
Inducible Pluripotent Stem Cells to Model and Treat Inherited Degenerative Diseases of the Outer Retina: 3D-Organoids Limitations and Bioengineering Solutions


Abstract
:
1. Introduction
1.1. Disease Modeling by hiPSC-Derived ROs and Drug Development for Retinal IRD
1.2. hiPSC-Derived ROs and Replacement Therapy for Retinal IRD
1.3. IRD Treatments: What Role for hiPSC-Derived ROs in Addressing Current Issues?
2. The Path from Retinal Development to ROs
2.1. Derivation of 3D ROs from iPSC
2.2. Morphological and Transcriptomic Limitations of iPSC-Derived ROs
2.3. Functional Limitations of hiPSC-Derived ROs
3. The SRS Microenvironment
3.1. SRS Structures
3.2. SRS Biochemical and Functional Features
3.3. Oxygen Levels in the SRS and the Outer Retina
4. Bioengineering the SRS Microenvironment
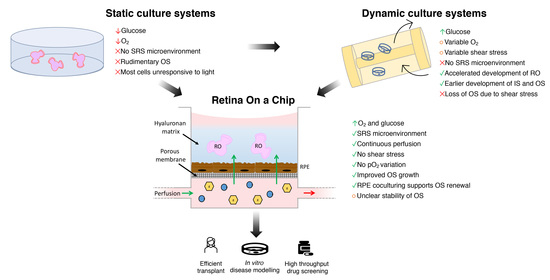
4.1. Limitations of Conventional Static Culture Systems
4.2. Dynamic Culture Systems
4.3. Biotechnologies and Human ROs-Derived Photoreceptor Precursors in Transplantation
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gollisch, T.; Meister, M. Eye smarter than scientists believed: Neural computations in circuits of the retina. Neuron 2010, 65, 150–164. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, G.J.; Rieke, F.; Shea-Brown, E.T. Nonlinear convergence boosts information coding in circuits with parallel outputs. Proc. Natl. Acad. Sci. USA 2021, 118, e1921882118. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The neuronal organization of the retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azeredo da Silveira, R.; Roska, B. Cell types, circuits, computation. Curr. Opin. Neurobiol. 2011, 21, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Wassle, H. Parallel processing in the mammalian retina. Nat. Rev. Neurosci. 2004, 5, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Grimes, W.N.; Songco-Aguas, A.; Rieke, F. Parallel Processing of Rod and Cone Signals: Retinal Function and Human Perception. Annu. Rev. Vis. Sci. 2018, 4, 123–141. [Google Scholar] [CrossRef]
- Iuliano, L.; Fogliato, G.; Corbelli, E.; Bandello, F.; Codenotti, M. Blind patients in end-stage inherited retinal degeneration: Multimodal imaging of candidates for artificial retinal prosthesis. Eye 2021, 35, 289–298. [Google Scholar] [CrossRef]
- Roska, B.; Sahel, J.A. Restoring vision. Nature 2018, 557, 359–367. [Google Scholar] [CrossRef]
- Ovando-Roche, P.; Georgiadis, A.; Smith, A.J.; Pearson, R.A.; Ali, R.R. Harnessing the Potential of Human Pluripotent Stem Cells and Gene Editing for the Treatment of Retinal Degeneration. Curr. Stem Cell Rep. 2017, 3, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Welby, E.; Li, T.; Swaroop, A. Retinal disease in ciliopathies: Recent advances with a focus on stem cell-based therapies. Transl. Sci. Rare Dis. 2019, 4, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Kruczek, K.; Swaroop, A. Pluripotent stem cell-derived retinal organoids for disease modeling and development of therapies. Stem Cells 2020, 38, 1206–1215. [Google Scholar] [CrossRef]
- Singh, R.K.; Nasonkin, I.O. Limitations and Promise of Retinal Tissue From Human Pluripotent Stem Cells for Developing Therapies of Blindness. Front. Cell. Neurosci. 2020, 14, 179. [Google Scholar] [CrossRef]
- Aasen, D.M.; Vergara, M.N. New Drug Discovery Paradigms for Retinal Diseases: A Focus on Retinal Organoids. J. Ocul. Pharmacol. Ther. 2020, 36, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.S.; Howden, S.E.; Wallace, K.A.; Verhoeven, A.D.; Wright, L.S.; Capowski, E.E.; Pinilla, I.; Martin, J.M.; Tian, S.; Stewart, R.; et al. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 2011, 29, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamm, D.M.; Phillips, M.J.; Singh, R. Modeling retinal degenerative diseases with human iPS-derived cells: Current status and future implications. Expert Rev. Ophthalmol. 2013, 8, 213–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Phillips, J.; Joseph Phillips, M.; Gamm, D.M. Mimicking Retinal Development and Disease With Human Pluripotent Stem Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, ORSFf1–ORSFf9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfitt, D.A.; Lane, A.; Ramsden, C.; Jovanovic, K.; Coffey, P.J.; Hardcastle, A.J.; Cheetham, M.E. Using induced pluripotent stem cells to understand retinal ciliopathy disease mechanisms and develop therapies. Biochem. Soc. Trans. 2016, 44, 1245–1251. [Google Scholar] [CrossRef]
- Mellough, C.B.; Collin, J.; Sernagor, E.; Wride, N.K.; Steel, D.H.; Lako, M. Lab generated retina: Realizing the dream. Vis. Neurosci. 2014, 31, 317–332. [Google Scholar] [CrossRef]
- Artero Castro, A.; Lukovic, D.; Jendelova, P.; Erceg, S. Concise Review: Human Induced Pluripotent Stem Cell Models of Retinitis Pigmentosa. Stem Cells 2018, 36, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Shimada, H.; Lu, Q.; Insinna-Kettenhofen, C.; Nagashima, K.; English, M.A.; Semler, E.M.; Mahgerefteh, J.; Cideciyan, A.V.; Li, T.; Brooks, B.P.; et al. In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations. Cell Rep. 2017, 20, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Achberger, K.; Probst, C.; Haderspeck, J.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. eLife 2019, 8, e46188. [Google Scholar] [CrossRef]
- Achberger, K.; Haderspeck, J.C.; Kleger, A.; Liebau, S. Stem cell-based retina models. Adv. Drug Deliv. Rev. 2019, 140, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Eintracht, J.; Toms, M.; Moosajee, M. The Use of Induced Pluripotent Stem Cells as a Model for Developmental Eye Disorders. Front. Cell Neurosci. 2020, 14, 265. [Google Scholar] [CrossRef] [PubMed]
- O’Hara-Wright, M.; Gonzalez-Cordero, A. Retinal organoids: A window into human retinal development. Development 2020, 147, dev189746. [Google Scholar] [CrossRef]
- Wright, L.S.; Phillips, M.J.; Pinilla, I.; Hei, D.; Gamm, D.M. Induced pluripotent stem cells as custom therapeutics for retinal repair: Progress and rationale. Exp. Eye Res. 2014, 123, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Foggia, V.; Makwana, P.; Ali, R.R.; Sowden, J.C. Induced Pluripotent Stem Cell Therapies for Degenerative Disease of the Outer Retina: Disease Modeling and Cell Replacement. J. Ocul. Pharmacol. Ther. 2016, 32, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef]
- Chang, G.Q.; Hao, Y.; Wong, F. Apoptosis: Final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron 1993, 11, 595–605. [Google Scholar] [CrossRef]
- Portera-Cailliau, C.; Sung, C.H.; Nathans, J.; Adler, R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1994, 91, 974–978. [Google Scholar] [CrossRef] [Green Version]
- Sanges, D.; Comitato, A.; Tammaro, R.; Marigo, V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 17366–17371. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.N.; Qian, H.; Parkhurst, C.N.; Gan, W.B.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef]
- Viringipurampeer, I.A.; Gregory-Evans, C.Y.; Metcalfe, A.L.; Bashar, E.; Moritz, O.L.; Gregory-Evans, K. Cell Death Pathways in Mutant Rhodopsin Rat Models Identifies Genotype-Specific Targets Controlling Retinal Degeneration. Mol. Neurobiol. 2019, 56, 1637–1652. [Google Scholar] [CrossRef]
- Kakavand, K.; Jobling, A.I.; Greferath, U.; Vessey, K.A.; de Iongh, R.U.; Fletcher, E.L. Photoreceptor Degeneration in Pro23His Transgenic Rats (Line 3) Involves Autophagic and Necroptotic Mechanisms. Front. Neurosci. 2020, 14, 581579. [Google Scholar] [CrossRef] [PubMed]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Power, M.; Das, S.; Schutze, K.; Marigo, V.; Ekstrom, P.; Paquet-Durand, F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog. Retin Eye Res. 2020, 74, 100772. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Zeiss, C.J.; Johnson, E.A. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Invest. Ophthalmol. Vis. Sci. 2004, 45, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.Y.; Zhu, X.A.; Zhang, C.; Yang, L.P.; Wu, L.M.; Tso, M.O. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2992–2999. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Xiao, J.; Wang, K.; So, K.F.; Tipoe, G.L.; Lin, B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef] [Green Version]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Zabel, M.K.; Zhao, L.; Zhang, Y.; Gonzalez, S.R.; Ma, W.; Wang, X.; Fariss, R.N.; Wong, W.T. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 2016, 64, 1479–1491. [Google Scholar] [CrossRef]
- Zhang, X.H.; Jin, Z.B. Patient iPSC-derived retinal organoids: Observable retinal diseases in-a-dish. Histol. Histopathol. 2021, 18307. [Google Scholar] [CrossRef]
- Phillips, M.J.; Perez, E.T.; Martin, J.M.; Reshel, S.T.; Wallace, K.A.; Capowski, E.E.; Singh, R.; Wright, L.S.; Clark, E.M.; Barney, P.M.; et al. Modeling human retinal development with patient-specific induced pluripotent stem cells reveals multiple roles for visual system homeobox 2. Stem Cells 2014, 32, 1480–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capowski, E.E.; Simonett, J.M.; Clark, E.M.; Wright, L.S.; Howden, S.E.; Wallace, K.A.; Petelinsek, A.M.; Pinilla, I.; Phillips, M.J.; Meyer, J.S.; et al. Loss of MITF expression during human embryonic stem cell differentiation disrupts retinal pigment epithelium development and optic vesicle cell proliferation. Hum. Mol. Genet. 2014, 23, 6332–6344. [Google Scholar] [CrossRef] [Green Version]
