
Glucagon Potentiates Insulin Secretion Via β-Cell GCGR at Physiological Concentrations of Glucose

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. IPGTT
2.3. Isolation of Mouse Islets and Dissociation of the Islets into Individual β-Cells or Cell Clusters
2.4. Detection of Hormone Secretion from Islets
2.5. Electrophysiology and Membrane Capacitance Recording
2.6. Fluorescence Imaging of cAMP
2.7. RNA Extraction and Quantitative Real-Time PCR
2.8. Pancreatic Sections and Immunohistochemistry
2.9. Statistics
3. Results
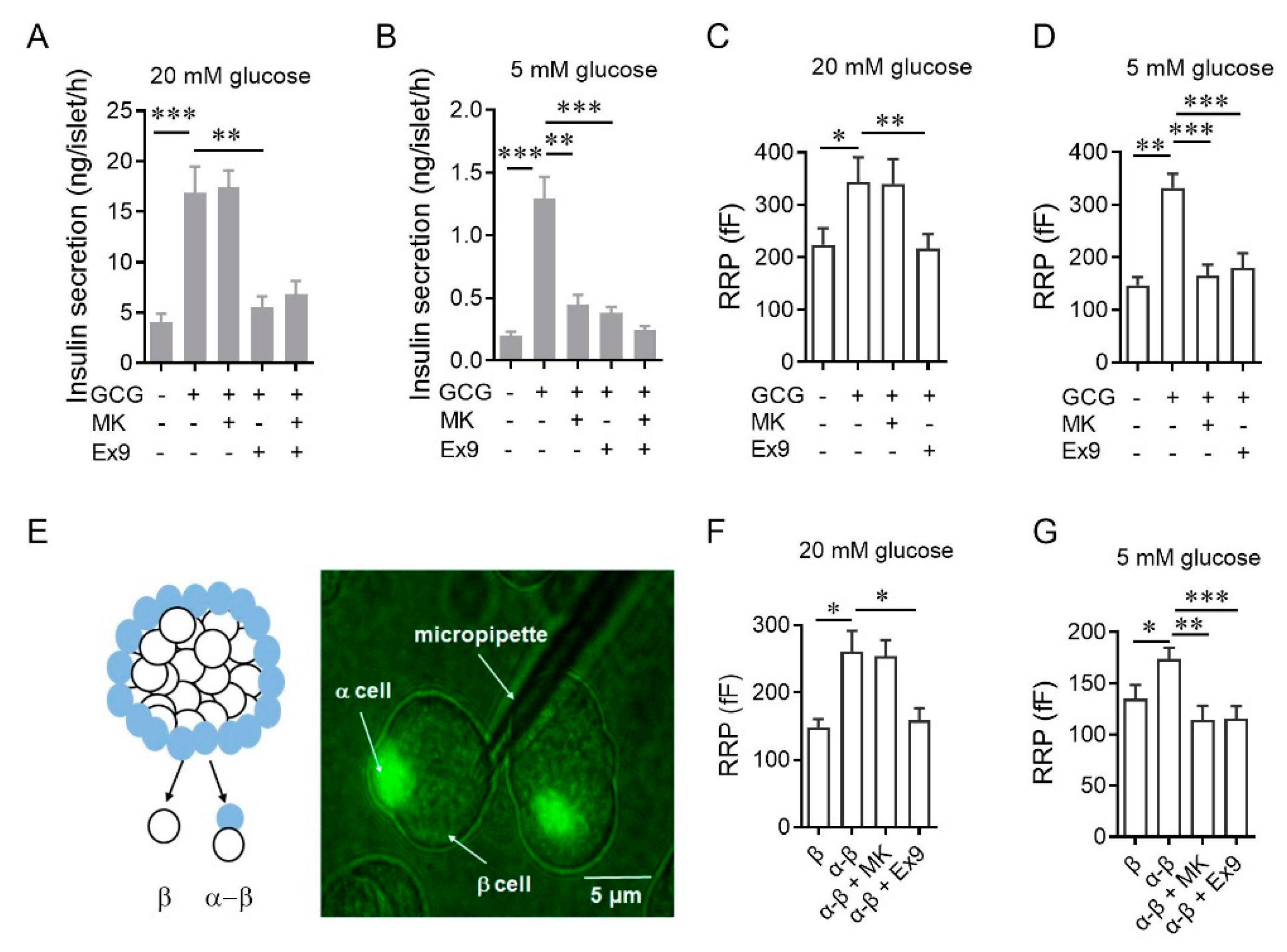
3.1. Glucagon Potentiates Insulin Secretion Via β-Cell GCGR at Physiological Concentrations of Glucose
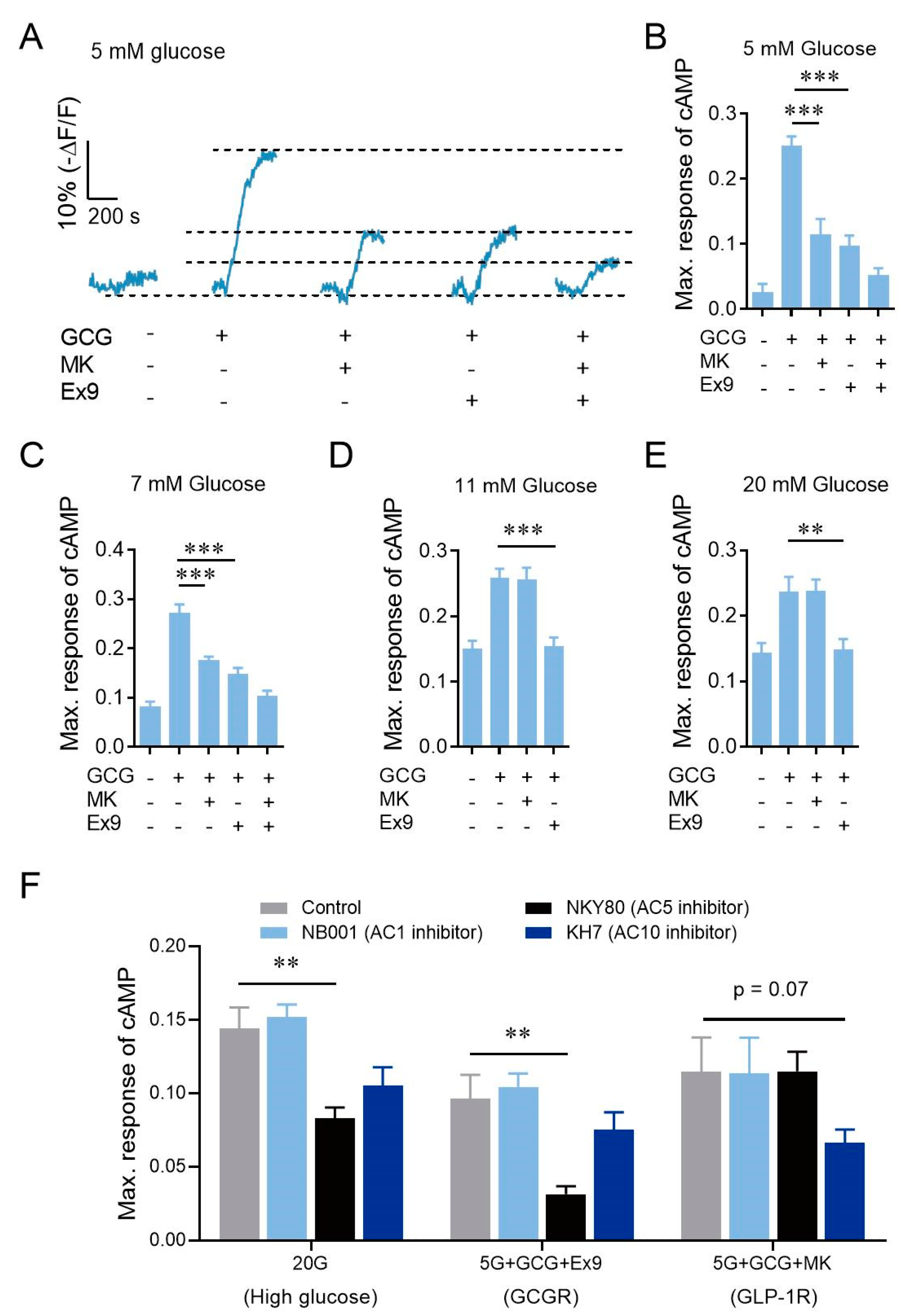
3.2. High Glucose Concentration Evokes cAMP Elevation via AC5 in β-Cells, Which Is Also Downstream of GCGR Activation, thus Bypassing GCGR in Promoting Insulin Secretion
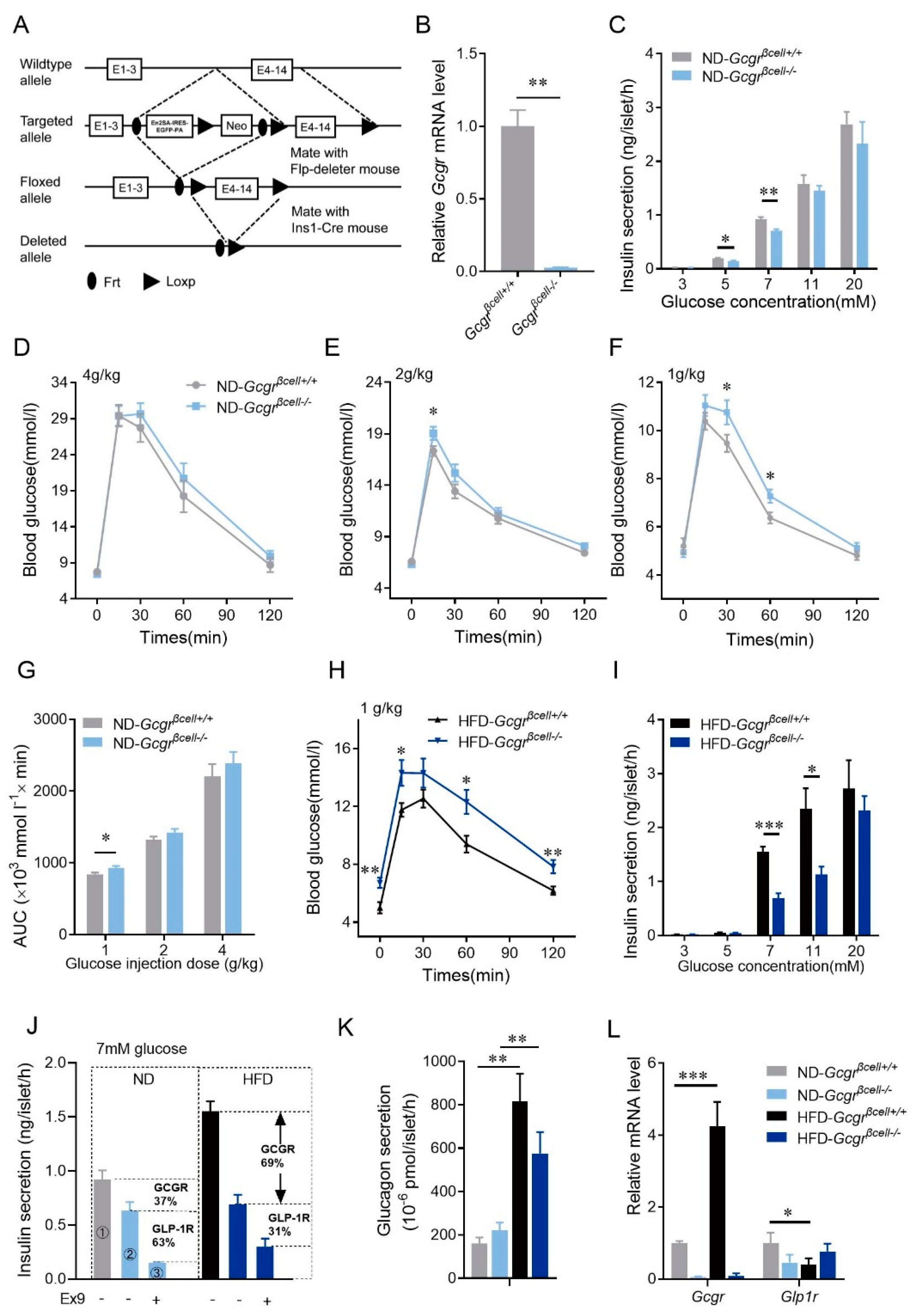
3.3. The Physiological Function of β-Cell GCGR Becomes Upregulated and More Essential to Glucose Homeostasis in Mice Fed HFD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Unger, R.H.; Aguilar-Parada, E.; Müller, W.A.; Eisentraut, A.M. Studies of pancreatic alpha cell function in normal and diabetic subjects. J. Clin. Investig. 1970, 49, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perley, M.J.; Kipnis, D.M. Plasma insulin responses to oral and intravenous glucose: Studies in normal and diabetic subjects. J. Clin. Investig. 1967, 46, 1954. [Google Scholar] [CrossRef] [PubMed]
- Dupre, J.; Ross, S.; Watson, D.; Brown, J. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef]
- Schmidt, W.; Siegel, E.; Creutzfeldt, W. Glucagon-like peptide-1 but not glucagon-like peptide-2 stimulates insulin release from isolated rat pancreatic islets. Diabetologia 1985, 28, 704–707. [Google Scholar] [CrossRef]
