar] [CrossRef]
- Burtscher, J.; Zangrandi, L.; Schwarzer, C.; Gnaiger, E. Differences in mitochondrial function in homogenated samples from healthy and epileptic specific brain tissues revealed by high-resolution respirometry. Mitochondrion 2015, 25, 104–112. [Google Scholar] [CrossRef]
- Risiglione, P.; Leggio, L.; Cubisino, S.A.M.; Reina, S.; Paterno, G.; Marchetti, B.; Magri, A.; Iraci, N.; Messina, A. High-Resolution Respirometry Reveals MPP(+) Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7809. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C. Dysfunction of mitochondria: Implications for Alzheimer’s disease. Int. Rev. Neurobiol. 2019, 145, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.A.; King, D.; Smith, C.; Gillingwater, T.H.; Henstridge, C.M.; Spires-Jones, T.L. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Contino, S.; Porporato, P.E.; Bird, M.; Marinangeli, C.; Opsomer, R.; Sonveaux, P.; Bontemps, F.; Dewachter, I.; Octave, J.N.; Bertrand, L.; et al. Presenilin 2-Dependent Maintenance of Mitochondrial Oxidative Capacity and Morphology. Front. Physiol. 2017, 8, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109 (Suppl. 1), 153–159. [Google Scholar] [CrossRef] [Green Version]
- Oliver, D.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Carmona, S.; Muhammad, A.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W., 2nd; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Invest. 2019, 129, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Leal, N.S.; Dentoni, G.; Schreiner, B.; Naia, L.; Piras, A.; Graff, C.; Cattaneo, A.; Meli, G.; Hamasaki, M.; Nilsson, P.; et al. Amyloid Beta-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models. Cells 2020, 9, 2552. [Google Scholar] [CrossRef]
- Yin, J.; Reiman, E.M.; Beach, T.G.; Serrano, G.E.; Sabbagh, M.N.; Nielsen, M.; Caselli, R.J.; Shi, J. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology 2020, 94, e2404–e2411. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; García, E.; Simón, D.; Avila, J.; García-Escudero, V. Mitophagy Failure in APP and Tau Overexpression Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 525–540. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Niesman, I.R.; Zemke, N.; Fridolfsson, H.N.; Haushalter, K.J.; Levy, K.; Grove, A.; Schnoor, R.; Finley, J.C.; Patel, P.M.; Roth, D.M.; et al. Caveolin isoform switching as a molecular, structural, and metabolic regulator of microglia. Mol. Cell Neurosci. 2013, 56, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.S.; Nisr, R.B.; Stretton, C.; Krasteva-Christ, G.; Hundal, H.S. Caveolin-3 deficiency associated with the dystrophy P104L mutation impairs skeletal muscle mitochondrial form and function. J. Cachexia Sarcopenia Muscle 2020, 11, 838–858. [Google Scholar] [CrossRef]
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2014, 33, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Lin, M.I.; Stan, R.V.; Bauer, P.M.; Yu, J.; Sessa, W.C. Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J. Biol. Chem. 2007, 282, 16631–16643. [Google Scholar] [CrossRef] [Green Version]
- Adebiyi, A.; Narayanan, D.; Jaggar, J.H. Caveolin-1 assembles type 1 inositol 1,4,5-trisphosphate receptors and canonical transient receptor potential 3 channels into a functional signaling complex in arterial smooth muscle cells. J. Biol. Chem. 2011, 286, 4341–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim. Biophys. Acta 2009, 1787, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, G.; Zhang, H.; Zhu, A.; Lin, Y.; Zhang, L.; Ye, B.; Cheng, J.; Shen, W.; Jin, L.; Liu, C.; et al. Treadmill exercise attenuates cerebral ischaemic injury in rats by protecting mitochondrial function via enhancement of caveolin-1. Life Sci. 2021, 264, 118634. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Ichinomiya, T.; Terada, Y.; Wang, D.; Patel, H.H.; Head, B.P. Synapsin-Promoted Caveolin-1 Overexpression Maintains Mitochondrial Morphology and Function in PSAPP Alzheimer’s Disease Mice. Cells 2021, 10, 2487. https://doi.org/10.3390/cells10092487
Wang S, Ichinomiya T, Terada Y, Wang D, Patel HH, Head BP. Synapsin-Promoted Caveolin-1 Overexpression Maintains Mitochondrial Morphology and Function in PSAPP Alzheimer’s Disease Mice. Cells. 2021; 10(9):2487. https://doi.org/10.3390/cells10092487
Chicago/Turabian StyleWang, Shanshan, Taiga Ichinomiya, Yuki Terada, Dongsheng Wang, Hemal H. Patel, and Brian P. Head. 2021. "Synapsin-Promoted Caveolin-1 Overexpression Maintains Mitochondrial Morphology and Function in PSAPP Alzheimer’s Disease Mice" Cells 10, no. 9: 2487. https://doi.org/10.3390/cells10092487
APA StyleWang, S., Ichinomiya, T., Terada, Y., Wang, D., Patel, H. H., & Head, B. P. (2021). Synapsin-Promoted Caveolin-1 Overexpression Maintains Mitochondrial Morphology and Function in PSAPP Alzheimer’s Disease Mice. Cells, 10(9), 2487. https://doi.org/10.3390/cells10092487