- Capowski, E.E.; Wright, L.S.; Liang, K.; Phillips, M.J.; Wallace, K.; Petelinsek, A.; Hagstrom, A.; Pinilla, I.; Borys, K.; Lien, J.; et al. Regulation of WNT Signaling by VSX2 During Optic Vesicle Patterning in Human Induced Pluripotent Stem Cells. Stem Cells 2016, 34, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef]
- Megaw, R.; Abu-Arafeh, H.; Jungnickel, M.; Mellough, C.; Gurniak, C.; Witke, W.; Zhang, W.; Khanna, H.; Mill, P.; Dhillon, B.; et al. Gelsolin dysfunction causes photoreceptor loss in induced pluripotent cell and animal retinitis pigmentosa models. Nat. Commun. 2017, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Arno, G.; Agrawal, S.A.; Eblimit, A.; Bellingham, J.; Xu, M.; Wang, F.; Chakarova, C.; Parfitt, D.A.; Lane, A.; Burgoyne, T.; et al. Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2016, 99, 1305–1315. [Google Scholar] [CrossRef]
- Schwarz, N.; Lane, A.; Jovanovic, K.; Parfitt, D.A.; Aguila, M.; Thompson, C.L.; da Cruz, L.; Coffey, P.J.; Chapple, J.P.; Hardcastle, A.J.; et al. Arl3 and RP2 regulate the trafficking of ciliary tip kinesins. Hum. Mol. Genet. 2017, 26, 3451. [Google Scholar] [CrossRef] [Green Version]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.F.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.M.; Buck, T.M.; Mulder, A.A.; Ohonin, C.; Alves, C.H.; Vos, R.M.; Bialecka, M.; van Herwaarden, T.; van Dijk, E.H.C.; Talib, M.; et al. Human iPSC-Derived Retinas Recapitulate the Fetal CRB1 CRB2 Complex Formation and Demonstrate that Photoreceptors and Muller Glia Are Targets of AAV5. Stem Cell Rep. 2019, 12, 906–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.C.; Wang, M.L.; Chen, S.J.; Kuo, J.C.; Wang, W.J.; Nhi Nguyen, P.N.; Wahlin, K.J.; Lu, J.F.; Tran, A.A.; Shi, M.; et al. Morphological and Molecular Defects in Human Three-Dimensional Retinal Organoid Model of X-Linked Juvenile Retinoschisis. Stem Cell Rep. 2019, 13, 906–923. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, S.E.; Fiorentino, A.; Ottaviani, D.; Fanucchi, S.; Melo, U.S.; Corral-Serrano, J.C.; Mulders, T.; Georgiou, M.; Rivolta, C.; Pontikos, N.; et al. Structural Variants Create New Topological-Associated Domains and Ectopic Retinal Enhancer-Gene Contact in Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2020, 107, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Vig, A.; Poulter, J.A.; Ottaviani, D.; Tavares, E.; Toropova, K.; Tracewska, A.M.; Mollica, A.; Kang, J.; Kehelwathugoda, O.; Paton, T.; et al. DYNC2H1 hypomorphic or retina-predominant variants cause nonsyndromic retinal degeneration. Genet. Med. 2020, 22, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Arno, G.; Fakin, A.; Parfitt, D.A.; Dhooge, P.P.A.; Albert, S.; Bax, N.M.; Duijkers, L.; Niblock, M.; Hau, K.L.; et al. Detailed Phenotyping and Therapeutic Strategies for Intronic ABCA4 Variants in Stargardt Disease. Mol. Ther. Nucleic Acids 2020, 21, 412–427. [Google Scholar] [CrossRef]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 2018, 9, 4234. [Google Scholar] [CrossRef]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 2005. [Google Scholar] [CrossRef]
- Diakatou, M.; Dubois, G.; Erkilic, N.; Sanjurjo-Soriano, C.; Meunier, I.; Kalatzis, V. Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3. Int. J. Mol. Sci. 2021, 22, 2607. [Google Scholar] [CrossRef]
- Dulla, K.; Aguila, M.; Lane, A.; Jovanovic, K.; Parfitt, D.A.; Schulkens, I.; Chan, H.L.; Schmidt, I.; Beumer, W.; Vorthoren, L.; et al. Splice-Modulating Oligonucleotide QR-110 Restores CEP290 mRNA and Function in Human c.2991+1655A>G LCA10 Models. Mol. Ther. Nucleic Acids 2018, 12, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Ohlemacher, S.K.; Sridhar, A.; Xiao, Y.; Hochstetler, A.E.; Sarfarazi, M.; Cummins, T.R.; Meyer, J.S. Stepwise Differentiation of Retinal Ganglion Cells from Human Pluripotent Stem Cells Enables Analysis of Glaucomatous Neurodegeneration. Stem Cells 2016, 34, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.J.; Ma, Y.; Jin, Z.B. The road to restore vision with photoreceptor regeneration. Exp. Eye Res. 2021, 202, 108283. [Google Scholar] [CrossRef]
- Gasparini, S.J.; Llonch, S.; Borsch, O.; Ader, M. Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog. Retin Eye Res. 2019, 69, 1–37. [Google Scholar] [CrossRef]
- Radtke, N.D.; Seiler, M.J.; Aramant, R.B.; Petry, H.M.; Pidwell, D.J. Transplantation of intact sheets of fetal neural retina with its retinal pigment epithelium in retinitis pigmentosa patients. Am. J. Ophthalmol. 2002, 133, 544–550. [Google Scholar] [CrossRef]
- Radtke, N.D.; Aramant, R.B.; Petry, H.M.; Green, P.T.; Pidwell, D.J.; Seiler, M.J. Vision improvement in retinal degeneration patients by implantation of retina together with retinal pigment epithelium. Am. J. Ophthalmol. 2008, 146, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Aramant, R.B.; Seiler, M.J. Progress in retinal sheet transplantation. Prog. Retin Eye Res. 2004, 23, 475–494. [Google Scholar] [CrossRef]
- McLelland, B.T.; Lin, B.; Mathur, A.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Transplanted hESC-Derived Retina Organoid Sheets Differentiate, Integrate, and Improve Visual Function in Retinal Degenerate Rats. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2586–2603. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; McLelland, B.T.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Retina Organoid Transplants Develop Photoreceptors and Improve Visual Function in RCS Rats With RPE Dysfunction. Investig. Ophthalmol. Vis. Sci. 2020, 61, 34. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.J.; Lin, R.E.; McLelland, B.T.; Mathur, A.; Lin, B.; Sigman, J.; De Guzman, A.T.; Kitzes, L.M.; Aramant, R.B.; Thomas, B.B. Vision Recovery and Connectivity by Fetal Retinal Sheet Transplantation in an Immunodeficient Retinal Degenerate Rat Model. Investig. Ophthalmol. Vis. Sci. 2017, 58, 614–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimoto, M.; Cheng, H.; Zhu, D.; Brzezinski, J.A.; Khanna, R.; Filippova, E.; Oh, E.C.; Jing, Y.; Linares, J.L.; Brooks, M.; et al. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc. Natl. Acad. Sci. USA 2006, 103, 3890–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaroop, A.; Xu, J.Z.; Pawar, H.; Jackson, A.; Skolnick, C.; Agarwal, N. A conserved retina-specific gene encodes a basic motif/leucine zipper domain. Proc. Natl. Acad. Sci. USA 1992, 89, 266–270. [Google Scholar] [CrossRef] [Green Version]
- MacLaren, R.E.; Pearson, R.A.; MacNeil, A.; Douglas, R.H.; Salt, T.E.; Akimoto, M.; Swaroop, A.; Sowden, J.C.; Ali, R.R. Retinal repair by transplantation of photoreceptor precursors. Nature 2006, 444, 203–207. [Google Scholar] [CrossRef]
- Gust, J.; Reh, T.A. Adult donor rod photoreceptors integrate into the mature mouse retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5266–5272. [Google Scholar] [CrossRef]
- Bartsch, U.; Oriyakhel, W.; Kenna, P.F.; Linke, S.; Richard, G.; Petrowitz, B.; Humphries, P.; Farrar, G.J.; Ader, M. Retinal cells integrate into the outer nuclear layer and differentiate into mature photoreceptors after subretinal transplantation into adult mice. Exp. Eye Res. 2008, 86, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.C.; Hippert, C.; Duran, Y.; West, E.L.; Bainbridge, J.W.; Warre-Cornish, K.; Luhmann, U.F.; Lakowski, J.; Sowden, J.C.; Ali, R.R.; et al. Repair of the degenerate retina by photoreceptor transplantation. Proc. Natl. Acad. Sci. USA 2013, 110, 354–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, R.A.; Hippert, C.; Graca, A.B.; Barber, A.C. Photoreceptor replacement therapy: Challenges presented by the diseased recipient retinal environment. Vis. Neurosci. 2014, 31, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.A.; Barber, A.C.; Rizzi, M.; Hippert, C.; Xue, T.; West, E.L.; Duran, Y.; Smith, A.J.; Chuang, J.Z.; Azam, S.A.; et al. Restoration of vision after transplantation of photoreceptors. Nature 2012, 485, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.S.; Charbel Issa, P.; Butler, R.; Martin, C.; Lipinski, D.M.; Sekaran, S.; Barnard, A.R.; MacLaren, R.E. Reversal of end-stage retinal degeneration and restoration of visual function by photoreceptor transplantation. Proc. Natl. Acad. Sci. USA 2013, 110, 1101–1106. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, Z.; Gu, P. Stem/progenitor cell-based transplantation for retinal degeneration: A review of clinical trials. Cell Death Dis. 2020, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Park, S.S.; Albini, T.A.; Canto-Soler, M.V.; Klassen, H.; MacLaren, R.E.; Takahashi, M.; Nagiel, A.; Schwartz, S.D.; Bharti, K. Retinal stem cell transplantation: Balancing safety and potential. Prog. Retin Eye Res. 2020, 75, 100779. [Google Scholar] [CrossRef] [PubMed]