- Kreymann, B.; Williams, G.; Ghatei, M.A.; Bloom, S.R. Glucagon-like peptide-1 7–36: A physiological incretin in man. Lancet 1987, 2, 1300–1304. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved Incretin Activity of Glucagon-Like Peptide-1 [7-36 Amide] but Not of Synthetic Human Gastric-Inhibitory Polypeptide in Patients with Type-2 Diabetes-Mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.P.; An, Z.; Wagner, C.; Lewis, A.G.; Cohen, E.B.; Li, B.; Mahbod, P.; Sandoval, D.; Perez-Tilve, D.; Tamarina, N.; et al. The role of beta cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab. 2014, 19, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Wideman, R.D.; Irene, L.; Webber, T.D.; Verchere, C.B.; Johnson, J.D.; Cheung, A.T.; Kieffer, T.J. Improving function and survival of pancreatic islets by endogenous production of glucagon-like peptide 1 (GLP-1). Proc. Natl. Acad. Sci. USA 2006, 103, 13468–13473. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Burcelin, R. GLP-1 effects on islets: Hormonal, neuronal, or paracrine? Diabetes Care 2013, 36, S145–S148. [Google Scholar] [CrossRef] [Green Version]
- Whalley, N.M.; Pritchard, L.E.; Smith, D.M.; White, A. Processing of proglucagon to GLP-1 in pancreatic alpha-cells: Is this a paracrine mechanism enabling GLP-1 to act on beta-cells? J. Endocrinol. 2011, 211, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Masur, K.; Tibaduiza, E.C.; Chen, C.; Ligon, B.; Beinborn, M. Basal receptor activation by locally produced glucagon-like peptide-1 contributes to maintaining beta-cell function. Mol. Endocrinol. 2005, 19, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Kilimnik, G.; Kim, A.; Steiner, D.F.; Friedman, T.C.; Hara, M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic alpha-cells in mouse models of ss-cell regeneration. Islets 2010, 2, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, A.P.; Sorrell, J.E.; Haller, A.; Roelofs, K.; Hutch, C.R.; Kim, K.S.; Gutierrez-Aguilar, R.; Li, B.; Drucker, D.J.; D’Alessio, D.A.; et al. The Role of Pancreatic Preproglucagon in Glucose Homeostasis in Mice. Cell Metab. 