- Barnea-Cramer, A.O.; Wang, W.; Lu, S.J.; Singh, M.S.; Luo, C.; Huo, H.; McClements, M.E.; Barnard, A.R.; MacLaren, R.E.; Lanza, R. Function of human pluripotent stem cell-derived photoreceptor progenitors in blind mice. Sci. Rep. 2016, 6, 29784. [Google Scholar] [CrossRef]
- Wahlin, K.J.; Maruotti, J.A.; Sripathi, S.R.; Ball, J.; Angueyra, J.M.; Kim, C.; Grebe, R.; Li, W.; Jones, B.W.; Zack, D.J. Photoreceptor Outer Segment-like Structures in Long-Term 3D Retinas from Human Pluripotent Stem Cells. Sci. Rep. 2017, 7, 766. [Google Scholar] [CrossRef]
- Gonzalez-Cordero, A.; Kruczek, K.; Naeem, A.; Fernando, M.; Kloc, M.; Ribeiro, J.; Goh, D.; Duran, Y.; Blackford, S.J.I.; Abelleira-Hervas, L.; et al. Recapitulation of Human Retinal Development from Human Pluripotent Stem Cells Generates Transplantable Populations of Cone Photoreceptors. Stem Cell Rep. 2017, 9, 820–837. [Google Scholar] [CrossRef] [Green Version]
- Lamba, D.A.; Gust, J.; Reh, T.A. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell 2009, 4, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagins, W.A.; Penn, R.D.; Yoshikami, S. Dark current and photocurrent in retinal rods. Biophys. J. 1970, 10, 380–412. [Google Scholar] [CrossRef] [Green Version]
- Gargini, C.; Demontis, G.C.; Cervetto, L.; Bisti, S. Analysis of pharmacologically isolated components of the ERG. Vision Res. 1999, 39, 1759–1766. [Google Scholar] [CrossRef] [Green Version]
- Breton, M.E.; Schueller, A.W.; Lamb, T.D.; Pugh, E.N., Jr. Analysis of ERG a-wave amplification and kinetics in terms of the G-protein cascade of phototransduction. Investig. Ophthalmol. Vis. Sci. 1994, 35, 295–309. [Google Scholar]
- Robson, J.G.; Frishman, L.J. The rod-driven a-wave of the dark-adapted mammalian electroretinogram. Prog. Retin Eye Res. 2014, 39, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.H.; Sanyal, S. Development and degeneration of retina in rds mutant mice: The electroretinogram. Neurosci. Lett. 1984, 48, 231–237. [Google Scholar] [CrossRef]
- Machida, S.; Kondo, M.; Jamison, J.A.; Khan, N.W.; Kononen, L.T.; Sugawara, T.; Bush, R.A.; Sieving, P.A. P23H rhodopsin transgenic rat: Correlation of retinal function with histopathology. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3200–3209. [Google Scholar]
- Pearson, R.A.; Gonzalez-Cordero, A.; West, E.L.; Ribeiro, J.R.; Aghaizu, N.; Goh, D.; Sampson, R.D.; Georgiadis, A.; Waldron, P.V.; Duran, Y.; et al. Donor and host photoreceptors engage in material transfer following transplantation of post-mitotic photoreceptor precursors. Nat. Commun. 2016, 7, 13029. [Google Scholar] [CrossRef] [Green Version]
- Santos-Ferreira, T.; Llonch, S.; Borsch, O.; Postel, K.; Haas, J.; Ader, M. Retinal transplantation of photoreceptors results in donor-host cytoplasmic exchange. Nat. Commun. 2016, 7, 13028. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.S.; Balmer, J.; Barnard, A.R.; Aslam, S.A.; Moralli, D.; Green, C.M.; Barnea-Cramer, A.; Duncan, I.; MacLaren, R.E. Transplanted photoreceptor precursors transfer proteins to host photoreceptors by a mechanism of cytoplasmic fusion. Nat. Commun. 2016, 7, 13537. [Google Scholar] [CrossRef] [Green Version]
- Ortin-Martinez, A.; Tsai, E.L.; Nickerson, P.E.; Bergeret, M.; Lu, Y.; Smiley, S.; Comanita, L.; Wallace, V.A. A Reinterpretation of Cell Transplantation: GFP Transfer From Donor to Host Photoreceptors. Stem Cells 2017, 35, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldron, P.V.; Di Marco, F.; Kruczek, K.; Ribeiro, J.; Graca, A.B.; Hippert, C.; Aghaizu, N.D.; Kalargyrou, A.A.; Barber, A.C.; Grimaldi, G.; et al. Transplanted Donor- or Stem Cell-Derived Cone Photoreceptors Can Both Integrate and Undergo Material Transfer in an Environment-Dependent Manner. Stem Cell Rep. 2018, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Decembrini, S.; Martin, C.; Sennlaub, F.; Chemtob, S.; Biel, M.; Samardzija, M.; Moulin, A.; Behar-Cohen, F.; Arsenijevic, Y. Cone Genesis Tracing by the Chrnb4-EGFP Mouse Line: Evidences of Cellular Material Fusion after Cone Precursor Transplantation. Mol. Ther. 2017, 25, 634–653. [Google Scholar] [CrossRef] [Green Version]
- Zou, T.; Gao, L.; Zeng, Y.; Li, Q.; Li, Y.; Chen, S.; Hu, X.; Chen, X.; Fu, C.; Xu, H.; et al. Organoid-derived C-Kit+/SSEA4− human retinal progenitor cells promote a protective retinal microenvironment during transplantation in rodents. Nat. Commun. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Gupta, T.; Kapoor, K.; Sahni, D.; Singh, B. Mapping the Time Line of Development in Each Layer of Human Foetal Retina. J. Clin. Diagn. Res. 2016, 10, AC04–AC07. [Google Scholar] [CrossRef]
- Hendrickson, A.; Bumsted-O’Brien, K.; Natoli, R.; Ramamurthy, V.; Possin, D.; Provis, J. Rod photoreceptor differentiation in fetal and infant human retina. Exp. Eye Res. 2008, 87, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberle, D.; Schubert, S.; Postel, K.; Corbeil, D.; Ader, M. Increased integration of transplanted CD73-positive photoreceptor precursors into adult mouse retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6462–6471. [Google Scholar] [CrossRef]
- Lakowski, J.; Han, Y.T.; Pearson, R.A.; Gonzalez-Cordero, A.; West, E.L.; Gualdoni, S.; Barber, A.C.; Hubank, M.; Ali, R.R.; Sowden, J.C. Effective transplantation of photoreceptor precursor cells selected via cell surface antigen expression. Stem Cells 2011, 29, 1391–1404. [Google Scholar] [CrossRef] [Green Version]
- Lakowski, J.; Gonzalez-Cordero, A.; West, E.L.; Han, Y.T.; Welby, E.; Naeem, A.; Blackford, S.J.; Bainbridge, J.W.; Pearson, R.A.; Ali, R.R.; et al. Transplantation of Photoreceptor Precursors Isolated via a Cell Surface Biomarker Panel From Embryonic Stem Cell-Derived Self-Forming Retina. Stem Cells 2015, 33, 2469–2482. [Google Scholar] [CrossRef] [Green Version]
- Lakowski, J.; Welby, E.; Budinger, D.; Di Marco, F.; Di Foggia, V.; Bainbridge, J.W.B.; Wallace, K.; Gamm, D.M.; Ali, R.R.; Sowden, J.C. Isolation of Human Photoreceptor Precursors via a Cell Surface Marker Panel from Stem Cell-Derived Retinal Organoids and Fetal Retinae. Stem Cells 2018, 36, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, J.; Procyk, C.A.; West, E.L.; O’Hara-Wright, M.; Martins, M.F.; Khorasani, M.M.; Hare, A.; Basche, M.; Fernando, M.; Goh, D.; et al. Restoration of visual function in advanced disease after transplantation of purified human pluripotent stem cell-derived cone photoreceptors. Cell Rep. 2021, 35, 109022. [Google Scholar] [CrossRef] [PubMed]
- Engle, S.J.; Vincent, F. Small molecule screening in human induced pluripotent stem cell-derived terminal cell types. J. Biol. Chem. 2014, 289, 4562–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elitt, M.S.; Barbar, L.; Tesar, P.J. Drug screening for human genetic diseases using iPSC models. Hum. Mol. Genet. 2018, 27, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Xie, B.; He, L.; Zhou, T.; Gao, G.; Liu, S.; Pan, G.; Ge, J.; Peng, F.; Zhong, X. Generation of Retinal Organoids with Mature Rods and Cones from Urine-Derived Human Induced Pluripotent Stem Cells. Stem Cells Int. 2018, 2018, 4968658. [Google Scholar] [CrossRef]
- Canto-Soler, V.; Flores-Bellver, M.; Vergara, M.N. Stem Cell Sources and Their Potential for the Treatment of Retinal Degenerations. Investig. Ophthalmol. Vis. Sci. 2016, 57, ORSFd1–ORSFd9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Cao, J.; Li, M.; Hoffmann, B.; Xu, K.; Chen, J.; Lu, X.; Guo, F.; Li, X.; Phillips, M.J.; et al. PAX6D instructs neural retinal specification from human embryonic stem cell-derived neuroectoderm. EMBO Rep. 2020, 21, e50000. [Google Scholar] [CrossRef]
- Gamm, D.M.; Clark, E.; Capowski, E.E.; Singh, R. The Role of FGF9 in the Production of Neural Retina and RPE in a Pluripotent Stem Cell Model of Early Human Retinal Development. Am. J. Ophthalmol. 2019, 206, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Corral-Serrano, J.C.; Lamers, I.J.C.; van Reeuwijk, J.; Duijkers, L.; Hoogendoorn, A.D.M.; Yildirim, A.; Argyrou, N.; Ruigrok, R.A.A.; Letteboer, S.J.F.; Butcher, R.; et al. PCARE and WASF3 regulate ciliary F-actin assembly that is required for the initiation of photoreceptor outer segment disk formation. Proc. Natl. Acad. Sci. USA 2020, 117, 9922–9931. [Google Scholar] [CrossRef] [Green Version]
- Paquet-Durand, F.; Hauck, S.M.; van Veen, T.; Ueffing, M.; Ekstrom, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef]
- Xu, J.; Morris, L.; Thapa, A.; Ma, H.; Michalakis, S.; Biel, M.; Baehr, W.; Peshenko, I.V.; Dizhoor, A.M.; Ding, X.Q. cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: Evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J. Neurosci. 2013, 33, 14939–14948. [Google Scholar] [CrossRef] [Green Version]
- Paquet-Durand, F.; Beck, S.; Michalakis, S.; Goldmann, T.; Huber, G.; Muhlfriedel, R.; Trifunovic, D.; Fischer, M.D.; Fahl, E.; Duetsch, G.; et al. A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet. 2011, 20, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Vighi, E.; Trifunovic, D.; Veiga-Crespo, P.; Rentsch, A.; Hoffmann, D.; Sahaboglu, A.; Strasser, T.; Kulkarni, M.; Bertolotti, E.; van den Heuvel, A.; et al. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2997–E3006. [Google Scholar] [CrossRef] [Green Version]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A look into retinal organoids: Methods, analytical techniques, and applications. Cell Mol. Life Sci. 2021, 1–28. [Google Scholar] [CrossRef]
- Assawachananont, J.; Mandai, M.; Okamoto, S.; Yamada, C.; Eiraku, M.; Yonemura, S.; Sasai, Y.; Takahashi, M. Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Rep. 2014, 2, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Shirai, H.; Mandai, M.; Matsushita, K.; Kuwahara, A.; Yonemura, S.; Nakano, T.; Assawachananont, J.; Kimura, T.; Saito, K.; Terasaki, H.; et al. Transplantation of human embryonic stem cell-derived retinal tissue in two primate models of retinal degeneration. Proc. Natl. Acad. Sci. USA 2016, 113, E81–E90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decembrini, S.; Koch, U.; Radtke, F.; Moulin, A.; Arsenijevic, Y. Derivation of traceable and transplantable photoreceptors from mouse embryonic stem cells. Stem Cell Rep. 2014, 2, 853–865. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Cordero, A.; West, E.L.; Pearson, R.A.; Duran, Y.; Carvalho, L.S.; Chu, C.J.; Naeem, A.; Blackford, S.J.I.; Georgiadis, A.; Lakowski, J.; et al. Photoreceptor precursors derived from three-dimensional embryonic stem cell cultures integrate and mature within adult degenerate retina. Nat. Biotechnol. 2013, 31, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scruggs, B.A.; Jiao, C.; Cranston, C.M.; Kaalberg, E.; Wang, K.; Russell, S.R.; Wiley, L.A.; Mullins, R.F.; Stone, E.M.; Tucker, B.A.; et al. Optimizing Donor Cellular Dissociation and Subretinal Injection Parameters for Stem Cell-Based Treatments. Stem Cells Transl. Med. 2019, 8, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Andreazzoli, M. Molecular regulation of vertebrate retina cell fate. Birth Defects Res. C Embryo Today 2009, 87, 284–295. [Google Scholar] [CrossRef]
- Stenkamp, D.L. Development of the Vertebrate Eye and Retina. Prog. Mol. Biol. Transl. Sci. 2015, 134, 397–414. [Google Scholar] [CrossRef] [Green Version]
- Lan, L.; Vitobello, A.; Bertacchi, M.; Cremisi, F.; Vignali, R.; Andreazzoli, M.; Demontis, G.C.; Barsacchi, G.; Casarosa, S. Noggin elicits retinal fate in Xenopus animal cap embryonic stem cells. Stem Cells 2009, 27, 2146–2152. [Google Scholar] [CrossRef]
- Viczian, A.S.; Solessio, E.C.; Lyou, Y.; Zuber, M.E. Generation of functional eyes from pluripotent cells. PLoS Biol. 2009, 7, e1000174. [Google Scholar] [CrossRef] [Green Version]
- Lamba, D.A.; Karl, M.O.; Ware, C.B.; Reh, T.A. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 12769–12774. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwahara, A.; Ozone, C.; Nakano, T.; Saito, K.; Eiraku, M.; Sasai, Y. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 2015, 6, 6286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.A.; Monville, C.; Goureau, O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. USA 2014, 111, 8518–8523. [Google Scholar] [CrossRef] [Green Version]
- Mellough, C.B.; Collin, J.; Khazim, M.; White, K.; Sernagor, E.; Steel, D.H.; Lako, M. IGF-1 Signaling Plays an Important Role in the Formation of Three-Dimensional Laminated Neural Retina and Other Ocular Structures From Human Embryonic Stem Cells. Stem Cells 2015, 33, 2416–2430. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiler, D.; Chen, X.; Hazen, J.; Kupriyanov, S.; Carroll, P.A.; Qu, C.; Xu, B.; Johnson, D.; Griffiths, L.; Frase, S.; et al. Quantification of Retinogenesis in 3D Cultures Reveals Epigenetic Memory and Higher Efficiency in iPSCs Derived from Rod Photoreceptors. Cell Stem Cell 2015, 17, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellough, C.B.; Collin, J.; Queen, R.; Hilgen, G.; Dorgau, B.; Zerti, D.; Felemban, M.; White, K.; Sernagor, E.; Lako, M. Systematic Comparison of Retinal Organoid Differentiation from Human Pluripotent Stem Cells Reveals Stage Specific, Cell Line, and Methodological Differences. Stem Cells Transl. Med. 2019, 8, 694–706. [Google Scholar] [CrossRef] [Green Version]
- Chichagova, V.; Hilgen, G.; Ghareeb, A.; Georgiou, M.; Carter, M.; Sernagor, E.; Lako, M.; Armstrong, L. Human iPSC differentiation to retinal organoids in response to IGF1 and BMP4 activation is line- and method-dependent. Stem Cells 2020, 38, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, C.S.; Renner, M.; De Gennaro, M.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikra, T.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e1634. [Google Scholar] [CrossRef]
- Mandai, M.; Fujii, M.; Hashiguchi, T.; Sunagawa, G.A.; Ito, S.I.; Sun, J.; Kaneko, J.; Sho, J.; Yamada, C.; Takahashi, M. iPSC-Derived Retina Transplants Improve Vision in rd1 End-Stage Retinal-Degeneration Mice. Stem Cell Rep. 2017, 8, 69–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkner, M.; Zschatzsch, M.; Rostovskaya, M.; Overall, R.W.; Busskamp, V.; Anastassiadis, K.; Karl, M.O. Retinal Organoids from Pluripotent Stem Cells Efficiently Recapitulate Retinogenesis. Stem Cell Rep. 2016, 6, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Kaya, K.D.; Chen, H.Y.; Brooks, M.J.; Kelley, R.A.; Shimada, H.; Nagashima, K.; de Val, N.; Drinnan, C.T.; Gieser, L.; Kruczek, K.; et al. Transcriptome-based molecular staging of human stem cell-derived retinal organoids uncovers accelerated photoreceptor differentiation by 9-cis retinal. Mol. Vis. 2019, 25, 663–678. [Google Scholar]
- Felemban, M.; Dorgau, B.; Hunt, N.C.; Hallam, D.; Zerti, D.; Bauer, R.; Ding, Y.; Collin, J.; Steel, D.; Krasnogor, N.; et al. Extracellular matrix component expression in human pluripotent stem cell-derived retinal organoids recapitulates retinogenesis in vivo and reveals an important role for IMPG1 and CD44 in the development of photoreceptors and interphotoreceptor matrix. Acta Biomater. 2018, 74, 207–221. [Google Scholar] [CrossRef]
- Dorgau, B.; Felemban, M.; Hilgen, G.; Kiening, M.; Zerti, D.; Hunt, N.C.; Doherty, M.; Whitfield, P.; Hallam, D.; White, K.; et al. Decellularised extracellular matrix-derived peptides from neural retina and retinal pigment epithelium enhance the expression of synaptic markers and light responsiveness of human pluripotent stem cell derived retinal organoids. Biomaterials 2019, 199, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.C.; Hallam, D.; Chichagova, V.; Steel, D.H.; Lako, M. The Application of Biomaterials to Tissue Engineering Neural Retina and Retinal Pigment Epithelium. Adv. Healthcare Mater. 2018, 7, e1800226. [Google Scholar] [CrossRef]
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Hallam, D.; Hilgen, G.; Dorgau, B.; Zhu, L.; Yu, M.; Bojic, S.; Hewitt, P.; Schmitt, M.; Uteng, M.; Kustermann, S.; et al. Human-Induced Pluripotent Stem Cells Generate Light Responsive Retinal Organoids with Variable and Nutrient-Dependent Efficiency. Stem Cells 2018, 36, 1535–1551. [Google Scholar] [CrossRef] [Green Version]
- Kruczek, K.; Gonzalez-Cordero, A.; Goh, D.; Naeem, A.; Jonikas, M.; Blackford, S.J.I.; Kloc, M.; Duran, Y.; Georgiadis, A.; Sampson, R.D.; et al. Differentiation and Transplantation of Embryonic Stem Cell-Derived Cone Photoreceptors into a Mouse Model of End-Stage Retinal Degeneration. Stem Cell Rep. 2017, 8, 1659–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, S.; Cepko, C.L. Fgf8 Expression and Degradation of Retinoic Acid Are Required for Patterning a High-Acuity Area in the Retina. Dev. Cell 2017, 42, 68–81.e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daum, J.M.; Keles, O.; Holwerda, S.J.; Kohler, H.; Rijli, F.M.; Stadler, M.; Roska, B. The formation of the light-sensing compartment of cone photoreceptors coincides with a transcriptional switch. eLife 2017, 6, e31437. [Google Scholar] [CrossRef]
- Brooks, M.J.; Chen, H.Y.; Kelley, R.A.; Mondal, A.K.; Nagashima, K.; De Val, N.; Li, T.; Chaitankar, V.; Swaroop, A. Improved Retinal Organoid Differentiation by Modulating Signaling Pathways Revealed by Comparative Transcriptome Analyses with Development In Vivo. Stem Cell Rep. 2019, 13, 891–905. [Google Scholar] [CrossRef] [Green Version]
- Busskamp, V.; Krol, J.; Nelidova, D.; Daum, J.; Szikra, T.; Tsuda, B.; Juttner, J.; Farrow, K.; Scherf, B.G.; Alvarez, C.P.; et al. miRNAs 182 and 183 are necessary to maintain adult cone photoreceptor outer segments and visual function. Neuron 2014, 83, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Zuzic, M.; Rojo Arias, J.E.; Wohl, S.G.; Busskamp, V. Retinal miRNA Functions in Health and Disease. Genes 2019, 10, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Krol, I.; Alvarez, C.P.; Fiscella, M.; Hierlemann, A.; Roska, B.; Filipowicz, W. A network comprising short and long noncoding RNAs and RNA helicase controls mouse retina architecture. Nat. Commun. 2015, 6, 7305. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.C.; Gamm, D.M. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.J.; Wallace, K.A.; Dickerson, S.J.; Miller, M.J.; Verhoeven, A.D.; Martin, J.M.; Wright, L.S.; Shen, W.; Capowski, E.E.; Percin, E.F.; et al. Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2007–2019. [Google Scholar] [CrossRef] [PubMed]