2017, 25, 927–934.e923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traub, S.; Meier, D.T.; Schulze, F.; Dror, E.; Nordmann, T.M.; Goetz, N.; Koch, N.; Dalmas, E.; Stawiski, M.; Makshana, V.; et al. Pancreatic alpha Cell-Derived Glucagon-Related Peptides Are Required for beta Cell Adaptation and Glucose Homeostasis. Cell Rep. 2017, 18, 3192–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, P.; Lupi, R.; Bugliani, M.; Kirkpatrick, C.L.; Sebastiani, G.; Grieco, F.A.; Del Guerra, S.; D’Aleo, V.; Piro, S.; Marselli, L.; et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 2012, 55, 3262–3272. [Google Scholar] [CrossRef] [Green Version]
- Samols, E.; Marri, G.; Marks, V. Promotion of insulin secretion by glucagon. Lancet 1965, 286, 415–416. [Google Scholar] [CrossRef]
- Moens, K.; Flamez, D.; Van Schravendijk, C.; Ling, Z.; Pipeleers, D.; Schuit, F. Dual glucagon recognition by pancreatic beta-cells via glucagon and glucagon-like peptide 1 receptors. Diabetes 1998, 47, 66–72. [Google Scholar] [CrossRef]
- Svendsen, B.; Larsen, O.; Gabe, M.B.N.; Christiansen, C.B.; Rosenkilde, M.M.; Drucker, D.J.; Holst, J.J. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep. 2018, 25, 1127–1134.e1122. [Google Scholar] [CrossRef] [Green Version]
- Capozzi, M.E.; Svendsen, B.; Encisco, S.E.; Lewandowski, S.L.; Martin, M.D.; Lin, H.; Jaffe, J.L.; Coch, R.W.; Haldeman, J.M.; MacDonald, P.E.; et al. beta Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 2019, 4, e126742. [Google Scholar] [CrossRef]
- Capozzi, M.E.; Wait, J.B.; Koech, J.; Gordon, A.N.; Coch, R.W.; Svendsen, B.; Finan, B.; D’Alessio, D.A.; Campbell, J.E. Glucagon lowers glycemia when beta-cells are active. JCI Insight 2019, 5, e129954. [Google Scholar] [CrossRef] [Green Version]
- Thorens, B.; Tarussio, D.; Maestro, M.A.; Rovira, M.; Heikkila, E.; Ferrer, J. Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia 2015, 58, 558–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, K.; Du, W.; Lu, J.; Li, F.; Yang, L.; Xue, Y.; Hille, B.; Chen, L. Alterations of the Ca2+ signaling pathway in pancreatic beta-cells isolated from db/db mice. Protein Cell 2014, 5, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Guo, J.; Candelore, M.R.; Liang, R.; Miller, C.; Dallas-Yang, Q.; Jiang, G.; McCann, P.E.; Qureshi, S.A.; Tong, X. Discovery of a Novel Glucagon Receptor Antagonist N-[(4-{(1 S)-1-[3-(3, 5-Dichlorophenyl)-5-(6-methoxynaphthalen-2-yl)-1 H-pyrazol-1-yl] ethyl} phenyl) carbonyl]-β-alanine (MK-0893) for the Treatment of Type II Diabetes. J. Med. Chem. 2012, 55, 6137–6148. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, R.H.; Richard, J.E.; Eerola, K.; Ferreras, L.L.; Nordbeck, E.B.; Hansson, C.; Nissbrandt, H.; Berqquist, F.; Gribble, F.M.; Reimann, F. Glucagon-like peptide-1 and its analogues act in the dorsal raphe and modulate central serotonin to reduce appetite and body weight. Diabetes 2017, 66, 1062–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marinis, Y.Z.; Salehi, A.; Ward, C.E.; Zhang, Q.; Abdulkader, F.; Bengtsson, M.; Braha, O.; Braun, M.; Ramracheya, R.; Amisten, S. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N-and L-type Ca2+ channel-dependent exocytosis. Cell Metab. 2010, 11, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Misler, S. Amperometric detection of quantal secretion from patch-clamped rat pancreatic beta-cells. J. Biol. Chem. 1996, 271, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Dou, H.Q.; Xu, Y.F.; Sun, J.P.; Shang, S.; Guo, S.; Zheng, L.H.; Chen, C.C.; Bruce, I.C.; Yu, X.; Zhou, Z. Thiopental-induced insulin secretion via activation of IP3-sensitive calcium stores in rat pancreatic beta-cells. Am. J. Physiol. Cell Physiol. 2012, 302, C796–C803. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.; Zhang, Y.; Roth, R.H.; Zhang, J.F.; Mo, A.; Tenner, B.; Huganir, R.L.; Zhang, J. Single-fluorophore biosensors for sensitive and multiplexed detection of signalling activities. Nat Cell Biol 2018, 20, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Alford, S.C.; Abdelfattah, A.S.; Ding, Y.; Campbell, R.E. A fluorogenic red fluorescent protein heterodimer. Chem. Biol. 2012, 19, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Han, C.; Zhu, W.; Wu, Z.; Liu, Y.; Chen, L. An optical method to evaluate both mass and functional competence of pancreatic α-and β-cells. J. Cell Sci. 2016, 129, 2462–2471. [Google Scholar] [CrossRef] [Green Version]
- Rorsman, P.; Renstrom, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef]