- Baylor, D.A.; Lamb, T.D.; Yau, K.W. The membrane current of single rod outer segments. J. Physiol. 1979, 288, 589–611. [Google Scholar] [CrossRef]
- Baylor, D.A.; Nunn, B.J.; Schnapf, J.L. The photocurrent, noise and spectral sensitivity of rods of the monkey Macaca fascicularis. J. Physiol. 1984, 357, 575–607. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.W.; Schneeweis, D.M.; Schnapf, J.L. Visual transduction in human rod photoreceptors. J. Physiol. 1993, 464, 747–765. [Google Scholar] [CrossRef]
- Biel, M.; Wahl-Schott, C.; Michalakis, S.; Zong, X. Hyperpolarization-activated cation channels: From genes to function. Physiol. Rev. 2009, 89, 847–885. [Google Scholar] [CrossRef] [Green Version]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [Green Version]
- Barravecchia, I.; Demontis, G.C. HCN1 channels: A versatile tool for signal processing by primary sensory neurons. Prog. Biophys. Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Seeliger, M.W.; Brombas, A.; Weiler, R.; Humphries, P.; Knop, G.; Tanimoto, N.; Muller, F. Modulation of rod photoreceptor output by HCN1 channels is essential for regular mesopic cone vision. Nat. Commun. 2011, 2, 532. [Google Scholar] [CrossRef]
- Frech, G.C.; VanDongen, A.M.; Schuster, G.; Brown, A.M.; Joho, R.H. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature 1989, 340, 642–645. [Google Scholar] [CrossRef]
- Czirják, G.; Tóth, Z.E.; Enyedi, P. Characterization of the Heteromeric Potassium Channel Formed by Kv2.1 and the Retinal Subunit Kv8.2 in Xenopus Oocytes. J. Neurophysiol. 2007, 98, 1213–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beech, D.J.; Barnes, S. Characterization of a voltage-gated K+ channel that accelerates the rod response to dim light. Neuron 1989, 3, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fehlhaber, K.E.; Sarria, I.; Cao, Y.; Ingram, N.T.; Guerrero-Given, D.; Throesch, B.; Baldwin, K.; Kamasawa, N.; Ohtsuka, T.; et al. The Auxiliary Calcium Channel Subunit alpha2delta4 Is Required for Axonal Elaboration, Synaptic Transmission, and Wiring of Rod Photoreceptors. Neuron 2017, 93, 1359–1374.e1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, C.A.; Jiang, D.; Mackin, R.D.; Samuel, M.A. Development and maintenance of vision’s first synapse. Dev. Biol. 2021, 476, 218–239. [Google Scholar] [CrossRef]
- Van Hook, M.J.; Nawy, S.; Thoreson, W.B. Voltage- and calcium-gated ion channels of neurons in the vertebrate retina. Prog. Retin Eye Res. 2019, 72, 100760. [Google Scholar] [CrossRef]
- Caputo, A.; Piano, I.; Demontis, G.C.; Bacchi, N.; Casarosa, S.; Della Santina, L.; Gargini, C. TMEM16A is associated with voltage-gated calcium channels in mouse retina and its function is disrupted upon mutation of the auxiliary alpha2delta4 subunit. Front. Cell Neurosci. 2015, 9, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslanidis, A.; Karlstetter, M.; Walczak, Y.; Jagle, H.; Langmann, T. RETINA-specific expression of Kcnv2 is controlled by cone-rod homeobox (Crx) and neural retina leucine zipper (Nrl). Adv. Exp. Med. Biol. 2014, 801, 31–41. [Google Scholar] [CrossRef]
- Sisak, S.; Banin, E.; Blumenthal, E.Z. A two-compartment model of the human retina. Med. Hypotheses 2004, 62, 808–816. [Google Scholar] [CrossRef]
- Zhang, P.; Shibata, B.; Peinado, G.; Zawadzki, R.J.; FitzGerald, P.; Pugh, E.N., Jr. Measurement of Diurnal Variation in Rod Outer Segment Length In Vivo in Mice With the OCT Optoretinogram. Investig. Ophthalmol. Vis. Sci. 2020, 61, 9. [Google Scholar] [CrossRef]
- Williams, D.S.; Arikawa, K.; Paallysaho, T. Cytoskeletal components of the adherens junctions between the photoreceptors and the supportive Muller cells. J. Comp. Neurol. 1990, 295, 155–164. [Google Scholar] [CrossRef]
- Farquhar, M.G.; Palade, G.E. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [Green Version]
- Paffenholz, R.; Kuhn, C.; Grund, C.; Stehr, S.; Franke, W.W. The arm-repeat protein NPRAP (neurojungin) is a constituent of the plaques of the outer limiting zone in the retina, defining a novel type of adhering junction. Exp. Cell Res. 1999, 250, 452–464. [Google Scholar] [CrossRef]
- Uga, S.; Smelser, G.K. Electron microscopic study of the development of retinal Mullerian cells. Investig. Ophthalmol. Vis. Sci. 1973, 12, 295–307. [Google Scholar]
- Den Hollander, A.I.; ten Brink, J.B.; de Kok, Y.J.; van Soest, S.; van den Born, L.I.; van Driel, M.A.; van de Pol, D.J.; Payne, A.M.; Bhattacharya, S.S.; Kellner, U.; et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat. Genet. 1999, 23, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Bunt-Milam, A.H.; Saari, J.C.; Klock, I.B.; Garwin, G.G. Zonulae adherentes pore size in the external limiting membrane of the rabbit retina. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1377–1380. [Google Scholar] [PubMed]
- Asayama, K. In vivo study on the absorption of the subretianl fluid. 2. Studies on an absorption of tracers (I125.human serum albumin and lanthanum nitrate) injected between the sensory retina and the pigment epithelium layer (author’s transl). Nippon Ganka Gakkai Zasshi 1976, 80, 598–607. [Google Scholar]
- Takeuchi, A.; Kricorian, G.; Marmor, M.F. Albumin movement out of the subretinal space after experimental retinal detachment. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1298–1305. [Google Scholar] [PubMed]
- Takeuchi, A.; Kricorian, G.; Yao, X.Y.; Kenny, J.W.; Marmor, M.F. The rate and source of albumin entry into saline-filled experimental retinal detachments. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3792–3798. [Google Scholar]
- Carter-Dawson, L.; Burroughs, M. Interphotoreceptor retinoid-binding protein in the cone matrix sheath. Electron microscopic immunocytochemical localization. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1584–1588. [Google Scholar]
- Chifflet, S.; Correa, V.; Nin, V.; Justet, C.; Hernandez, J.A. Effect of membrane potential depolarization on the organization of the actin cytoskeleton of eye epithelia. The role of adherens junctions. Exp. Eye Res. 2004, 79, 769–777. [Google Scholar] [CrossRef]
- Van de Pavert, S.A.; Kantardzhieva, A.; Malysheva, A.; Meuleman, J.; Versteeg, I.; Levelt, C.; Klooster, J.; Geiger, S.; Seeliger, M.W.; Rashbass, P.; et al. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J. Cell Sci. 2004, 117, 4169–4177. [Google Scholar] [CrossRef] [Green Version]
- Koike, C.; Nishida, A.; Akimoto, K.; Nakaya, M.A.; Noda, T.; Ohno, S.; Furukawa, T. Function of atypical protein kinase C lambda in differentiating photoreceptors is required for proper lamination of mouse retina. J. Neurosci. 2005, 25, 10290–10298. [Google Scholar] [CrossRef] [PubMed]
- Daniele, L.L.; Adams, R.H.; Durante, D.E.; Pugh, E.N., Jr.; Philp, N.J. Novel distribution of junctional adhesion molecule-C in the neural retina and retinal pigment epithelium. J. Comp. Neurol. 2007, 505, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Omri, S.; Omri, B.; Savoldelli, M.; Jonet, L.; Thillaye-Goldenberg, B.; Thuret, G.; Gain, P.; Jeanny, J.C.; Crisanti, P.; Behar-Cohen, F. The outer limiting membrane (OLM) revisited: Clinical implications. Clin. Ophthalmol. 2010, 4, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Kantardzhieva, A.; Gosens, I.; Alexeeva, S.; Punte, I.M.; Versteeg, I.; Krieger, E.; Neefjes-Mol, C.A.; den Hollander, A.I.; Letteboer, S.J.; Klooster, J.; et al. MPP5 recruits MPP4 to the CRB1 complex in photoreceptors. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2192–2201. [Google Scholar] [CrossRef] [Green Version]
- Gosens, I.; den Hollander, A.I.; Cremers, F.P.; Roepman, R. Composition and function of the Crumbs protein complex in the mammalian retina. Exp. Eye Res. 2008, 86, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Pellikka, M.; Tanentzapf, G.; Pinto, M.; Smith, C.; McGlade, C.J.; Ready, D.F.; Tepass, U. Crumbs, the Drosophila homologue of human CRB1/RP12, is essential for photoreceptor morphogenesis. Nature 2002, 416, 143–149. [Google Scholar] [CrossRef]
- Tepass, U. Crumbs, a component of the apical membrane, is required for zonula adherens formation in primary epithelia of Drosophila. Dev. Biol. 1996, 177, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.J.; Wijnholds, J. Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective. Genes 2019, 10, 987. [Google Scholar] [CrossRef] [Green Version]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Reports 2018, 10, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Volkner, M.; Kurth, T.; Schor, J.; Ebner, L.J.A.; Bardtke, L.; Kavak, C.; Hackermuller, J.; Karl, M.O. Mouse Retinal Organoid Growth and Maintenance in Longer-Term Culture. Front. Cell Dev. Biol. 2021, 9, 645704. [Google Scholar] [CrossRef]
- Lukovic, D.; Artero Castro, A.; Kaya, K.D.; Munezero, D.; Gieser, L.; Davo-Martinez, C.; Corton, M.; Cuenca, N.; Swaroop, A.; Ramamurthy, V.; et al. Retinal Organoids derived from hiPSCs of an AIPL1-LCA Patient Maintain Cytoarchitecture despite Reduced levels of Mutant AIPL1. Sci. Rep. 2020, 10, 5426. [Google Scholar] [CrossRef]
- Meschede, I.P.; Ovenden, N.C.; Seabra, M.C.; Futter, C.E.; Votruba, M.; Cheetham, M.E.; Burgoyne, T. Symmetric arrangement of mitochondria:plasma membrane contacts between adjacent photoreceptor cells regulated by Opa1. Proc. Natl. Acad. Sci. USA 2020, 117, 15684–15693. [Google Scholar] [CrossRef]
- Hanein, S.; Perrault, I.; Gerber, S.; Tanguy, G.; Barbet, F.; Ducroq, D.; Calvas, P.; Dollfus, H.; Hamel, C.; Lopponen, T.; et al. Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum. Mutat. 2004, 23, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yzer, S.; Leroy, B.P.; De Baere, E.; de Ravel, T.J.; Zonneveld, M.N.; Voesenek, K.; Kellner, U.; Ciriano, J.P.; de Faber, J.T.; Rohrschneider, K.; et al. Microarray-based mutation detection and phenotypic characterization of patients with Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1167–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, G.J.; Clarke, S.; Davis, J.A.; Simpson, D.A.; Silvestri, G. Pigmented paravenous chorioretinal atrophy is associated with a mutation within the crumbs homolog 1 (CRB1) gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, A.G.; Aartsen, W.M.; Meuleman, J.; Klooster, J.; Malysheva, A.; Versteeg, I.; Arsanto, J.P.; Le Bivic, A.; Wijnholds, J. Pals1/Mpp5 is required for correct localization of Crb1 at the subapical region in polarized Muller glia cells. Hum. Mol. Genet. 2006, 15, 2659–2672. [Google Scholar] [CrossRef]
- Mehalow, A.K.; Kameya, S.; Smith, R.S.; Hawes, N.L.; Denegre, J.M.; Young, J.A.; Bechtold, L.; Haider, N.B.; Tepass, U.; Heckenlively, J.R.; et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet. 2003, 12, 2179–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Weng, K.; Liang, J.; Zhou, Y.; Hao, Q.; Hao, Y.; Yao, K.; Zou, J. The extracellular and intracellular regions of Crb2a play distinct roles in guiding the formation of the apical zonula adherens. Biomed. Pharmacother. 2020, 125, 109942. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Alves, C.H.; Quinn, P.M.; Vos, R.M.; Tanimoto, N.; Lundvig, D.M.; Dudok, J.J.; Hooibrink, B.; Richard, F.; Beck, S.C.; et al. Targeted ablation of CRB1 and CRB2 in retinal progenitor cells mimics Leber congenital amaurosis. PLoS Genet. 2013, 9, e1003976. [Google Scholar] [CrossRef] [PubMed]
- Caceres, P.S.; Rodriguez-Boulan, E. Retinal pigment epithelium polarity in health and blinding diseases. Curr. Opin. Cell Biol. 2020, 62, 37–45. [Google Scholar] [CrossRef]
- Naylor, A.; Hopkins, A.; Hudson, N.; Campbell, M. Tight Junctions of the Outer Blood Retina Barrier. Int. J. Mol. Sci. 2019, 21, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzoni, G.; Martinez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef] [Green Version]
- Farjood, F.; Vargis, E. Physical disruption of cell-cell contact induces VEGF expression in RPE cells. Mol. Vis. 2017, 23, 431–446. [Google Scholar] [PubMed]
- Marneros, A.G.; Fan, J.; Yokoyama, Y.; Gerber, H.P.; Ferrara, N.; Crouch, R.K.; Olsen, B.R. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.B.; Xu, T.; Peng, S.; Singh, D.; Ghiassi-Nejad, M.; Adelman, R.A.; Rizzolo, L.J. Disease-associated mutations of claudin-19 disrupt retinal neurogenesis and visual function. Commun. Biol. 2019, 2, 113. [Google Scholar] [CrossRef] [PubMed]
- Konrad, M.; Schaller, A.; Seelow, D.; Pandey, A.V.; Waldegger, S.; Lesslauer, A.; Vitzthum, H.; Suzuki, Y.; Luk, J.M.; Becker, C.; et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am. J. Hum. Genet. 2006, 79, 949–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Geathers, J.S.; Grillo, S.L.; Weber, S.R.; Wang, W.; Zhao, Y.; Sundstrom, J.M. Role of Epithelial-Mesenchymal Transition in Retinal Pigment Epithelium Dysfunction. Front. Cell Dev. Biol. 2020, 8, 501. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Li, Y.; Thompson, D.A.; Weir, J.; Orth, U.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Vollrath, D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic reliance promotes anabolism in photoreceptors. eLife 2017, 6, e25946. [Google Scholar] [CrossRef] [PubMed]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6, e28899. [Google Scholar] [CrossRef]
- Jonnal, R.S.; Besecker, J.R.; Derby, J.C.; Kocaoglu, O.P.; Cense, B.; Gao, W.; Wang, Q.; Miller, D.T. Imaging outer segment renewal in living human cone photoreceptors. Opt. Express 2010, 18, 5257–5270. [Google Scholar] [CrossRef] [Green Version]
- Winkler, B.S. Glycolytic and oxidative metabolism in relation to retinal function. J. Gen. Physiol. 1981, 77, 667–692. [Google Scholar] [CrossRef] [Green Version]
- Linsenmeier, R.A. Effects of light and darkness on oxygen distribution and consumption in the cat retina. J. Gen. Physiol. 1986, 88, 521–542. [Google Scholar] [CrossRef] [Green Version]
- Winkler, B.S.; Starnes, C.A.; Twardy, B.S.; Brault, D.; Taylor, R.C. Nuclear magnetic resonance and biochemical measurements of glucose utilization in the cone-dominant ground squirrel retina. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4613–4619. [Google Scholar] [CrossRef] [Green Version]
- Nickla, D.L.; Wallman, J. The multifunctional choroid. Prog. Retin Eye Res. 2010, 29, 144–168. [Google Scholar] [CrossRef] [Green Version]
- Linsenmeier, R.A.; Zhang, H.F. Retinal oxygen: From animals to humans. Prog. Retin Eye Res. 2017, 58, 115–151. [Google Scholar] [CrossRef] [Green Version]
- Swarup, A.; Samuels, I.S.; Bell, B.A.; Han, J.Y.S.; Du, J.; Massenzio, E.; Abel, E.D.; Boesze-Battaglia, K.; Peachey, N.S.; Philp, N.J. Modulating GLUT1 expression in retinal pigment epithelium decreases glucose levels in the retina: Impact on photoreceptors and Muller glial cells. Am. J. Physiol. Cell Physiol. 2019, 316, C121–C133. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J.; Wood, J.P.; Han, G.; Kittipassorn, T.; Peet, D.J.; Chidlow, G. M-Type Pyruvate Kinase Isoforms and Lactate Dehydrogenase A in the Mammalian Retina: Metabolic Implications. Investig. Ophthalmol. Vis. Sci. 2016, 57, 66–80. [Google Scholar] [CrossRef]
- Rueda, E.M.; Johnson, J.E., Jr.; Giddabasappa, A.; Swaroop, A.; Brooks, M.J.; Sigel, I.; Chaney, S.Y.; Fox, D.A. The cellular and compartmental profile of mouse retinal glycolysis, tricarboxylic acid cycle, oxidative phosphorylation, and ~P transferring kinases. Mol. Vis. 2016, 22, 847–885. [Google Scholar] [PubMed]
- Ait-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clerin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, X.; Chaffiol, A.; Kole, C.; Yang, Y.; Millet-Puel, G.; Clerin, E.; Ait-Ali, N.; Bennett, J.; Dalkara, D.; Sahel, J.A.; et al. The Thioredoxin Encoded by the Rod-Derived Cone Viability Factor Gene Protects Cone Photoreceptors Against Oxidative Stress. Antioxid Redox Signal. 2016, 24, 909–923. [Google Scholar] [CrossRef]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrustegui, J.; Hurley, J.B. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demontis, G.C.; Ratto, G.M.; Bisti, S.; Cervetto, L. Effect of blocking the Na+/K+ ATPase on Ca2+ extrusion and light adaptation in mammalian retinal rods. Biophys. J. 1995, 69, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Demontis, G.C.; Longoni, B.; Gargini, C.; Cervetto, L. The energetic cost of photoreception in retinal rods of mammals. Arch. Ital. Biol. 1997, 135, 95–109. [Google Scholar] [PubMed]
- Okawa, H.; Sampath, A.P.; Laughlin, S.B.; Fain, G.L. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr. Biol. 2008, 18, 1917–1921. [Google Scholar] [CrossRef] [Green Version]
- Petit, L.; Ma, S.; Cipi, J.; Cheng, S.Y.; Zieger, M.; Hay, N.; Punzo, C. Aerobic Glycolysis Is Essential for Normal Rod Function and Controls Secondary Cone Death in Retinitis Pigmentosa. Cell Rep. 2018, 23, 2629–2642. [Google Scholar] [CrossRef] [Green Version]
- Rajala, R.V.S. Aerobic Glycolysis in the Retina: Functional Roles of Pyruvate Kinase Isoforms. Front. Cell Dev. Biol. 2020, 8, 266. [Google Scholar] [CrossRef]
- Rajala, R.V.; Rajala, A.; Kooker, C.; Wang, Y.; Anderson, R.E. The Warburg Effect Mediator Pyruvate Kinase M2 Expression and Regulation in the Retina. Sci. Rep. 2016, 6, 37727. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Yang, H.J.; Brooks, M.J.; Zelinger, L.; Karakulah, G.; Gotoh, N.; Boleda, A.; Gieser, L.; Giuste, F.; Whitaker, D.T.; et al. NRL-Regulated Transcriptome Dynamics of Developing Rod Photoreceptors. Cell Rep. 2016, 17, 2460–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratto, G.M.; Robinson, D.W.; Yan, B.; McNaughton, P.A. Development of the light response in neonatal mammalian rods. Nature 1991, 351, 654–657. [Google Scholar] [CrossRef]
- Ashizawa, K.; Willingham, M.C.; Liang, C.M.; Cheng, S.Y. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J. Biol. Chem. 1991, 266, 16842–16846. [Google Scholar] [CrossRef]