- Quoix, N.; Cheng-Xue, R.; Guiot, Y.; Herrera, P.L.; Henquin, J.C.; Gilon, P. The GluCre-ROSA26EYFP mouse: A new model for easy identification of living pancreatic alpha-cells. FEBS Lett. 2007, 581, 4235–4240. [Google Scholar] [CrossRef]
- Moens, K.; Berger, V.; Ahn, J.M.; Van Schravendijk, C.; Hruby, V.J.; Pipeleers, D.; Schuit, F. Assessment of the role of interstitial glucagon in the acute glucose secretory responsiveness of in situ pancreatic beta-cells. Diabetes 2002, 51, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, H.; Winzell, M.S.; Brand, C.L.; Fosgerau, K.; Gelling, R.W.; Nishimura, E.; Ahren, B. Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes 2006, 55, 3463–3469. [Google Scholar] [CrossRef] [Green Version]
- Vuguin, P.M.; Kedees, M.H.; Cui, L.; Guz, Y.; Gelling, R.W.; Nejathaim, M.; Charron, M.J.; Teitelman, G. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology 2006, 147, 3995–4006. [Google Scholar] [CrossRef]
- Longuet, C.; Robledo, A.M.; Dean, E.D.; Dai, C.H.; Ali, S.; McGuinness, I.; de Chavez, V.; Vuguin, P.M.; Charron, M.J.; Powers, A.C.; et al. Liver-Specific Disruption of the Murine Glucagon Receptor Produces alpha-Cell Hyperplasia Evidence for a Circulating alpha-Cell Growth Factor. Diabetes 2013, 62, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Hodson, D.J.; Mitchell, R.K.; Marselli, L.; Pullen, T.J.; Brias, S.G.; Semplici, F.; Everett, K.L.; Cooper, D.M.; Bugliani, M.; Marchetti, P. ADCY5 couples glucose to insulin secretion in human islets. Diabetes 2014, 63, 3009–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leech, C.A.; Castonguay, M.A.; Habener, J.F. Expression of adenylyl cyclase subtypes in pancreatic β-cells. Biochem. Biophys. Res. Commun. 1999, 254, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents-SI. Nat. Chem. Biol. 2009, 5, 749–757. [Google Scholar] [CrossRef]