- Dombrauckas, J.D.; Santarsiero, B.D.; Mesecar, A.D. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005, 44, 9417–9429. [Google Scholar] [CrossRef]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, H.P.; O’Reilly, F.J.; Wear, M.A.; O’Neill, J.R.; Fothergill-Gilmore, L.A.; Hupp, T.; Walkinshaw, M.D. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc. Natl. Acad. Sci. USA 2013, 110, 5881–5886. [Google Scholar] [CrossRef] [Green Version]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Semenza, G.L. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2011, 2, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Kinkl, N.; Hageman, G.S.; Sahel, J.A.; Hicks, D. Fibroblast growth factor receptor (FGFR) and candidate signaling molecule distribution within rat and human retina. Mol. Vis. 2002, 8, 149–160. [Google Scholar]
- Valter, K.; van Driel, D.; Bisti, S.; Stone, J. FGFR1 expression and FGFR1-FGF-2 colocalisation in rat retina: Sites of FGF-2 action on rat photoreceptors. Growth Factors 2002, 20, 177–188. [Google Scholar] [CrossRef]
- Rajala, A.; Wang, Y.; Brush, R.S.; Tsantilas, K.; Jankowski, C.S.R.; Lindsay, K.J.; Linton, J.D.; Hurley, J.B.; Anderson, R.E.; Rajala, R.V.S. Pyruvate kinase M2 regulates photoreceptor structure, function, and viability. Cell Death Dis. 2018, 9, 240. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Cui, J.; Du, J.; Wei, D.; Jia, Z.; Zhang, J.; Zhu, Z.; Gao, Y.; Xie, K. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin. Cancer Res. 2014, 20, 4370–4380. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [Green Version]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J. Biol. Chem. 1995, 270, 21021–21027. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 1985 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.Y.; Cringle, S.J. Oxygen distribution in the mouse retina. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [Green Version]
- Wubben, T.J.; Pawar, M.; Smith, A.; Toolan, K.; Hager, H.; Besirli, C.G. Photoreceptor metabolic reprogramming provides survival advantage in acute stress while causing chronic degeneration. Sci. Rep. 2017, 7, 17863. [Google Scholar] [CrossRef] [Green Version]
- Barben, M.; Ail, D.; Storti, F.; Klee, K.; Schori, C.; Samardzija, M.; Michalakis, S.; Biel, M.; Meneau, I.; Blaser, F.; et al. Hif1a inactivation rescues photoreceptor degeneration induced by a chronic hypoxia-like stress. Cell Death Differ. 2018, 25, 2071–2085. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Guerin, C.J.; Lewis, G.P.; Fisher, S.K.; Anderson, D.H. Recovery of photoreceptor outer segment length and analysis of membrane assembly rates in regenerating primate photoreceptor outer segments. Investig. Ophthalmol. Vis. Sci. 1993, 34, 175–183. [Google Scholar] [PubMed]
- Wang, S.; Linsenmeier, R.A. Hyperoxia improves oxygen consumption in the detached feline retina. Invest. Ophthalmol. Vis. Sci. 2007, 48, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- McMurtrey, R.J. Analytic Models of Oxygen and Nutrient Diffusion, Metabolism Dynamics, and Architecture Optimization in Three-Dimensional Tissue Constructs with Applications and Insights in Cerebral Organoids. Tissue Eng. Part. C Methods 2016, 22, 221–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinstrup, P.; Stahl, N.; Mellergard, P.; Uski, T.; Ungerstedt, U.; Nordstrom, C.H. Intracerebral microdialysis in clinical practice: Baseline values for chemical markers during wakefulness, anesthesia, and neurosurgery. Neurosurgery 2000, 47, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guerrero, A.; Mur-Bonet, G.; Vidal-Jorge, M.; Gandara-Sabatini, D.; Chocron, I.; Cordero, E.; Poca, M.A.; Mullen, K.; Sahuquillo, J. Reappraisal of the reference levels for energy metabolites in the extracellular fluid of the human brain. J. Cereb. Blood Flow Metab. 2017, 37, 2742–2755. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, L.; Pace, R.; D’Arros, C.; Rethore, G.; Guicheux, J.; Le Visage, C.; Weiss, P. Assessing glucose and oxygen diffusion in hydrogels for the rational design of 3D stem cell scaffolds in regenerative medicine. J. Tissue Eng. Regen. Med. 2018, 12, 1238–1246. [Google Scholar] [CrossRef]
- Decembrini, S.; Hoehnel, S.; Brandenberg, N.; Arsenijevic, Y.; Lutolf, M.P. Hydrogel-based milliwell arrays for standardized and scalable retinal organoid cultures. Sci. Rep. 2020, 10, 10275. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Kaya, K.D.; Dong, L.; Swaroop, A. Three-dimensional retinal organoids from mouse pluripotent stem cells mimic in vivo development with enhanced stratification and rod photoreceptor differentiation. Mol. Vis. 2016, 22, 1077–1094. [Google Scholar] [PubMed]
- Gao, L.; Chen, X.; Zeng, Y.; Li, Q.; Zou, T.; Chen, S.; Wu, Q.; Fu, C.; Xu, H.; Yin, Z.Q. Intermittent high oxygen influences the formation of neural retinal tissue from human embryonic stem cells. Sci. Rep. 2016, 6, 29944. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.W.; Hamby, A.M.; Swanson, R.A. Hypoglycemia, brain energetics, and hypoglycemic neuronal death. Glia 2007, 55, 1280–1286. [Google Scholar] [CrossRef]
- Hodgkin, A.L.; Nunn, B.J. Control of light-sensitive current in salamander rods. J. Physiol. 1988, 403, 439–471. [Google Scholar] [CrossRef] [PubMed]
- Frings, S.; Seifert, R.; Godde, M.; Kaupp, U.B. Profoundly different calcium permeation and blockage determine the specific function of distinct cyclic nucleotide-gated channels. Neuron 1995, 15, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Kaupp, U.B. Family of cyclic nucleotide gated ion channels. Curr. Opin. Neurobiol. 1995, 5, 434–442. [Google Scholar] [CrossRef]
- Cervetto, L.; Lagnado, L.; Perry, R.J.; Robinson, D.W.; McNaughton, P.A. Extrusion of calcium from rod outer segments is driven by both sodium and potassium gradients. Nature 1989, 337, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Schneeweis, D.M.; Schnapf, J.L. Photovoltage of rods and cones in the macaque retina. Science 1995, 268, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Demontis, G.C.; Longoni, B.; Barcaro, U.; Cervetto, L. Properties and functional roles of hyperpolarization-gated currents in guinea-pig retinal rods. J. Physiol. 1999, 515, 813–828. [Google Scholar] [CrossRef]
- Demontis, G.C.; Moroni, A.; Gravante, B.; Altomare, C.; Longoni, B.; Cervetto, L.; DiFrancesco, D. Functional characterisation and subcellular localisation of HCN1 channels in rabbit retinal rod photoreceptors. J. Physiol. 2002, 542, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Demontis, G.C.; Gargini, C.; Paoli, T.G.; Cervetto, L. Selective Hcn1 channels inhibition by ivabradine in mouse rod photoreceptors. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1948–1955. [Google Scholar] [CrossRef]
- Cerani, A.; Tetreault, N.; Menard, C.; Lapalme, E.; Patel, C.; Sitaras, N.; Beaudoin, F.; Leboeuf, D.; De Guire, V.; Binet, F.; et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab. 2013, 18, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Tosini, G.; Menaker, M. Circadian rhythms in cultured mammalian retina. Science 1996, 272, 419–421. [Google Scholar] [CrossRef]
- Tosini, G.; Menaker, M. The clock in the mouse retina: Melatonin synthesis and photoreceptor degeneration. Brain Res. 1998, 789, 221–228. [Google Scholar] [CrossRef]
- Robinson, D.W.; Ratto, G.M.; Lagnado, L.; McNaughton, P.A. Temperature dependence of the light response in rat rods. J. Physiol. 1993, 462, 465–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ovando-Roche, P.; West, E.L.; Branch, M.J.; Sampson, R.D.; Fernando, M.; Munro, P.; Georgiadis, A.; Rizzi, M.; Kloc, M.; Naeem, A.; et al. Use of bioreactors for culturing human retinal organoids improves photoreceptor yields. Stem Cell Res. Ther. 2018, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Giandomenico, S.L.; Mierau, S.B.; Gibbons, G.M.; Wenger, L.M.D.; Masullo, L.; Sit, T.; Sutcliffe, M.; Boulanger, J.; Tripodi, M.; Derivery, E.; et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019, 22, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.P.; Goodwin, T.J.; Wolf, D.A. Cell culture for three-dimensional modeling in rotating-wall vessels: An application of simulated microgravity. J. Tissue Cult. Methods 1992, 14, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.G.; Hammond, J.M. Optimized suspension culture: The rotating-wall vessel. Am. J. Physiol. Renal Physiol. 2001, 281, F12–F25. [Google Scholar] [CrossRef]
- Dutt, K.; Scott, M.; Wang, M.; Semple, E.; Sharma, G.P.; Srinivasan, A. Establishment of a human retinal cell line by transfection of SV40 T antigen gene with potential to undergo neuronal differentiation. DNA Cell Biol. 1994, 13, 909–921. [Google Scholar] [CrossRef]
- Dutt, K.; Ezeonu, I.; Scott, M.; Semple, E.; Srinivasan, A. Proto-oncogene expression in cAMP and TPA-mediated neuronal differentiation in a human retinal cell line KGLDMSM. Curr. Eye Res. 1996, 15, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Ezeonu, I.; Derrickson, B.; Dutt, K. Cell fate decisions in a human retinal precursor cell line: Basic fibroblast growth factor- and transforming growth factor-alpha-mediated differentiation. DNA Cell Biol. 2000, 19, 527–537. [Google Scholar] [CrossRef]
- Dutt, K.; Harris-Hooker, S.; Ellerson, D.; Layne, D.; Kumar, R.; Hunt, R. Generation of 3D retina-like structures from a human retinal cell line in a NASA bioreactor. Cell Transplant. 2003, 12, 717–731. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.A.; Bernstein, P.S.; Bok, D.; Turner, J.; Nachtigal, M.; Hunt, R.C. A human retinal pigment epithelial cell line that retains epithelial characteristics after prolonged culture. Investig. Ophthalmol. Vis. Sci. 1995, 36, 955–964. [Google Scholar] [PubMed]
- Hunter, C.J.; Matyas, J.R.; Duncan, N.A. The notochordal cell in the nucleus pulposus: A review in the context of tissue engineering. Tissue Eng. 2003, 9, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmor, M.F. Control of subretinal fluid: Experimental and clinical studies. Eye 1990, 4, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Villarroel, M.; Corraliza, L.; Hernandez, C.; Garcia-Ramirez, M. The retinal pigment epithelium: Something more than a constituent of the blood-retinal barrier--implications for the pathogenesis of diabetic retinopathy. J. Biomed. Biotechnol. 2010, 2010, 190724. [Google Scholar] [CrossRef]
- Jung, Y.H.; Phillips, M.J.; Lee, J.; Xie, R.; Ludwig, A.L.; Chen, G.; Zheng, Q.; Kim, T.J.; Zhang, H.; Barney, P.; et al. 3D Microstructured Scaffolds to Support Photoreceptor Polarization and Maturation. Adv. Mater. 2018, 30, e1803550. [Google Scholar] [CrossRef]
- Lee, I.K.; Ludwig, A.L.; Phillips, M.J.; Lee, J.; Xie, R.; Sajdak, B.S.; Jager, L.D.; Gong, S.; Gamm, D.M.; Ma, Z. Ultrathin micromolded 3D scaffolds for high-density photoreceptor layer reconstruction. Sci. Adv. 2021, 7, eabf0344. [Google Scholar] [CrossRef] [PubMed]
- Demontis, G.C.; Aruta, C.; Comitato, A.; De Marzo, A.; Marigo, V. Functional and molecular characterization of rod-like cells from retinal stem cells derived from the adult ciliary epithelium. PLoS ONE 2012, 7, e33338. [Google Scholar] [CrossRef]
- Li, L.; Zhao, H.; Xie, H.; Akhtar, T.; Yao, Y.; Cai, Y.; Dong, K.; Gu, Y.; Bao, J.; Chen, J.; et al. Electrophysiological characterization of photoreceptor-like cells in human inducible pluripotent stem cell-derived retinal organoids during in vitro maturation. Stem Cells 2021, 39, 959–974. [Google Scholar] [CrossRef]
- Lamb, T.D.; Pugh, E.N., Jr. A quantitative account of the activation steps involved in phototransduction in amphibian photoreceptors. J. Physiol. 1992, 449, 719–758. [Google Scholar] [CrossRef]
- Lamb, T.D.; Pugh, E.N., Jr. Phototransduction, dark adaptation, and rhodopsin regeneration the proctor lecture. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5137–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepko, C. Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci. 2014, 15, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Cepko, C.L. The Determination of Rod and Cone Photoreceptor Fate. Annu. Rev. Vis. Sci. 2015, 1, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Yan, R.T.; He, L.; Zhan, W.; Wang, S.Z. Induction of ectopic retina-like tissue by transgenic expression of neurogenin. PLoS ONE 2015, 10, e0116171. [Google Scholar] [CrossRef] [Green Version]





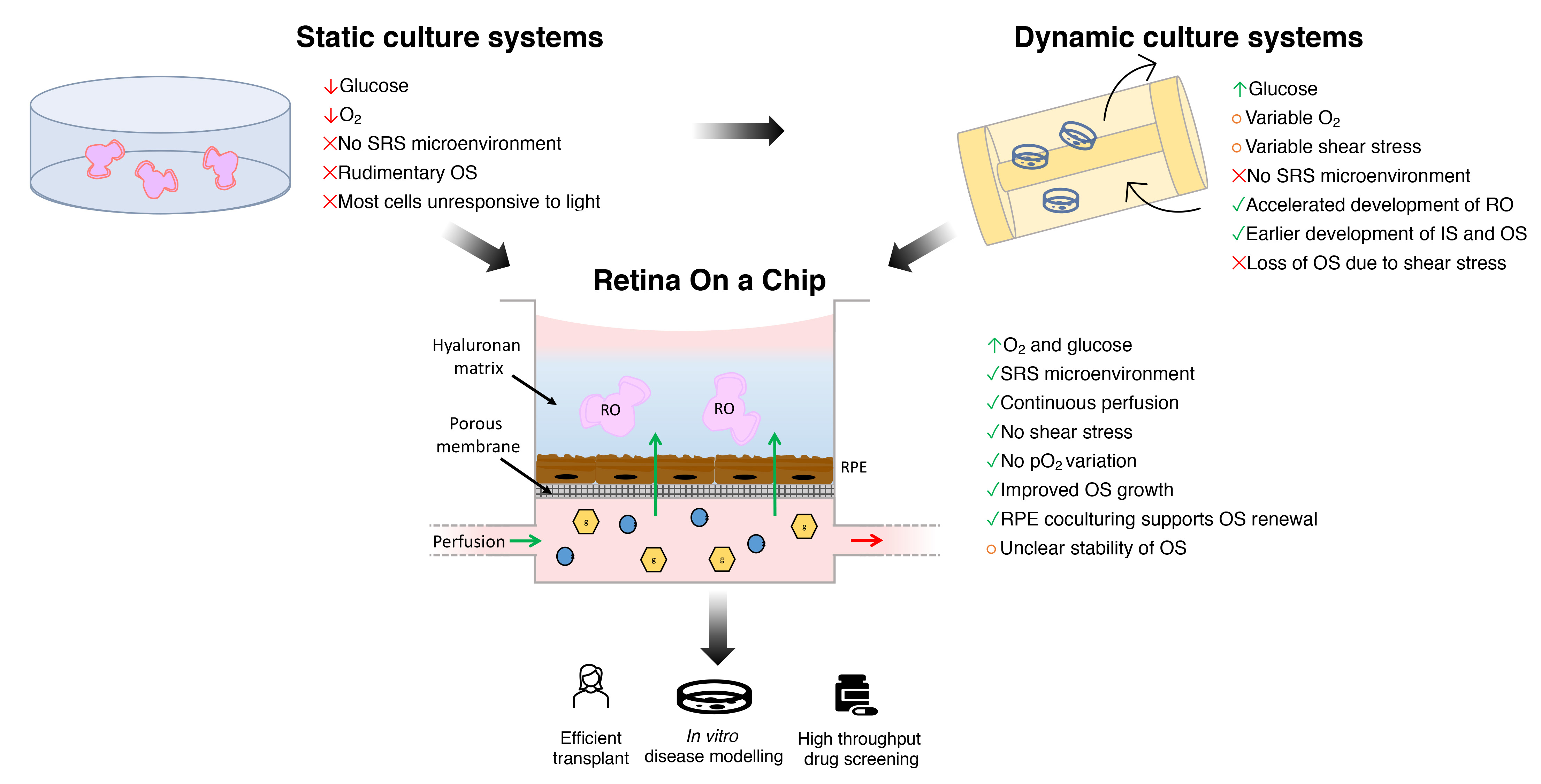{kind=link}
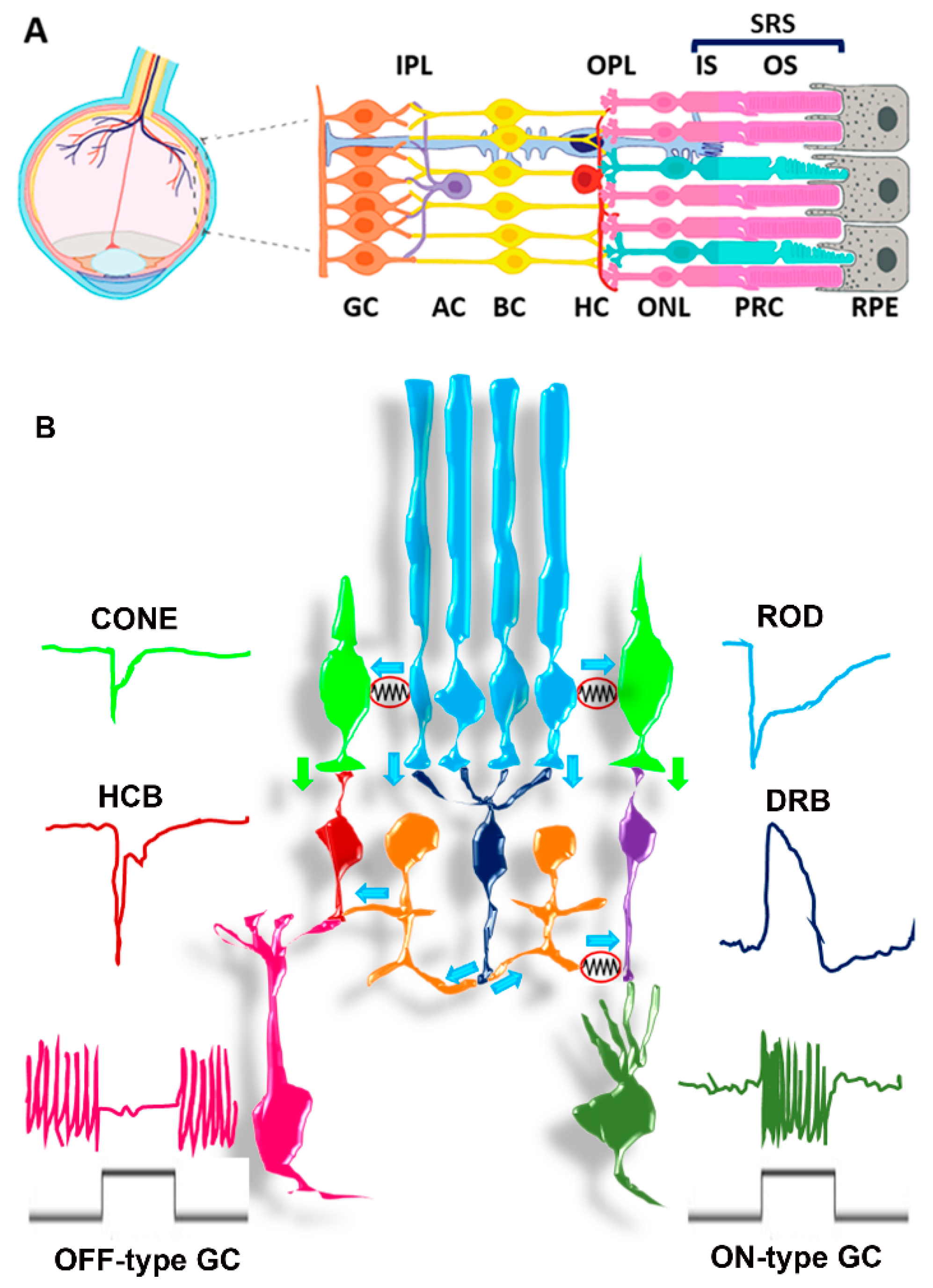{kind=link}
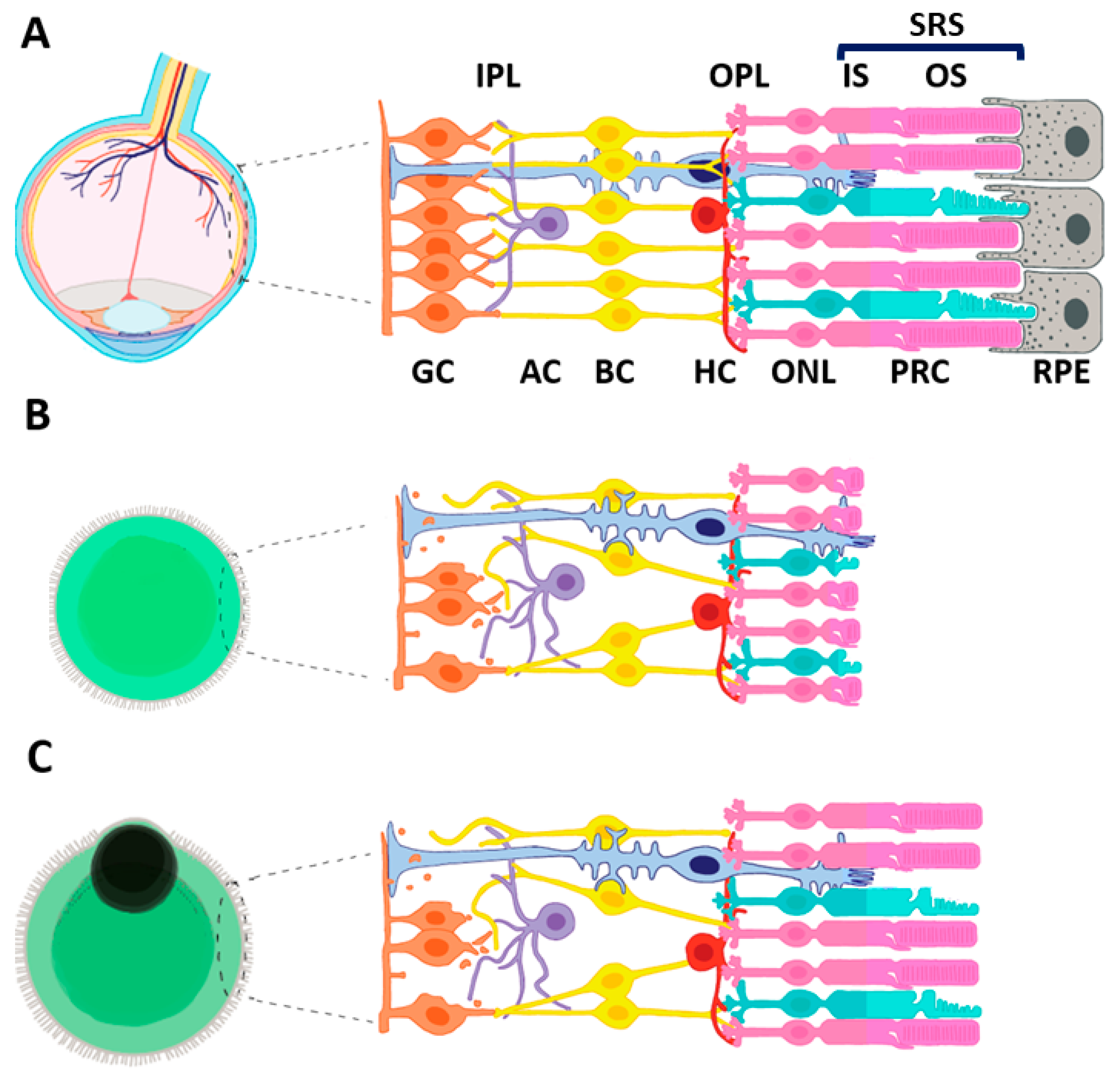{kind=link}
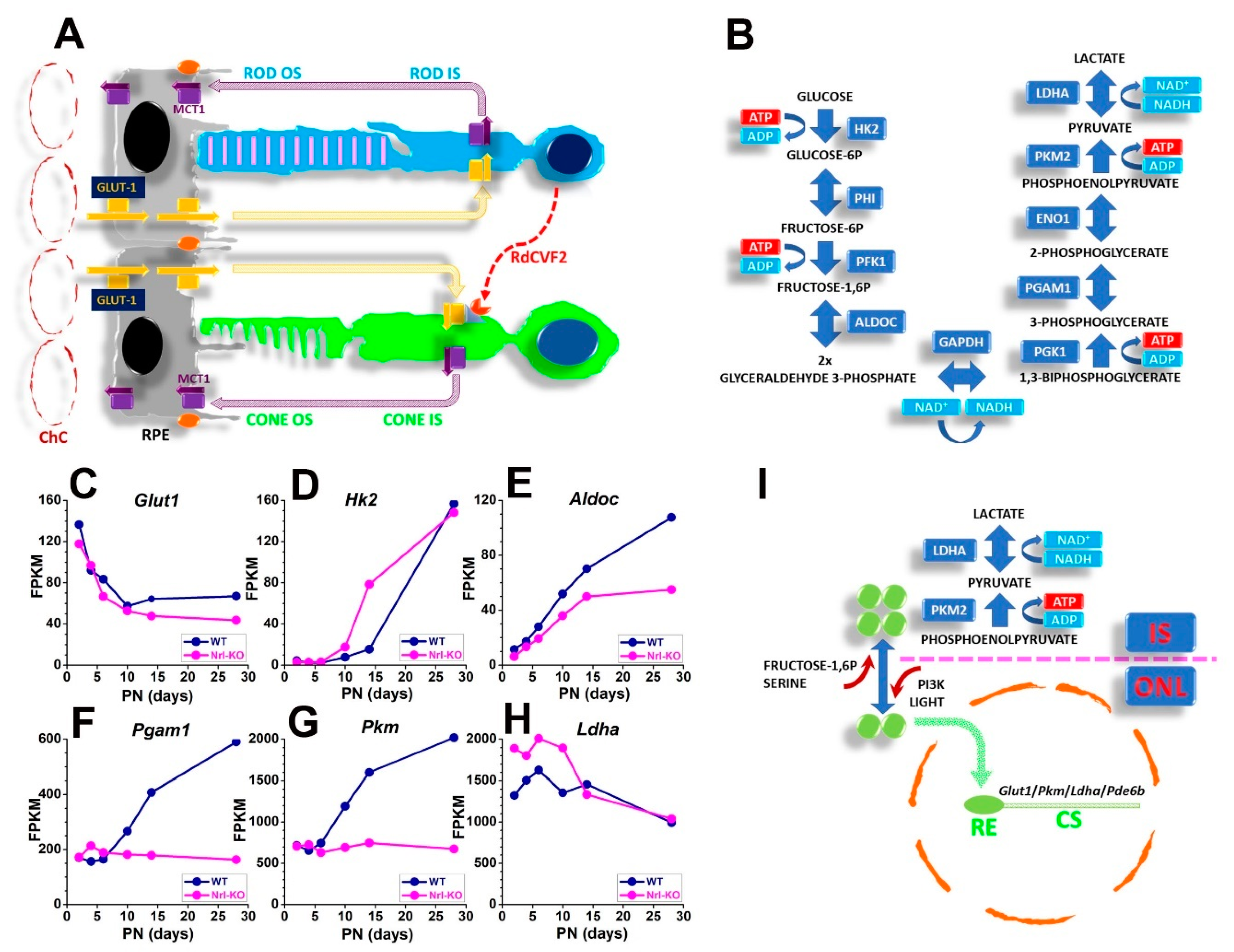{kind=link}
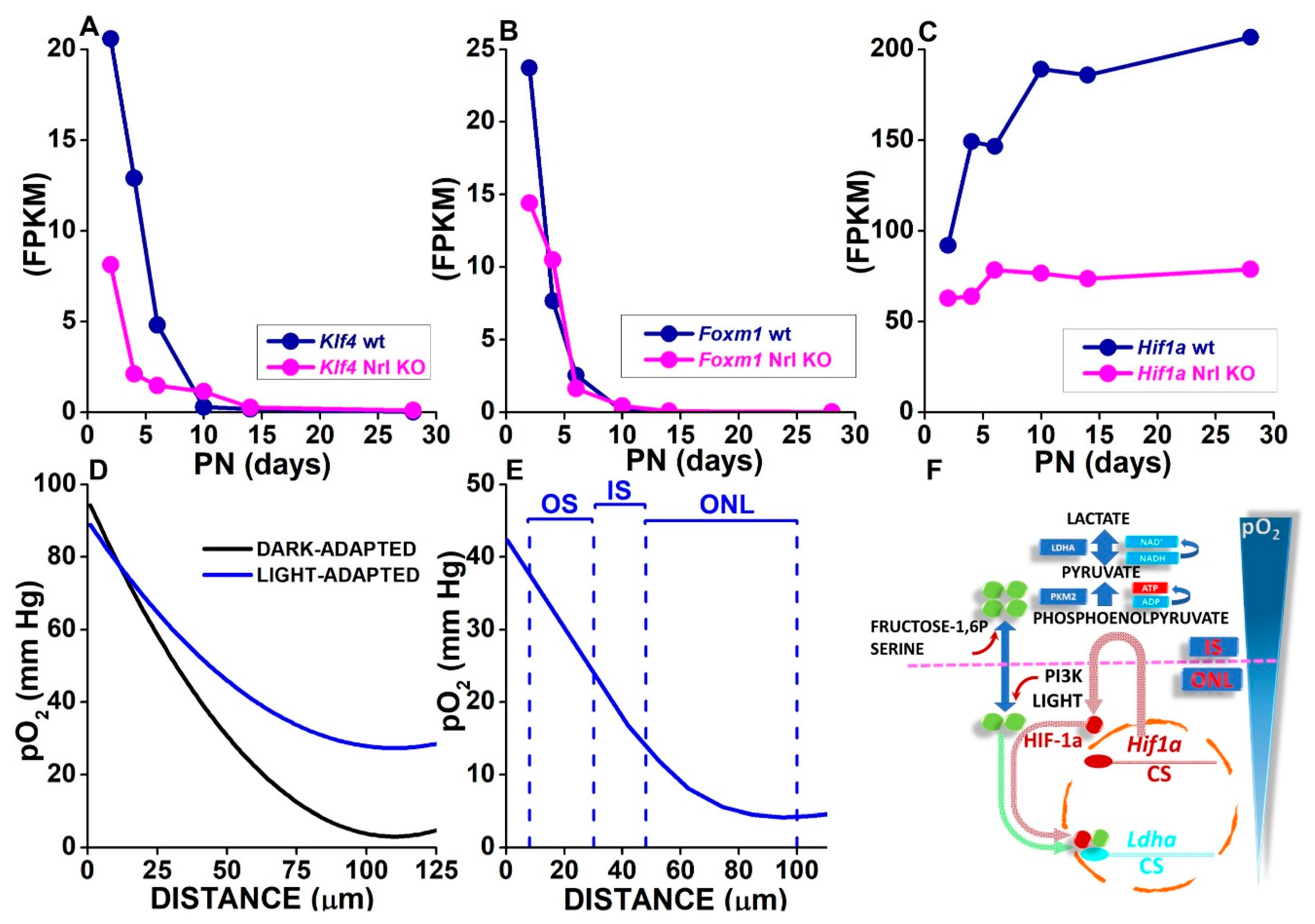{kind=link}
| Diseases Category | Inheritance Mode | Loci | Genes |
|---|---|---|---|
| retinitis pigmentosa | dominant | 23 | 22 |
| retinitis pigmentosa | recessive | 43 | 41 |
| retinitis pigmentosa | x-linked | 5 | 2 |
| subtotal | 71 | 65 | |
| leber congenital amaurosis | dominant | 1 | 1 |
| leber congenital amaurosis | recessive | 13 | 13 |
| subtotal | 14 | 14 | |
| cone or cone-rod dystrophy | dominant | 9 | 5 |
| cone or cone-rod dystrophy | recessive | 17 | 16 |
| cone or cone-rod dystrophy | x-linked | 1 | 0 |
| subtotal | 27 | 21 | |
| macular degeneration | dominant | 14 | 10 |
| macular degeneration | recessive | 4 | 4 |
| subtotal | 18 | 14 | |
| overall total | 130 | 114 |
| Acronym | Full Name |
|---|---|
| ABCA4 | ATP Binding Cassette Subfamily A Member 4 |
| AIF | Apoptosis-Inducing Factor |
| ALDOC | Aldolase Isoform C |
| ARL2BP | ADP Ribosylation Factor Like GTPase 2 Binding Protein |
| ARL6 | ADP-Ribosylation Factor-Like GTPase 6 |
| BMP | Bone Morphogenetic Protein |
| C-KIT | KIT Proto-Oncogene, Receptor Tyrosine Kinase (CD117) |
| C2ORF71 | Photoreceptor Cilium Actin Regulator |
| CACNA1F | Calcium Channel A Subunit Subfamily Xx Isoform 1F (Cav1.4) |
| CACNA2D4 | Calcium Channel A2d Subunit Isoform 4 (Cava2d4) |
| CACNAB2 | Calcium Channel Β Subunit Isoform 2 (Cav2.2) |
| CD24 | Cluster of Differentiation 24 |
| CD44 | Cluster of Differentiation 44 |
| CD73 | Cluster of Differentiation 73 |
| CEP290 | Centrosomal Protein 290 |
| CLDN19 | Claudin 19 |
| CNGA1 | A Subunit Of The Rod Cyclic-Nucleotide-Gated-Channel |
| CNGB1 | Β Subunit of The Rod Cyclic-Nucleotide-Gated-Channel |
| CRB1 | Crumbs Homolog 1 |
| CX3CL1 | C-X3-C Motif Chemokine Receptor 1 Ligand |
| CX3CR1 | C-X3-C Motif Chemokine Receptor 1 |
| FGF1 | Fibroblast Growth Factor 1 |
| FGF9 | Fibroblast Growth Factor 9 |
| FOXM1 | Forkhead Box M1 |
| GLUT1 | Glucose Transporter Isoform 1 |
| GNAT1 | G Protein Subunit A Transducin |
| HCN1 | Hyperpolarization-Activated Cyclic Nucleotide-Gated Isoform 1 |
| HIF-1alpha | Hypoxia Inducible-Factor Subunit Alpha Isoform 1 |
| HIF-1beta | Hypoxia Inducible-Factor Subunit Beta Isoform 1 |
| HK2 | Hexokinase Isoform 2 |
| IGF1 | Insuline-like Growth Factor 1 |
| IMPG1 | Interphotoreceptor Matrix Proteoglycan 1 |
| JAM | Junctional Adhesion Molecules |
| KCNB1 | Potassium Channel Subfamily B Isoform 1 (Kv2.1) |
| KCNV2 | Potassium Channel Subfamily V Isoform 2 (Kv8.2) |
| KLF4 | Kruppel-Like Factor 4 |
| LDHA | Lactic Dehydrogenase A |
| MCT1 | Mono Carboxylate Transporter Isoform 1 |
| MITF | Melanocyte Inducing Transcription Factor |
| Mupp1 | Multiple PDZ Proteins 1 |
| NEUROG1 | Neurongenin1 |
| NEUROG2 | Neurogenin 2 |
| NRL | Neural Retina Leucine Zipper |
| OCLN | Occludin |
| PAX6D | Paired Box 6 Isoform D |
| PDE6A | Phospodiesterase 6A |
| PDE6B | Phospodiesterase 6B |
| PDE6G | Phospodiesterase 6G |
| PGAM1 | Phosphoglyceraldehyde Mutase Isoform 1 |
| Pgk1 | Phosphoglycerate Kinase 1 |
| PKM | Pyruvate Kinase |
| RdCVF | Rod-Derived Cone Viability Factor |
| REEP6 | Receptor Expression Enhancing Protein 6 |
| RETGC1 | Retinal Guanylate Cyclase 1 |
| RHO | Rhodopsin |
| RP1 | Oxygen- Regulated Prein-1 (ORP1) |
| RP2 | Retinitis Pigmentosa 2 |
| RPE65 | Retinoid Isomerohydrolase 65 |
| RPGR | Retinitis Pigmentosa GTPase Regulator |
| RS1 | Retinoschisin 1 |
| SAG | S-Antigen (Arrestin) |
| SSEA4 | Stage-Specific Embryonic Antigen-4 |
| TMEM16B | Transmembrane Channel Family 16 Isoform B |
| TTC8 | Tetratricopeptide Repeat Domain-Containing Protein 8 |
| VEGF | Vascular Endothelial Growth Factor |
| VHL | Von Hippel Lindau |
| VSX2 | Visual System Homeobox 2 |
| WNT | Wingless and Int1 |
| ZO1 | Zonula Occludens 1 |
| Medium Volume (ml) | D (Diffusion Coefficient) | C0 (pO2 in Air × Solubility Coefficient) | Q (O2 Consumption Rate × Retina Volume) | LMAX (µm) | Medium Height (mm) |
|---|---|---|---|---|---|
| 2 (Dark) | 2.8 × 10−9 m2 s−1 | 2.09 × 10−4 Moles L−1 | 47 µMoles O2 s−1 L−1 | 158 | 2830 |
| 2 (Light) | 2.8 × 10−9 m2 s−1 | 2.09 × 10−4 Moles L−1 | 20 µMoles O2 s−1 L−1 | 241 | 2830 |
| 1 (Dark) | 2.8 × 10−9 m2 s−1 | 2.09 × 10−4 Moles L−1 | 47 µMoles O2 s−1 L−1 | 158 | 2610 |
| 1 (Light) | 2.8 × 10−9 m2 s−1 | 2.09 × 10−4 Moles L−1 | 20 µMoles O2 s−1 L−1 | 241 | 2610 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreazzoli, M.; Barravecchia, I.; De Cesari, C.; Angeloni, D.; Demontis, G.C. Inducible Pluripotent Stem Cells to Model and Treat Inherited Degenerative Diseases of the Outer Retina: 3D-Organoids Limitations and Bioengineering Solutions. Cells 2021, 10, 2489. https://doi.org/10.3390/cells10092489
Andreazzoli M, Barravecchia I, De Cesari C, Angeloni D, Demontis GC. Inducible Pluripotent Stem Cells to Model and Treat Inherited Degenerative Diseases of the Outer Retina: 3D-Organoids Limitations and Bioengineering Solutions. Cells. 2021; 10(9):2489. https://doi.org/10.3390/cells10092489
Chicago/Turabian StyleAndreazzoli, Massimiliano, Ivana Barravecchia, Chiara De Cesari, Debora Angeloni, and Gian Carlo Demontis. 2021. "Inducible Pluripotent Stem Cells to Model and Treat Inherited Degenerative Diseases of the Outer Retina: 3D-Organoids Limitations and Bioengineering Solutions" Cells 10, no. 9: 2489. https://doi.org/10.3390/cells10092489
APA StyleAndreazzoli, M., Barravecchia, I., De Cesari, C., Angeloni, D., & Demontis, G. C. (2021). Inducible Pluripotent Stem Cells to Model and Treat Inherited Degenerative Diseases of the Outer Retina: 3D-Organoids Limitations and Bioengineering Solutions. Cells, 10(9), 2489. https://doi.org/10.3390/cells10092489