- Sadry, S.A.; Drucker, D.J. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nat. Rev. Endocrinol. 2013, 9, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.A.; McCullough, K.A.; Field, B.C.; Minnion, J.S.; Martin, N.M.; Ghatei, M.A.; Bloom, S.R. Glucagon and GLP-1 inhibit food intake and increase c-fos expression in similar appetite regulating centres in the brainstem and amygdala. Int. J. Obes. 2013, 37, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G.; et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissova, M.; Fowler, M.J.; Nicholson, W.E.; Chu, A.; Hirshberg, B.; Harlan, D.M.; Powers, A.C. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 2005, 53, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Diaz, R.; Molano, R.D.; Weitz, J.R.; Abdulreda, M.H.; Berman, D.M.; Leibiger, B.; Leibiger, I.B.; Kenyon, N.S.; Ricordi, C.; Pileggi, A.; et al. Paracrine Interactions within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab. 2018, 27, 549–558.e544. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Reagent | Source | Identifier |
|---|---|---|
| glucagon | Sigma | G2044 |
| MK0893 | Medchem Express | HY-50663 |
| Exendin 9-39 | Sigma | E7269 |
| Reagent | Source | Identifier |
|---|---|---|
| NB001 | MedChemExpress | HY-14425 |
| NKY80 | Cayman | 17,777 |
| KH7 | Cayman | 13,243 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Han, C.; Zhu, W.; Yang, G.; Peng, X.; Mehta, S.; Zhang, J.; Chen, L.; Liu, Y. Glucagon Potentiates Insulin Secretion Via β-Cell GCGR at Physiological Concentrations of Glucose. Cells 2021, 10, 2495. https://doi.org/10.3390/cells10092495
Zhang Y, Han C, Zhu W, Yang G, Peng X, Mehta S, Zhang J, Chen L, Liu Y. Glucagon Potentiates Insulin Secretion Via β-Cell GCGR at Physiological Concentrations of Glucose. Cells. 2021; 10(9):2495. https://doi.org/10.3390/cells10092495
Chicago/Turabian StyleZhang, Yulin, Chengsheng Han, Wenzhen Zhu, Guoyi Yang, Xiaohong Peng, Sohum Mehta, Jin Zhang, Liangyi Chen, and Yanmei Liu. 2021. "Glucagon Potentiates Insulin Secretion Via β-Cell GCGR at Physiological Concentrations of Glucose" Cells 10, no. 9: 2495. https://doi.org/10.3390/cells10092495
APA StyleZhang, Y., Han, C., Zhu, W., Yang, G., Peng, X., Mehta, S., Zhang, J., Chen, L., & Liu, Y. (2021). Glucagon Potentiates Insulin Secretion Via β-Cell GCGR at Physiological Concentrations of Glucose. Cells, 10(9), 2495. https://doi.org/10.3390/cells10092495